

Abstract
Meningococcal factor H-binding protein (fHbp) is a promising vaccine antigen. Previous studies described three fHbp antigenic variant groups and identified amino acid residues between 100 and 255 as important targets of variant-specific bactericidal antibodies. We investigated residues affecting expression of an epitope recognized by a murine IgG2a anti-fHbp mAb, designated JAR 4, which cross-reacted with fHbps in variant groups 1 or 2 (95% of strains), and elicited human complement-mediated, cooperative bactericidal activity with other non-bactericidal anti-fHbp mAbs with epitopes involving residues between 121 and 216. From filamentous bacteriophage libraries containing random peptides that were recognized by JAR 4, we identified a consensus tripeptide, DHK, that matched residues 25-27 in the N-terminal domain of fHbp. Since DHK was present in both JAR 4-reactive and non-reactive fHbps, the tripeptide was necessary but not sufficient for reactivity. Based on site-directed mutagenesis studies, the JAR 4 epitope could either be knocked out of a reactive variant 1 fHbp, or introduced into a non-reactive variant 3 protein. Collectively, the data indicated that the JAR 4 epitope was discontinuous and involved DHK residues beginning at position 25; YGN residues beginning at position 57; and a KDN tripeptide that was present in variant 3 proteins beginning at position 67 that negatively affected expression of the epitope. Thus, the region of fHbp encompassing residues 25 to 59 in the N-terminal domain is important for eliciting antibodies that can cooperate with other anti-fHbp antibodies for cross-reactive bactericidal activity against strains expressing fHbp from different antigenic variant groups.
Keywords: Neisseria meningitidis, fHbp, mAb epitope, peptide phage display, vaccine, bactericidal
Introduction
Meningococcal factor H-binding protein (fHbp) is a promising vaccine candidate that is part of two vaccines in clinical development (Jiang et al., 2008; Rappuoli, 2008). The protein binds factor H (fH) (Granoff et al., 2009; Madico et al., 2006), which is a down-regulatory molecule in the complement pathway (Schneider et al., 2007). The presence of fH on the Neisseria meningitidis bacterial surface is critical for the organism to circumvent innate host defenses (Madico et al., 2006; Schneider et al., 2006; Welsch et al., 2008). In the absence of bound fH, the organism becomes susceptible to unregulated alternative complement activation and bacteriolysis (Granoff et al., 2009; Seib et al., 2008; Welsch et al., 2008). Recently, binding of fH was demonstrated to be specific for human fH (low for chimpanzee and negligible for baboon and rhesus monkey), which adds to a list of mechanisms by which N. meningitidis only infects humans (Granoff et al., 2009). Antibodies against fHbp both activate the classical complement pathway and also block binding of fH to the surface of the bacteria (Beernink et al., 2008; Welsch et al., 2008).
Among different strains of N. meningitidis, fHbp exists in several antigenic variant groups based on antibody cross-reactivity and amino acid sequence identity. These fHbp groups have been designated variant 1, 2 or 3 by Masignani et al (Masignani et al., 2003), or sub-families A and B by Fletcher et al (Fletcher et al., 2004). Sub-family A includes strains with fHbp in the variant 2 or 3 groups as defined by Masignani, and sub-family B includes strains with fHbp in the variant 1 group. JAR 4 is an IgG2a mAb that was isolated from a mouse immunized with recombinant fHbp in the variant 1 group (Welsch et al., 2004). The mAb was not bactericidal with human complement when tested alone, even against strains with relatively high fHbp expression (Welsch et al., 2008; Welsch et al., 2004). However, the mAb activated C3b deposition on strains expressing fHbp in the variant 1 or 2 groups (Welsch et al., 2004) and elicited cooperative, complement-mediated bactericidal activity with other non-bactericidal mAbs specific for fHbp in the variant 1 or 2 groups (Beernink et al., 2008; Welsch et al., 2008).
The reasons why JAR 4 individually was not bactericidal are not understood. JAR 4 did not inhibit binding of fH (Beernink et al., 2008; Welsch et al., 2008). For anti-fHbp mAbs that inhibited fH binding, the absence of bound fH on the bacterial surface would be expected to result in unregulated C3 activation and enhance susceptibility to bactericidal activity (Granoff et al., 2009)(See also Discussion section). The lack of JAR 4 bactericidal activity also may be the result of insufficient amounts of JAR 4 immune complex at the bacterial surface to engage C1q and activate the classical complement pathway (Beernink et al., 2008; Welsch et al., 2008). In contrast, the combination of JAR 4 along with other anti-fHbp mAbs may provide sufficient immune complex.
In a previous study, we mapped the locations of residues involved in the epitopes recognized by eight anti-fHbp mAbs using a combination of sequence alignments and site-specific mutagenesis (Beernink et al., 2008). However, this approach was not successful for mapping the JAR 4 epitope. The objective of the present study was to identify amino acid residues in fHbp affecting the JAR 4 epitope. The long-term goals were to identify the portion of the fHbp molecular capable of eliciting cross-reactive bactericidal antibodies against strains with fHbp from antigenic groups and to increase our understanding of the basis of cooperative cross-reactive anti-fHbp mAb bactericidal activity.
