

Abstract
Structure-function studies of membrane proteins present a unique challenge to researchers due to the numerous technical difficulties associated with their expression, purification and structural characterization. In the absence of structural information, rational identification of putative functionally important residues/regions is difficult. Phylogenetic relationships could provide valuable information about the functional significance of a particular residue or region of a membrane protein. Evolutionary Trace (ET) analysis is a method developed to utilize this phylogenetic information to predict functional sites in proteins. In this method, residues are ranked according to conservation or divergence through evolution, based on the hypothesis that mutations at key positions should coincide with functional evolutionary divergences. This information can be used as the basis for a systematic mutational analysis of identified residues, leading to the identification of functionally important residues and/or domains in membrane proteins, in the absence of structural information apart from the primary amino acid sequence. This approach is potentially useful in the context of the auditory system, as several key processes in audition involve the action of membrane proteins, many of which are novel and not well characterized structurally or functionally to date.
Keywords: phylogenetic, membrane protein, structure-function, site-directed mutagenesis, membrane expression
1. Introduction
There is usually minimal or no secondary and tertiary structural information for membrane-associated protein families in general, due to the technical difficulties associated with the study of membrane proteins. Attempts to locate specific functional domains and/or residues essential for function in membrane proteins are therefore extremely challenging, and more often than not employ shotgun approaches such as mutating all charged residues, putative phosphorylation or glycosylation sites, consensus/conserved motifs, etc. An alternative approach is to utilize evolutionary relationships in protein families to obtain structural and functional information. The computational method of Evolutionary Trace (ET) ranks individual residues in proteins by correlating amino acid variation patterns with functional divergences in protein families (1). ET analysis thus combines the power of computational analysis with the insights offered by evolutionary and phylogenetic comparison of protein sequences.
The key hypothesis upon which ET analysis rests is that substitutions at residues that are more important to function should necessarily be linked with greater evolutionary divergences than substitutions at residues of lesser importance (1, 2). Therefore, in order to estimate the functional importance of a residue, ET looks for specific patterns of amino acid variations in multiple sequence alignments of a family of ancestrally related proteins (homologs). Specifically, ET looks for conservation within subgroup partitions of the entire family, where the partitions are defined by successive divergences at every node of the evolutionary tree. Thus partition 1, or the root partition is the entire family, partition 2 consists of two subgroups, each defined by one of the first two branches, partition 3 consists of three subgroups, each defined by one of the first three branches, and so on until partition n, where each one of the n sequences is in its own singlet subgroup (Fig. 1). For each residue, ET systematically tests every partition starting from partition 1 to find the first, say partition i, at which a residue’s position becomes invariant in everyone of its i subgroups. This partition number, i, is the ET rank of evolutionary importance for that sequence alignment position.
Fig. 1.
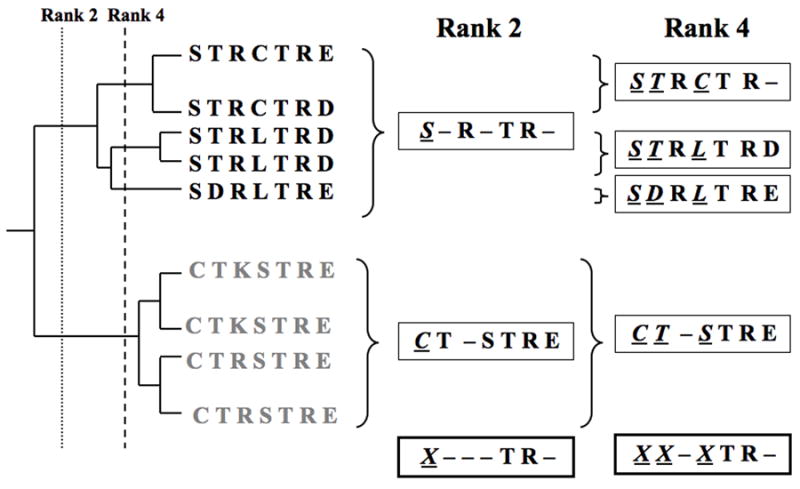
A schematic representation of the residue ranking system used in Evolutionary Trace analysis. Trace residues are invariant within a given branch but may vary between them. Rank 2 residues (position 1, underlined and italicized), defined by the second branchpoint, are invariant within a subgroup (black) but vary from the other subgroup (gray). Note: the first branchpoint is the root and includes the entire family. Rank 4 residues are invariant within subgroups defined by the fourth branchpoint. Trace residue positions are denoted by Xs, underlined and italicized, at the bottom of the alignment in this schematic.
The top-ranked residues (with ranks of 1, 2, 3, …) are called trace residues and they have important characteristics. First, except for the residues of rank 1 which must be invariant, other residues must vary among some homologs, and the variations may be non-conservative. Second, these variations occur only among major evolutionary branches of trace residues, so that top-ranked trace residues are strongly linked with the functional divergences of the corresponding subfamilies. Third, clusters of top-ranked residues are a common feature of protein tertiary structure and they usually indicate functional sites or molecular recognition surfaces in proteins (3, 4). Residues identified by ET analysis can be then subjected to systematic mutational and functional studies, including evaluation of protein stability, trafficking, membrane expression, and appropriate functional assays (5–8). Using these techniques, we have shown that residues in the region of the sulfate anion transporter domain of the outer hair cell membrane protein prestin are essential for its function (9). This analysis provided insight into the local structure and packing of the transmembrane helices associated with the sulfate anion transporter domain.
2. Materials
2.1. Evolutionary trace analysis
Primary amino acid sequence of membrane protein of interest.
2.2. Generation of mutants
Quikchange site-directed mutagenesis kit (Stratagene, La Jolla, CA).
Qiagen Spin Miniprep kit (Qiagen, Valencia, CA).
Appropriate mutagenesis primers (see Subheading 3.2 step 2 for primer design). Primers can be obtained from a commercial DNA synthesis service such as Sigma-Genosys (the minimum synthesis scale –3 OD, and basic purification scale is sufficient. Since primers are supplied as lyophilized powder, dissolve primers in water to a stock concentration of 1 μg/μL, and store at −20°C until further use.
Relevant wild type (Wt) gene cloned into a mammalian expression vector.
Thin-walled PCR tubes (200 μL capacity).
PCR thermocycler.
Super Optimal broth – Catabolite repression (SOC) medium (per liter): 20 g Bacto-tryptone, 5 g Bacto-yeast extract, 0.5 g NaCl, pH 7.0. Autoclave and allow to cool. Add glucose to a 20 mM final concentration and store at 4 °C.
Falcon disposable culture tubes with caps (BD Biosciences, San Jose, CA).
Luria Bertani (LB) broth (per liter): 10 g Bacto-tryptone, 5 g Bacto-yeast extract, 10 g NaCl, pH 7.0. Autoclave and store at 4 °C.
Plasmid mini-prep kit (Qiagen, Valencia, CA).
Appropriate LB agar plates (per liter): 10 g Bacto-tryptone, 5 g Bacto-yeast extract, 10 g NaCl, 1.8 g agar, pH 7.0. Autoclave, cool to about 50 °C, add appropriate selection antibiotic and pour 20–25 ml into each 10 cm plate. Store at 4 °C.
XL-1 blue competent E. coli cells (Stratagene, San Diego, CA).
DpnI restriction enzyme.
2.3. Evaluation of membrane expression of mutants
HEK 293 or other appropriate mammalian cell line.
12 mm circular glass cover slips.
Dulbecco’s Modified Eagle Medium (DMEM; Hyclone, Ogden, UT) supplemented with 10% fetal bovine serum (FBS; Invitrogen, Carlsbad, CA) and 1% penicillin/streptomycin (Invitrogen). Store at 4 °C.
Fugene 6 transfection reagent (Roche, Indianapolis, IN).
Poly-d-lysine coated 24-well plates (Greiner Bio-One, Monroe, NC).
Trypsin (Hyclone).
Wild Type (Wt) and mutant DNA plasmids.
Phosphate-buffered saline (PBS): 137 mM NaCl, 10 mM Phosphate, 2.7 mM KCl, pH 7.4.