Experimental
Materials and Methods
Gene cloning and site-specific mutagenesis
The methods used for cloning of fHbp variant 1, 2 or 3 in the expression plasmid pET21b (Novagen, Inc., Madison, WI) were described previously (Beernink et al., 2008; Masignani et al., 2003). Plasmids encoding fHbp with single or multiple amino acid substitutions were generated using the QuikChange II kit (Stratagene, La Jolla, CA) and the manufacturer’s protocols. The mutagenesis reactions were performed using 10 ng of plasmid template and a PTC-200 thermal cycler (MJ Research, Waltham, MA). The forward mutagenic primers were: MC58 D25A 5’-AACCGCACCGCTCGCCCATAAAGACAAAGG-3’; MC58 H26A 5’-AACCGCACCGCTCGACGCTAAAGACAAAGGTTTGC-3’; MC58 K27A 5’-GCACCGCTCGACCATGCAGACAAAGGTTTGCAG-3’; MC58 ins KDN 5’-CTTATGGAAACGGTGACAAAGACAACAGCCTCAATACGGGC-3’; M1239 ΔKDN 5’-TTCAAAGCCGGCGACAGCCTCAACACGG-3’; M1239 FKA->YGN CACAAGGTGCGGAAAAAACTTACGGAAACGGCGACAGCCTCAACACGGG-3’, in which the underlined sequences denote mutated codons. The reverse primers were the respective antiparallel sequences. All oligonucleotides were synthesized commercially (Integrated DNA Technologies, Coralville, IA). Plasmids encoding wildtype or mutant fHbp were verified by DNA sequence determination (Davis Sequencing, Davis, CA) using primers described previously (Masignani et al., 2003).
Protein purification
Recombinant fHbps representative of the variant 1, 2 and 3 groups (cloned from the genes from strains MC58, 8047 and M1239; Genbank accession numbers NC_003112, FJ422922 and DQ523569, respectively) were expressed with C-terminal hexahistidine tags in E. coli strain BL21(DE3). Cultures were grown at 37 °C in Super Broth (30 g/l Bacto-tryptone (BD Biosciences, San Jose, CA), 20 g/l yeast extract (BD Biosciences), 10 g/l MOPS (3-N-morpholinopropanesulfonic acid; Sigma-Aldrich, St. Louis, MO), pH adjusted to 7.0 with NaOH). Once the cultures reached an optical density at 600 nm of 0.6, fHbp expression was induced with 0.5 mM IPTG for 3 h. The proteins were purified by metal chelate chromatography as described previously (Beernink and Granoff, 2008), dialyzed against PBS (Roche Applied Science, Indianapolis, IN), sterilized using 0.45 μm syringe-tip filters (Millipore, Billerica, MA) and stored at 4 °C prior to use.
Phage library preparation and screening
Peptides binding to JAR 4 mAb were selected by panning four phage libraries constructed in the two-gene/phagemid vector pC89 (Felici et al., 1991) by cassette mutagenesis. The libraries carried random inserts encoding peptides of various sizes fused into the N-terminal region of the major coat protein (pVIII) of filamentous phage. The pVIII-9aa and pVIII-12aa libraries were composed of random 9-mers and 12-mers, respectively, whereas the pVIII-9aa.Cys and pVIII-Cys.Cys libraries had random inserts, each containing two cysteine residues (Luzzago and Felici, 1998).
Specific phage clones were isolated from the libraries by two rounds of affinity selection. In the first round the JAR 4 mAb (1 μg/ml) was incubated with magnetic beads conjugated with protein G (50 μl, protein G-Dynabeads®, Dynal, Norway) for 1 h at room temperature under agitation. The beads were washed 3 times with washing solution (PBS, 0.5% Tween-20) and approximately 1010 ampicillin-transducing units of library preparation (~1011 phage particles) in a volume of 100 μl were added to 900 μl of blocking solution (PBS, 5% non-fat dry milk, 0.05% Tween 20) and agitated for 3-4 h at room temperature. The beads were washed 10 times with 1 ml of washing solution, and bound phages were eluted with 500 μl of 0.1 M HCl, adjusted to pH 2.2 with glycine, and 10 mg/ml BSA. The solution was neutralized with 60 μl of 2 M Tris pH 9.6, and the phages were amplified by infecting E. coli strain TG1. The second round of panning was carried out in the same way, but using 1010 ampicillin-transducing units obtained from the first round of amplified phage pools.
Positive phage clones were identified through immunoscreening, performed as described (Luzzago et al., 1993). In brief, phages obtained from the affinity selection were mixed with 2 ml of a culture of E. coli TG1 (OD600 ~ 0.4). Five hundred μl of this culture was infected with M13KO7 helper phage (109 pfu) in a 1.5 ml tube, and incubated for 15 min at 37 °C and an additional 15 min with shaking. Serial dilutions of infected bacteria were plated on Luria-Bertani (LB) agar plates containing ampicillin, kanamycin and IPTG, and were incubated overnight at 37 °C. Nitrocellulose filters (Protran BA85, 0.45 mm, Schleicher & Schuell, Keene, NH) were layered on the plates containing 50-200 colonies, and left at room temperature for 1 h. Filters were blocked for 1 h with blocking solution, and incubated for 2 h at room temperature with mAb JAR 4 (1 μg/ml in blocking solution), and then for 1 h at room temperature with an AP-conjugated anti-mouse IgG secondary Ab (Sigma, St. Louis, MO, 1:5000) in the same solution. Filters were washed in washing solution, and developed with nitro-blue-tetrazolium and 5-bromo-4-chloro-3-indolyl-phosphate (Sigma). The positive colonies were resuspended in 50 μl of 1X PBS. The bacterial cells were killed by heating at 70°C for 15 min. After centrifugation for 5 min in a microcentrifuge, the supernatants containing the positive phage particles were collected.