PCM buffer: Mix PBS with 1 mM CaCl2 and 0.5 mM MgCl2 just before use.
4% paraformaldehyde (PFA): Heat 50 mL water to 75 °C using a hotplate or microwave. Add 4 g paraformaldehyde and 10 μl NaOH while stirring. After the PFA dissolves, allow solution to cool to room temperature and filter using Whatman Qualitative Grade 5 filter paper (Maidstone, UK). Add 10 mL PBS and make up to 100 mL using Milli-Q (18MΩ) H2O.
TritonX-100: 10% stock solution in Milli-Q H2O.
Bovine Serum Albumin (BSA), Immunology grade – IgG free (Jackson ImmunoResearch Inc., West Grove, PA).
AlexaFluor350 labeled concanavalin-A (Invitrogen).
Appropriate primary and AlexaFluor594-labeled secondary antibodies for protein of interest.
Fluoromount™ antifade reagent (Electron Microscopy Sciences, Hatfield, PA); clear nail polish/lacquer.
Small forceps; glass microscopy slides (25 ×75 ×1 mm).
3. Methods
3.1 Evolutionary trace analysis
Identify sequence homologs to the sequence of interest, using a BLAST (Basic Local Alignment Search Tool) search in conjunction with NCBI’s (National Center for Biotechnology Information) non-redundant protein sequence database, the blosum62 substitution matrix and default parameters.
Retrieve and list the complete sequences of the top 100 homologs (see Note 1), using the following criteria: e-value > 0.05, and covering at least 50% of the sequence of interest. Sequences may be retrieved from NCBI’s protein database using the search engine.
Generate an initial multiple sequence alignment using the CLUSTALW alignment tool (http://ca.expasy.org/). Use the full alignment option and default parameters (gap open penalty 10, gap extension penalty 0.05).
Group the sequences into sub-families based on phylogenetic and evolutionary considerations (i.e., based on the species of the source organism and its position in evolutionary history), using this initial alignment as the starting point (see Note 2).
Refine this initial alignment. Remove evolutionary outliers (protein sequences with remote evolutionary connection to the target sequence; see Note 2) from the sequence list and delete partial sequence fragments in the multiple alignment.
Gaps in the sequence are treated as a 21st amino acid, denoted by an X, in evolutionary trace analysis. This is merely a computational device and not a biophysically relevant assumption. This convention serves to indicate that a deletion or insertion took place that was then conserved in all protein descendants, suggesting some functional importance at the location of those gaps.
Assign residues trace ranks based on the minimum number of branches into which the protein family tree must be partitioned for that residue to be invariant within each branch. A residue of rank i is invariant within each of the first i branches of the tree (starting from the root), but variable within one of the first (i − 1) branches. In general, the higher the rank of a trace residue, the more strongly it is likely to be linked to the specific function of the subfamily within which it is conserved. Also, residues can be ranked by measuring the information entropy within each branch and sub-branch as described in Mihalek et al (10).
Identify clusters of highly ranked trace residues in the primary sequence of the protein of interest; such clusters may indicate functionally important regions or domains. These residues may be used for targeting site-specific mutations to identify functional regions, residues or domains in the protein. (see Notes 3–7).
Two ET web servers (11, 12) are available at http://mammoth.bcm.tmc.edu/. (see Note 8)
3.2 Generation of mutants
Design mutations at each trace residue position identified to elicit maximum structural and functional information about the residue at that position. Therefore, create mutants using systematic variations in the size, charge and/or hydrophobicity of each trace residue.
-
Design appropriate mutagenesis primers using the Wt nucleotide sequence as a template. Guidelines for primer design are as follows:
Design primers between 25 and 45 bases in length.
Choose a codon for mutation that is approximately in the middle of the sequence, with 10–15 bases on either side; the sequence should incorporate the replacement codon in place of the original Wt codon.
Make sure that the annealing temperature (Tm) of the primer is greater than 78°C.
Design a reverse primer that is complementary to the forward primer.
Design forward and reverse primers with at least 40% GC content and design so that they end in one or more GC bases as a 3′ clamp.