ELISA
Binding of mAbs to recombinant fHbp was performed in a direct binding ELISA, using purified fHbp (2 μg/ml) as the antigen on the plate, and performed as described previously (Beernink and Granoff, 2008). Binding of JAR 4 to peptides displayed by the phage-library selected clones was assessed by ELISA (Dente et al., 1994). Ninety-six well plates were coated with 100 μl per well of a rat anti-pIII (coat protein III) mAb (1 μg /ml in 50 mM NaHCO3, 0.02% (w/v) NaN3, pH 9.6) and incubated overnight at 4°C. The plates were washed 8 times with TBST (50 mM Tris HCl, 150 mM NaCl, pH 7.5, 0.05% (v/v) Tween-20). One hundred μl per well of cleared phage supernatant were added and the plates were incubated for 1 h at 37 °C. After washing, the mAb was added (1 μg/ml in blocking buffer), incubated for 2 h at 37 °C, and mAb binding was detected by AP-conjugated goat anti-mouse IgG antibody (Sigma, 1:5000) using p-nitrophenyl phosphate substrate tablets (Sigma).
Anti-fHbp mAb cooperative bactericidal activity
The ability of anti-fHbp mAb, JAR 4, to elicit cooperative bactericidal activity with other non-bactericidal anti-fHbp mAbs was measured using human complement as previously described (Beernink et al., 2008). We used four test strains: H44/76-SL (B.15.P1.7,16, ST-32) from an epidemic in Norway; NZ98/254 (B:4;P1.7-2,4, ST-41/44 clonal complex) from a recent epidemic in New Zealand; 8047 (B:2b:P1.5-1, 2-2, ST- 8) from a patient hospitalized in the U.S., and BuFa 1/03 (W-135:nd:P1.5,2 and ST-11) from an epidemic in Burkina Faso. Strains H44/76 and NZ98/254 had fHbp in the variant 1 group and strains 8047 and BuFa 1/03 had fHbp in the variant 2 group. The respective fHbp peptide identification numbers as assigned in the fHbp peptide database were 1, 14, 77 and 23 (http://neisseria.org/perl/agdbnet/agdbnet.pl?file=nm_fhbp.xml&page=browse&locus=public_FHBP_p).
Results
By ELISA, the JAR 4 mAb cross-reacted with fHbp in the variant 1 and 2 groups, but did not react with fHbp in the variant 3 group (Fig. 1A). In contrast, the binding of most other anti-fHbp mAbs, for example JAR 5 (Fig. 1B), was specific for fHbps within individual variant groups. JAR 4 individually was not bactericidal with human complement but was bactericidal against representative strains with fHbp in the variant 1 or 2 groups when tested in combination with certain other non-bactericidal mAbs (Table 1). The epitopes of two of the mAbs with cooperative activity (JAR 3 and 5) involved amino acid residues 121 and 122 (Beernink et al., 2008) in the N-domain (Cantini et al., 2009; Mascioni et al., 2009) of fHbp, whereas the epitopes of five other mAbs with cooperative activity (mAb 502 (Giuliani et al., 2005), and JAR 11, 13, 32 and 35 (Beernink et al., 2008)) resided in the C-domain in the region between amino acids 174 and 216. The two mAbs, JAR 10 and 33, that lacked JAR 4 cooperative bactericidal activity had epitopes that involved K or R, respectively at position 192 and (for both mAbs) E180, in the C-domain of fHbp (Beernink et al., 2008).
Figure 1.

Binding of anti-fHbp mAbs to purified recombinant fHbp as measured by ELISA. The proteins were expressed from genes from strains MC58 (variant 1), 8047 (variant 2) and M1239 (variant 3), respectively. A, Binding of mAb JAR 4 to antigenic variant 1 and 2 proteins but not with a variant 3 protein. B, Binding of a control mAb, anti-fHbp, JAR 5, which recognizes a variant 1-specific epitope in the N-domain involving amino acid residues 121 and 122 (Beernink et al., 2008).
Table 1.
Cooperative bactericidal activity of anti-fHbp mAb, JAR 4 (IgG2a), when tested together with second anti-fHbp mAbs and human complementa
| Second mAb | Ig Isotype | fH Inhibitionb | Location of Residues | Distance (Å)c | Strain (fHbp variant)d | Combination BC50, μg/mle | |
|---|---|---|---|---|---|---|---|
| D25 | G58 | ||||||
| mAb 502 | G2a | - | R204 | 27 | 32 | H44/76 (1) | 0.5 |
| JAR 5 | G2b | ++ | G121, K122 | 42 | 24-26 | H44/76 (1) | 0.4 |
| JAR 3f | G3 | ++ | G121, K122 | 42 | 24-26 | NZ98/254 (1) | 1 |
| JAR 5f | G2b | ++ | G121, K122 | 42 | 24-26 | NZ98/254 (1) | 4 |
| JAR 10 | G1 | - | E180 | 36 | 41 | 8047 (2) | >50 |
| JAR 10 | G1 | - | K192 | 39 | 43 | 8047 (2) | >50 |
| JAR 11f | G2a | + | A174 | 39 | 47 | 8047 (2) | 5 |
| JAR 13f | G2a | ++ | S216 | 47 | 47 | 8047 (2) | 4 |
| JAR 32 | G2a | ++ | K174 | 39 | 47 | BuFa 1/03 (2) | 10 |
| JAR 33 | G2a | - | E180 | 36 | 41 | BuFa 1/03 (2) | >50 |
| JAR 33 | G2a | - | R192 | 39 | 43 | BuFa 1/03 (2) | >50 |
| JAR 35 | G2b | ++ | K174 | 39 | 47 | BuFa 1/03 (2) | 20 |
None of the mAbs was individually bactericidal against the strains shown (BC50>50 μg/ml).
ELISA inhibition was performed as described (Beernink et al., 2008) and defined by ++,>75%; +, 25 to 45%; -, <15%.
Distances were measured between the alpha-carbon of JAR 4 epitope residue and that of the residue affecting the epitope of the second mAb using SwissPDB Viewer/DeepView (http://spdbv.vital-it.ch/) using coordinates from the NMR solution structure of the entire of fHbp molecule (antigenic variant 1, strain MC58) (Cantini et al., 2009).
The four test strains each expressed different fHbp peptides that were recognized by JAR 4. The strains were selected because they also bound with the respective second mAbs tested.
Minimal combined concentration of the two mAbs sufficient for bactericidal activity (50 percent killing after 1 hr of incubation with human complement)
Data reported previously (Beernink et al., 2008).
JAR 4 did not bind to overlapping 13-mer peptides that spanned the entire fHbp molecule; nor did the mAb bind to recombinant fragments of fHbp from the antigenic variant 1 group extending from amino acids 8-100, 101-164, 165-255, or 101-255 (data not shown). These negative results suggested that the JAR 4 epitope was conformational and/or discontinuous. In an attempt to identify residues affecting the JAR 4 epitope, we screened filamentous bacteriophage libraries displaying random peptides (9- or 12-mer) fused to coat protein VIII for their ability to bind with JAR 4. We identified a number of phage clones with strong JAR 4 binding (Table 2). The majority of the recombinant pVIII genes from these clones encoded peptides containing a tripeptide consensus sequence, DHK, which also was present in the N-domain of fHbp beginning at residue 25 in the v.1 and v.2 proteins, and at residue 30 in fHbp v.3 (Table 3).
Table 2.
Amino acid sequences of the phage-displayed peptides mimicking the JAR 4 epitope
| Clonea | Sequenceb | Number of clones | ELISA ODc |
|---|---|---|---|
| 1 | H D H K L E G T E | 5 | |
| 2 | C G G V Y D D K T G C A | 3 | 2.0–3.5 |
| 3 | H D H K T Q L D P | 1 | |
| 4 | W T L A V F D H K A Q T | 2 | 1.0–1.9 |
| 5 | G C M G Y D H R S G C V | 1 | |
| 6 | F H D H K T A N Q | 1 | 0.5–0.9 |
| 7 | H D H R I W P L D V T A | 1 |
The positive phage clones, ranked by their reactivity with JAR 4.
Deduced amino acid sequences of the peptide inserts displayed through pVIII fusion on the phage library clones positive for JAR 4 binding. The most conserved residues are indicated in bold.
mAb JAR 4 reactivity of isolated clones, determined by ELISA.
Table 3.
Amino acid alignments of fHbp in the antigenic variant 1, 2 and 3 groupsa
| Antigenic Variant Group | Amino Acid Range | Amino Acid Sequenceb | ||
|---|---|---|---|---|
| 1 | 22–51 | APLDHKDKGL | QSLTLDQSVR | KNEKLKLAAQ |
| 2 | 22–51 | APLDHKDKGK | QSLTLDQSVR | KNEKLKLAAQ |
| 3 | 27–56 | APLDHKDKGL | KSLTLEDSIP | QNGTLTLSAQ |
| ********* | ||||
| 1 | 52–79 | GAEKTYGNGD | ---SLNTGKL | KNDKVSRFDF |
| 2 | 52–79 | GAEKTYGNGD | ---SLNTGKL | KNDKVSRFDF |
| 3 | 57–87 | GAEKTFKAGD | KDNSLNTGKL | KNDKISRFDF |
| ****** | ||||
Alignment of a region of the N-domains of fHbps in the antigenic variant 1, 2 and 3 groups (strains MC58, 8047 and M1239, respectively).
The positions that differed among JAR 4-postive fHbp variant 1 or 2 wildtype proteins and JAR-4 negative variant 3 wildtype proteins are indicated with an asterisk below the sequences. The consensus tripeptide sequence DHK was identified by phage display (Table 2). The DHK residues and the KDN and YGN tripeptides affecting JAR-4 binding were identified by site-specific mutagenesis (Figures 2, 3 and 4, respectively). The respective tripeptides are shown in bold.
When the individual residues at positions 25 to 27 in recombinant fHbp from the antigenic variant 1 group were mutated to alanine, binding of JAR 4 was either decreased (D25A or H26A) or eliminated (K27A) (Fig. 2A). These mutations did not destabilize the protein since binding by a control anti-fHbp mAb, JAR 5, which was specific for another epitope in the N-domain (Beernink et al., 2008), was not affected by the mutations (Fig. 2B). Nor did the mutations significantly affect binding of fH to fHbp as measured by ELISA (data not shown). The DHK residues that affected the expression of the JAR 4 epitope are located on a surface-exposed loop of the N-domain of the fHbp molecule as illustrated on a surface rendering of the three-dimensional structure of fHbp in the variant 1 group using coordinates from a recent NMR solution structure of the entire of molecule (Cantini et al., 2009).
Figure 2.

Effect of alanine substitutions at positions 25-27 on ELISA binding of JAR 4 to recombinant mutant fHbp from the variant 1 group (wild type gene from strain MC58). A, Binding of mAb JAR 4. B, Binding of a control anti-fHbp mAb, JAR 5 (see legend to Figure 1).