Avoid secondary structure in designed primers, specifically at their 3′ ends.
Analyze primer sequences for secondary structure (hairpins, loops etc), GC content and Tm using free primer analysis software available online at http://www.premierbiosoft.com/netprimer/netprlaunch/netprlaunch.html.
Dilute Wt plasmid template stock to a working concentration of 10 ng/μL using double-distilled, deionized water. Also, dilute each primer to a working concentration of 125 ng/μl.
-
Assemble a PCR reaction mix in a thin-walled 200 μl microcentrifuge PCR tube. Add each component carefully as individual droplets to the wall of the PCR tube in order to visually verify the addition of each small volume.
1–3 μL Wt DNA template.
1 μL forward primer and 1 μL reverse primer.
1 μL dNTPs (supplied with kit).
Add 35 to 37 μL distilled RNAse-free water, followed by 5 μL of the supplied 10X buffer, and 3 μL of the supplied Quiksolution (Quickchange II kit). Tap or flick the tube gently to facilitate mixing of the components, then place in the heating block of the PCR thermocycler.
-
Program the thermocycler to include the following steps:
Initial ‘hot-start’ phase (95°C - 1 min). Add Pfu polymerase at the end of the ‘hot-start’ phase to ensure complete melting of template DNA and any primer dimers prior to the annealing and extension phases; this improves PCR efficiency
Melting (95°C – 1 min)
Annealing (60°C – 1 min)
Extension (68°C – 1 min/kb of plasmid length).
Repeat steps b through d 17 times, for a total of 18 cycles.
Final extension phase of 68°C for 7 min.
Following PCR amplification, add 1 μL of DpnI restriction enzyme to the PCR tube, and incubate at 37°C for 60 to 90 min.
Use 1 to 2 μL of the DpnI-treated amplification product to transform XL-1 blue competent E. coli cells (TOP10 cells from Invitrogen may be substituted) by adding the DNA to 50 μL of competent cells in a pre-chilled 15 mL Falcon tube and incubate on ice for 30 min. Heat-shock the cells for 30 to 35 s at 42°C followed by incubation on ice for 2 min. Add 250 μL of room temperature SOC medium and plate 50 to 100 μL onto appropriate antibiotic selection LB agar plates. Incubate overnight at 37°C.
Pick one or two colonies per plate for overnight growth in 2–5 mL of liquid LB medium supplemented with appropriate antibiotics.
Perform a plasmid preparation using the Qiagen Spin Miniprep kit (Qiagen) according to the manufacturer’s instructions. Verify presence of desired mutations by sequencing.
3.3 Evaluating membrane expression of mutants
Day 1
Sterilize glass cover slips under UV light (1 h) and place in wells of a sterile 24-well plate.
Add 500 μL of medium (DMEM supplemented with 10% FBS and 1% penicillin/streptomycin) to each well.
Add an appropriate amount (12 μL if starting from a confluent 10 cm plate, or 40 μL from a 60 mm plate) of trypsinized HEK 293 cells from a pre-existing cell line into each well for 20% confluence
Incubate at 37°C overnight, to allow the cells to reach 50 to 70% confluence.
Day 2
Mix 225.6 μL of DMEM (without supplements) and 14.4 μL of Fugene 6. Incubate for 5 min at room temperature.
Divide the mix into 12 microcentrifuge tubes at a concentration of 20 μL each.
Add 400 ng DNA to each tube. Incubate 30 min at room temperature.
Add 10 μL into each well. Incubate plate at 37°C (see Note 8).
Day 4
Remove plate from incubator, remove medium by aspiration.
Wash each well 2 times with 500 μL PCM buffer for 5 min at room temperature.
Add 300 μL of a 100 μg/mL concentration of Concanavalin-A-Alexa 350 in PCM buffer. Incubate 1 h on ice. (see Note 9).
Wash each well 2 times with 500 μL PCM buffer for 5 min at RT.
Fix with 300 μL 4% PFA for 10 min at room temperature.
Wash each well 2 times with 500 μL PBS buffer for 5 min at room temperature.
Permeabilize with 500 μL PBS containing 0.1% Triton for 3 min at room temperature.
Wash 1 time with 500 μL PBS for 5 min at room tmeperature.
Cover with 1% BSA in PBS (500 μL) and incubate for 1 h at room temperature.
Incubate cells with 300 μL of the appropriate primary antibody in PBS for 2 h at 37°C, or overnight at 4°C. Make sure cells in each well are covered by buffer.
Wash each well 2 times with 500 μL PBS buffer for 5 min.
Cover cells with appropriate secondary antibody labeled with AlexaFluor594 in PBS for 1 h at room temperature.
Wash 3 times with 500 μL PBS.
Remove coverslips from wells using forceps, blot excess liquid from the edges onto lint free tissue paper (e.g. Kimwipe, Kimberly-Clark, Dallas, TX).
Add a drop of Fluoromount reagent on a clean dry slide. Invert cover slip (cell side down) onto the drop.
Seal the edges of the cover slip with clear nail polish. Allow to dry 2–3 h at room temperature before storing slides in the refrigerator at 4°C.
View slides under deconvolution microscope at 63× magnification using appropriate wavelength filters. Analyze images for colocalization of concanavalin-A immunofluorescence with that of AlexaFluor 594, which has been used to label the protein under study.
4. Notes
ET analysis can be done for any sample size of sequence homologs. If a structure is available, the statistical significance of structural clustering among top-ranked residues can be computed (13) to judge the results reliability. When a structure is not available, a minimum number of 15–20 sequences will typically produce useful evolutionary trace rankings, but this also depends on the evolutionary spread of these sequences.
A comprehensive evolutionary and phylogenetic tree encompassing the majority of known organisms can be found at the Tree of Life web project http://www.tolweb.org. This information, combined with the taxonomical information included with the protein sequence in NCBI’s database, may be used when ordering the sequences for computational analysis. Evolutionary outliers may also be determined and discarded based on this information.
Further information on ET analysis (1–4) and its applications to several diverse systems (5, 6, 14–16) is readily found in the literature.
Other computational methods based on similar evolutionary and phylogenetic considerations have been developed (17–20). Such phylogeny-based computational analyses have been used in at least two studies to predict functional regions in proteins involved in audition (9, 21).
Evolutionary Trace residues have been shown to exhibit regular periodicity in transmembrane helices and can, therefore, be used to confirm and/or refine the results of a hydrophobicity-based transmembrane topology prediction (9).
We have used Evolutionary Trace analysis, in conjunction with electrophysiological measurements of non-linear capacitance, as a readout of prestin activity, to pinpoint residues in a conserved motif region of the prestin protein that are essential for its function. Further, by analyzing the effects of charge and size substitutions, we have been able to conclude that tight packing of the transmembrane helices in this region is essential for prestin activity. Results of functional assays of individual mutants are shown in Fig. 2 below, and a more detailed account of the study can be found in Rajagopalan et al., 2006 (9).
An automated Evolutionary Trace analysis can be now performed at the ET servers at http://mammoth.bcm.tmc.edu/server.html. However, a step-by-step protocol has been provided to facilitate a thorough understanding of the method, and to allow manual refinement of the automated results when necessary.
These volumes are for an experiment where you use 12 different mutants and all 24 wells, with duplicates of each mutant. Change volumes appropriately depending on your experiment.
Concanavalin-A binds to carbohydrates on the cell surface, and is therefore used as a marker for cell membranes. Con-A labeled with AlexaFluor can be used in immunohistochemical analyses to label membranes, which can then be analyzed microscopically for colocalization to various labeled proteins to evaluate membrane expression of these proteins.
Fig. 2.

Normalized membrane capacitance of prestin single mutants. (A) Five single substitutions at A100: A100S (□), A102G (○), A102V (X), A102L (△) and A102W (□). A100V, L and W (clustered at bottom) have minimal voltage dependence, similar to untransfected or mock-transfected controls. (B) Five single substitutions at A102: A102S (□), A102G (○), A102V (X), A102L (△) and A102W (□). A102V, L and W have minimal voltage dependence, similar to untransfected or mock-transfected controls. (C) Three single substitutions at L104: L104V (□), L104I (△) and L104W (■). L104W has minimal voltage dependence, similar to untransfected or mock-transfected controls. (D) Capacitance plots of L113W (▲) and F117W (□). Representative plots, normalized relative to C (Vpkc) and Clin, from single cells are shown. In all panels, a representative plot of WT prestin is shown for comparison (●). Differences in magnitude of NLC have been ignored in this representation.