Since the DHK sequence was present in a fHbp from the variant 3 group that did not bind with JAR 4 (Fig. 1A), the results indicated that the DHK tripeptide was necessary but not sufficient for JAR 4 reactivity with the protein from the variant 1 and 2 groups. Comparison of the respective amino acid sequence alignments of JAR 4-reactive fHbps in the variant 1 and 2 groups with a JAR 4-non-reactive fHbp in the variant 3 group indicated the presence of additional polymorphisms downstream from the DHK tripeptide (Table 3). In the variant 3 protein, the loop containing DHK was adjacent to a surface-exposed region that contained a three amino acid insertion (KDN) that was not present in fHbp from the variant 1 or 2 groups (Table 3, residues 67-69 of fHbp v.3; and Fig. 3, shown by the arrow). To determine the possible influence of the KDN residues on expression of the JAR 4 epitope, we introduced the tripeptide into a fHbp from the variant 1 group (Fig. 3, arrow), and deleted the three residues from a fHbp in the variant 3 group. When we tested the resulting mutant variant 1 protein containing the KDN insertion for JAR 4 mAb reactivity by ELISA, JAR 4 binding was eliminated (Fig. 4A). In contrast, the reactivity of a control anti-fHbp mAb, JAR 5, was retained (Fig 4B). Deletion of the KDN residues in a variant 3 fHbp, however, failed to introduce the JAR 4 epitope (Fig. 5A, shaded triangle symbols).
Figure 3.
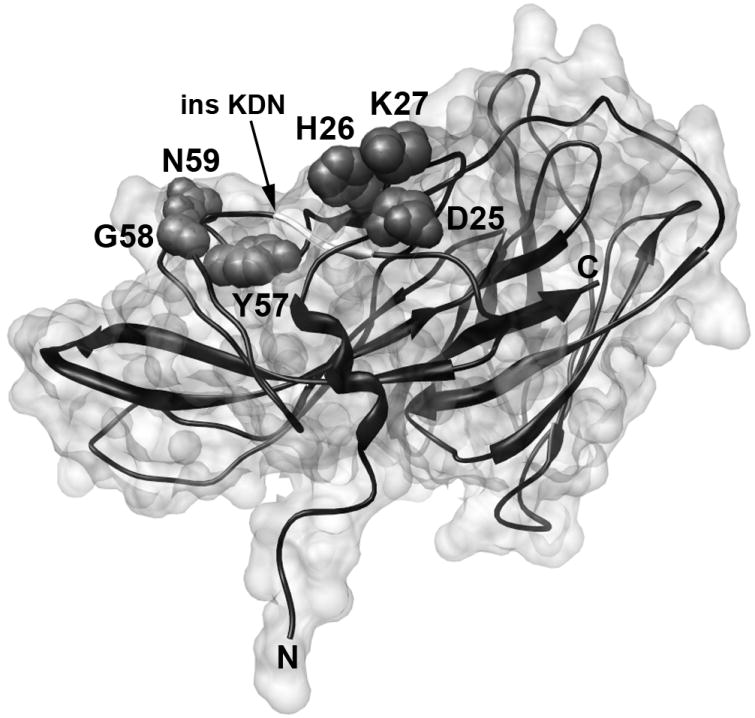
Surface rendering of fHbp variant 1 using coordinates from the NMR solution structure of the entire of fHbp molecule (antigenic variant 1, strain MC58) (Cantini et al., 2009). The relative positions of the DHK and YGN tripeptides that affect binding of JAR 4 are shown. The location of amino acids 67-69, which in the variant 3 protein contains the KDN tripeptide that negatively affects JAR 4 binding and is not present in variant 1 or 2 proteins (See Table 3), is shown as a white insert in the ribbon next to the arrow.
Figure 4.

Effect of insertion of a KDN tripeptide in a fHbp from the antigenic variant 1 group (v.1) on reactivity with anti-fHbp mAb JAR 4. The KDN sequence at residues 67-69 was naturally present only in fHbp in the variant 3 group. A, Binding of JAR 4. B, Binding of a control anti-fHbp mAb, JAR 5 (see Legend to Figure 1).
Figure 5.

Anti-fHbp mAb binding to mutants of a fHbp in the variant 3 group as measured by ELISA. Open squares, control WT fHbp in the variant 1 group (positive for JAR 4; negative for JAR 31); filled circles, control WT fHbp in the variant 3 group (negative for JAR 4; positive for JAR 31); shaded triangles, ΔKDN mutant of fHbp in the variant 3 group, which did not result in JAR 4 binding; asterisks, a double mutant of fHbp in the variant 3 group with ΔKDN and FKA->YGN, which resulted in partial expression of the JAR 4 epitope. A, Binding of JAR 4. B, Binding of a control anti-fHbp mAb, JAR 31, which recognizes an epitope in the C-domain (Beernink and Granoff, 2008). JAR 31 bound to WT fHbp in the variant 3 group and with the two mutants, but not with the WT fHbp in the variant 1 group.
Three additional residues immediately upstream from the KDN tripeptide also differed between JAR 4-reactive fHbp in the variant 1 and 2 groups and JAR 4-non-reactive fHbps in the variant 3 group (YGN and FKA, respectively; Table 3). The position of the YGN tripeptide in a variant 1 fHbp in relation to the DHK tripeptide is seen graphically in the space-filling model in Fig. 3. We constructed a double mutant of a variant 3 protein in which both the KDN residues were deleted and the FKA tripeptide was changed to YGN to mimic the amino acid sequence of a fHbp in the variant 1 or 2 group. By ELISA, JAR 4 bound to the double mutant of the variant 3 protein (asterisks with dashed line, Fig. 5A). A control anti-fHbp mAb, JAR 31, showed similar binding to the double mutant of the variant 3 protein and a WT fHbp from the variant 3 group (Fig. 5B). By ELISA, the double mutant also bound fH with a comparable signal as those obtained with the respective wildtype variant 1 and 3 fHbp control proteins (data not shown). Note that binding of JAR 4 to the double variant 3 mutant required ~100-fold higher concentrations of the mAb for equivalent binding to the WT variant 1 protein (2 μg/ml vs. 0.02 μg/ml, Fig. 5A). Therefore, other amino acids not replaced in the mutant protein that differed between JAR 4-positive fHbp from the variant 1 or 2 groups and JAR 4-non-reactive fHbp from the variant 3 group contribute to the JAR 4 epitope (e.g., likely one or more of the other 8 amino acids residues that differed between fHbp variant 1 or 2 and variant 3 in the region between residues 42 to 56 downstream from the DHK tripeptide, Table 3).
Discussion
In previous studies, the region of fHbp comprising amino acids 100 to 255 appeared to be important for eliciting bactericidal antibodies that were specific for different antigenic variants (Beernink and Granoff, 2008; Beernink et al., 2008; Giuliani et al., 2005;Cantini et al., 2009; Mascioni et al., 2009). While several broadly reactive mAbs recently were recently reported by Mascioni et al to map to the N-terminal domain, none was described as being bactericidal (Mascioni et al., 2009). The authors concluded that the linear epitopes recognized by these mAbs were not surface-exposed since the mAbs did not bind to bacterial cells in flow cytometry experiments. In contrast, in the present study we identified a region of the N-domain that contained a DHK tripeptide at residues 25-27 as being important for binding to JAR 4. This tripeptide was in the same region as that of the epitope of one of these previously described mAbs said not to bind to the bacterial surface, and overlapped the epitope of the second mAb that encompassed residues G15 to L34. Based on our data, this region of fHbp is surface-exposed since in our previous studies JAR 4 bound to live encapsulated bacterial cells by flow cytometry (Welsch et al., 2008), elicited C3b deposition on the bacterial surface (Welsch et al., 2008), and had cooperative bactericidal activity with certain other non-bactericidal anti-fHbp mAbs (Welsch et al., 2008) (Table 1). Our JAR 4 studies showing surface-accessibility of this region of fHbp used live bacteria whereas the negative mAb binding studies performed by Mascioni et al (Mascioni et al., 2009) used formaldehyde-killed organisms, which may have interfered with anti-fHbp mAb binding.
Using site-directed mutagenesis, we identified additional residues downstream from the DHK tripeptide in the N-domain that affected expression of the JAR 4 epitope. These included a KDN tripeptide at residues 67-69 of a variant 3 fHbp, which was absent in variant 1 or 2 fHbps and when inserted into a variant 1 protein eliminated JAR 4 binding. There also was a YGN tripeptide comprising residues 57-59 of variant 1 and 2 fHbps, which was not present in a variant 3 protein. When the FKA tripeptide in a variant 3 protein was replaced by the YGN tripeptide along with deletion of the KDN tripeptide, the resulting mutant variant 3 protein was recognized by JAR 4. Our studies did not define all of the contact residues required for expression of this complex epitope. However, the data help explain the specificity of JAR 4 for binding with fHbps in the variant 1 and 2 groups, and indicated that the N-terminal region is important for eliciting cross-reactive bactericidal antibodies against strains expressing fHbp from different antigenic variant groups.
The entire fHbp molecule containing amino acids 1 to 255 in variant 1 and 2 proteins expressed the JAR 4 epitope while the corresponding molecule from a variant 3 protein did not. Although specific site-directed amino acid substitutions at residues between 25 and 62 in the variant 1 protein (between residues 30 and 69 in the variant 3 protein) affected expression of the JAR 4 epitope, a polypeptide containing amino acids 8-100 from a JAR 4-reactive wildtype variant 1 protein was not sufficient for expression of the epitope. Thus, a fHbp polypeptide larger than amino acids 8-100 is needed for expression of the epitope, which may coincide with the entire N-domain from amino acids 1 to 136, as recently defined by NMR studies (Cantini et al., 2009; Mascioni et al., 2008).
Although the JAR 4 epitope appeared to be discontinuous in the native fHbp, the mAb bound strongly to certain small DHK-containing peptides expressed by phage particles. The specific amino acids that flanked the DHK residues in the phage peptides, and contributed to JAR 4 binding to the phages, remain undefined. Conceivably, one or more of these antigenic mimetic peptides also could function as immunogens and induce anti-peptide antibodies that elicit cooperative cross-reactive anti-fHbp bactericidal antibody. If true, the peptide mimetics would be useful additions to chimeric fHbp vaccines capable of stimulating broad cross-reactive serum anti-fHbp bactericidal antibodies (Beernink and Granoff, 2008).
In a previous study, we observed that pairs of anti-fHbp mAbs were bactericidal when the distances between residues affecting the epitopes were 16 to 20 Å, whereas there was no bactericidal activity when the distances were ≤14 or ≥27 Å (Beernink et al., 2008). However, the new data with JAR 4 indicated that cooperative bactericidal activity occurred with second anti-fHbp mAbs with epitopes located on the N- or C-domains of fHbp, and that the respective distances between amino acids affecting the JAR 4 epitope and those of the epitopes of the second mAb frequently exceeded 27 Å (Table 1). Thus, with JAR 4, there did not appear to be a relationship between the distances of the respective residues affecting the JAR 4 epitope and the epitope of the second mAb and the ability to elicit cooperative bactericidal antibody.
We have also observed that for all pairs of anti-fHbp mAbs with bactericidal activity, at least one member of the pair inhibited binding of fH (Beernink et al., 2008). In the absence of bound fH on the bacterial surface of N. meningitidis, C3 activation was unregulated (Beernink et al., 2008; Granoff et al., 2009; Madico et al., 2006; Schneider et al., 2006; Schneider et al., 2007; Welsch et al., 2008) and the organism was more susceptible to amplification of the alternative complement pathway and complement-mediated bacteriolysis (Granoff et al., 2009; Madico et al., 2006; Schneider et al., 2007; Welsch et al., 2008). JAR 4 did not inhibit binding of fH (Beernink et al., 2008). In the present study there was no cooperative bactericidal activity between JAR 4 and second anti-fHbp mAbs that also did not inhibit binding of fH (Table 1). Further, all but one of the anti-fHbp mAbs that elicited cooperative bactericidal activity with JAR 4 inhibited binding of fH to fHbp (Table 1). The exception was mAb 502, which had an estimated distance of 27-32 Å between the respective amino acid residues affecting expression of the JAR 4 and mAb 502 epitopes (Table 1). Thus, with the close proximity of the respective epitopes, binding by these two mAbs may have resulted in sufficient immune complex to engage C1q and activate complement-mediated bactericidal activity (Michaelsen et al., 1991; Welsch et al., 2008; Weynants et al., 2007) even in the absence of inhibition of binding of fH. However, additional studies are needed to elucidate the mechanism(s) underlying JAR cooperative bactericidal activity.
Conclusions
The region of fHbp comprising amino acid residues 25 to 59 is important for eliciting antibodies that can cooperate with other anti-fHbp antibodies for cross-reactive, complement-mediated bactericidal activity. Although a truncated fHbp containing only residues 101-255 was reported to elicit bactericidal antibodies against strains expressing fHbp from the homologous antigenic variant group (Giuliani et al., 2005), residues between 25 and 59 are important for expression of the JAR 4 epitope and for eliciting cross-reactive bactericidal anti-fHbp antibodies against strains from different antigenic variant groups.
Acknowledgments
This work was supported by Public Health Service grants R01 AI 46464 (to D.M.G.) and R01 AI 70955 (to P.T.B.) from the National Institute of Allergy and Infectious Diseases, NIH. The work at Children’s Hospital Oakland Research Institute was performed in a facility funded by Research Facilities Improvement Program grant number C06 RR 16226 from the National Center for Research Resources, NIH.
We are grateful to Dr. Maria Scarselli, Novartis Vaccines, Siena Italy, for providing calculations of the respective distances between pairs of the mAbs based on the coordinates of the NMR solution structure of the entire fHbp of an antigenic variant 1 strain (strain MC58), and for preparing Fig. 3. We thank Ryan Palapaz for technical assistance.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Beernink PT, Granoff DM. Bactericidal antibody responses induced by meningococcal recombinant chimeric factor H-binding protein vaccines. Infect Immun. 2008;76:2568–75. doi: 10.1128/IAI.00033-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beernink PT, Welsch JA, Bar-Lev M, Koeberling O, Comanducci M, Granoff DM. Fine antigenic specificity and cooperative bactericidal activity of monoclonal antibodies directed at the meningococcal vaccine candidate, factor H-binding protein. Infect Immun. 2008;76:4232–40. doi: 10.1128/IAI.00367-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantini F, Veggi D, Dragonetti S, Savino S, Scarselli M, Romagnoli G, Pizza M, Banci L, Rappuoli R. Solution structure of the factor H binding protein, a survival factor and protective antigen of Neisseria meningitidis. J Biol Chem. 2009 doi: 10.1074/jbc.C800214200. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dente L, Cesareni G, Micheli G, Felici F, Folgori A, Luzzago A, Monaci P, Nicosia A, Delmastro P. Monoclonal antibodies that recognise filamentous phage: tools for phage display technology. Gene. 1994;148:7–13. doi: 10.1016/0378-1119(94)90227-5. [DOI] [PubMed] [Google Scholar]
- Felici F, Castagnoli L, Musacchio A, Jappelli R, Cesareni G. Selection of antibody ligands from a large library of oligopeptides expressed on a multivalent exposition vector. J Mol Biol. 1991;222:301–10. doi: 10.1016/0022-2836(91)90213-p. [DOI] [PubMed] [Google Scholar]
- Fletcher LD, Bernfield L, Barniak V, Farley JE, Howell A, Knauf M, Ooi P, Smith RP, Weise P, Wetherell M, Xie X, Zagursky R, Zhang Y, Zlotnick GW. Vaccine potential of the Neisseria meningitidis 2086 lipoprotein. Infect Immun. 2004;72:2088–100. doi: 10.1128/IAI.72.4.2088-2100.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giuliani MM, Santini L, Brunelli B, Biolchi A, Arico B, Di Marcello F, Cartocci E, Comanducci M, Masignani V, Lozzi L, Savino S, Scarselli M, Rappuoli R, Pizza M. The region comprising amino acids 100 to 255 of Neisseria meningitidis lipoprotein GNA 1870 elicits bactericidal antibodies. Infect Immun. 2005;73:1151–60. doi: 10.1128/IAI.73.2.1151-1160.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Granoff DM, Welsch JA, Ram S. Binding of complement factor H (fH) to Neisseria meningitidis is specific for human fH and inhibits complement activation by rat and rabbit sera. Infect Immun. 2009;77:764–9. doi: 10.1128/IAI.01191-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang H-Q, Harris SL, Tan C, Scott A, Alexander K, Mason K, DaSilva I, Mack M, Zhao X-J, Guttmann S, Liu D, Wong P, McNeil L, Pride M, Arora A, Zhu D, Hoiseth SK, Mininni TL, Hagen M, Jones T, Jansen KU, Zlotnick GW. Prediction of broad vaccine coverage for a bivalent rLP2086 based vaccine which elicits serum bactericidal activity against a diverse collection of serogroup B Meningococci. In: van Alphen L, van Ley P, van den Dobbelsteen G, editors. 16th International Pathogenic Neisseria Conference; Rotterdam, The Netherlands. 2008. [Google Scholar]
- Luzzago A, Felici F. Construction of disulfide-constrained random peptide libraries displayed on phage coat protein VIII. Methods Mol Biol. 1998;87:155–64. doi: 10.1385/0-89603-392-9:155. [DOI] [PubMed] [Google Scholar]
- Luzzago A, Felici F, Tramontano A, Pessi A, Cortese R. Mimicking of discontinuous epitopes by phage-displayed peptides, I. Epitope mapping of human H ferritin using a phage library of constrained peptides. Gene. 1993;128:51–7. doi: 10.1016/0378-1119(93)90152-s. [DOI] [PubMed] [Google Scholar]
- Madico G, Welsch JA, Lewis LA, McNaughton A, Perlman DH, Costello CE, Ngampasutadol J, Vogel U, Granoff DM, Ram S. The meningococcal vaccine candidate GNA1870 binds the complement regulatory protein factor H and enhances serum resistance. J Immunol. 2006;177:501–10. doi: 10.4049/jimmunol.177.1.501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mascioni A, Bentley BE, Camarda R, Dilts DA, Fink P, Gusarova V, Hoiseth S, Jacob J, Lin SL, Malakian K, McNeil LK, Mininni T, Moy F, Murphy E, Novikova E, Sigethy S, Wen Y, Zlotnick GW, Tsao DH. Structural basis for the immunogenic properties of the meningococcal vaccine candidate LP2086. J Biol Chem. 2009 doi: 10.1074/jbc.M808831200. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masignani V, Comanducci M, Giuliani MM, Bambini S, Adu-Bobie J, Arico B, Brunelli B, Pieri A, Santini L, Savino S, Serruto D, Litt D, Kroll S, Welsch JA, Granoff DM, Rappuoli R, Pizza M. Vaccination against Neisseria meningitidis using three variants of the lipoprotein GNA1870. J Exp Med. 2003;197:789–99. doi: 10.1084/jem.20021911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michaelsen TE, Garred P, Aase A. Human IgG subclass pattern of inducing complement-mediated cytolysis depends on antigen concentration and to a lesser extent on epitope patchiness, antibody affinity and complement concentration. Eur J Immunol. 1991;21:11–6. doi: 10.1002/eji.1830210103. [DOI] [PubMed] [Google Scholar]
- Rappuoli R. The application of reverse vaccinology, Novartis MenB vaccine developed by design. In: van Alphen L, van Ley P, van den Dobbelsteen G, editors. 16th International Pathogenic Neisseria Conference. Rotterdam; Netherlands: 2008. [Google Scholar]
- Schneider MC, Exley RM, Chan H, Feavers I, Kang YH, Sim RB, Tang CM. Functional significance of factor H binding to Neisseria meningitidis. J Immunol. 2006;176:7566–75. doi: 10.4049/jimmunol.176.12.7566. [DOI] [PubMed] [Google Scholar]
- Schneider MC, Exley RM, Ram S, Sim RB, Tang CM. Interactions between Neisseria meningitidis and the complement system. Trends Microbiol. 2007;15:233–40. doi: 10.1016/j.tim.2007.03.005. [DOI] [PubMed] [Google Scholar]
- Seib KL, Serruto D, Oriente F, Delany I, Adu-Bobie J, Veggi D, Arico B, Rappuoli R, Pizza M. Factor H-binding protein is important for meningococcal survival in human whole blood and serum, and in the presence of the antimicrobial peptide LL-37. Infect Immun. 2008;77:292–99. doi: 10.1128/IAI.01071-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welsch JA, Ram S, Koeberling O, Granoff DM. Complement-dependent synergistic bactericidal activity of antibodies against factor H-binding protein, a sparsely distributed meningococcal vaccine antigen. J Infect Dis. 2008;197:1053–61. doi: 10.1086/528994. [DOI] [PubMed] [Google Scholar]
- Welsch JA, Rossi R, Comanducci M, Granoff DM. Protective activity of monoclonal antibodies to genome-derived neisserial antigen 1870, a Neisseria meningitidis candidate vaccine. J Immunol. 2004;172:5606–15. doi: 10.4049/jimmunol.172.9.5606. [DOI] [PubMed] [Google Scholar]
- Weynants VE, Feron CM, Goraj KK, Bos MP, Denoel PA, Verlant VG, Tommassen J, Peak IR, Judd RC, Jennings MP, Poolman JT. Additive and synergistic bactericidal activity of antibodies directed against minor outer membrane proteins of Neisseria meningitidis. Infect Immun. 2007;75:5434–42. doi: 10.1128/IAI.00411-07. [DOI] [PMC free article] [PubMed] [Google Scholar]