Acknowledgments
Preparation of these guidelines was supported in part by fellowship support from the Keck Center for Interdisciplinary Bioscience Training to LR. The authors thank Dr. Ivana Mihalek for helpful comments and suggestions.
References
- 1.Lichtarge O, Bourne HR, Cohen FE. An evolutionary trace method defines binding surfaces common to protein families. J Mol Biol. 1996;257:342–358. doi: 10.1006/jmbi.1996.0167. [DOI] [PubMed] [Google Scholar]
- 2.Lichtarge O, Sowa ME. Evolutionary predictions of binding surfaces and interactions. Curr Opin Struct Biol. 2002;12:21–27. doi: 10.1016/s0959-440x(02)00284-1. [DOI] [PubMed] [Google Scholar]
- 3.Madabushi S, Yao H, Marsh M, Kristensen DM, Philippi A, Sowa ME, Lichtarge O. Structural clusters of evolutionary trace residues are statistically significant and common in proteins. J Mol Biol. 2002;8:139–154. doi: 10.1006/jmbi.2001.5327. [DOI] [PubMed] [Google Scholar]
- 4.Yao H, Kristensen DM, Mihalek I, Sowa ME, Shaw C, Kimmel M, Kavraki L, Lichtarge O. An accurate, sensitive, and scalable method to identify functional sites in protein structures. J Mol Biol. 2003;326:255–261. doi: 10.1016/s0022-2836(02)01336-0. [DOI] [PubMed] [Google Scholar]
- 5.Sowa ME, He W, Slep KC, Kercher MA, Lichtarge O, Wensel TG. Prediction and confirmation of a site critical for effector regulation of RGS domain activity. Nat Struct Biol. 2001;8:234–237. doi: 10.1038/84974. [DOI] [PubMed] [Google Scholar]
- 6.Madabushi S, Gross AK, Philippi A, Meng EC, Wensel TG, Lichtarge O. Evolutionary trace of G protein-coupled receptors reveals clusters of residues that determine global and class-specific functions. J Biol Chem. 2004;279:8126–8132. doi: 10.1074/jbc.M312671200. [DOI] [PubMed] [Google Scholar]
- 7.Ribes-Zamora A, Mihalek I, Lichtarge O, Bertuch AA. Distinct faces of the Ku heterodimer mediate DNA repair and telomeric functions. Nat Struct Mol Biol. 2007;14:301–7. doi: 10.1038/nsmb1214. [DOI] [PubMed] [Google Scholar]
- 8.Shenoy SK, Drake MT, Nelson CD, Houtz DA, Xiao K, Madabushi S, Reiter E, Premont RT, Lichtarge O, Lefkowitz RJ. beta-arrestin-dependent, G protein-independent ERK1/2 activation by the beta2 adrenergic receptor. J Biol Chem. 2006;281:1261–73. doi: 10.1074/jbc.M506576200. [DOI] [PubMed] [Google Scholar]
- 9.Rajagopalan L, Patel N, Madabushi S, Goddard JA, Anjan V, Lin F, Shope C, Farrell B, Lichtarge O, Davidson AL, Brownell WE, Pereira FA. Essential helix interactions in the anion transporter domain of prestin revealed by evolutionary trace analysis. J Neurosci. 2006;26:12727–34. doi: 10.1523/JNEUROSCI.2734-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mihalek I, Res I, Lichtarge O. A family of evolution-entropy hybrid methods for ranking protein residues by importance. J Mol Biol. 2004;336:1265–82. doi: 10.1016/j.jmb.2003.12.078. [DOI] [PubMed] [Google Scholar]
- 11.Morgan DH, Kristensen DM, Mittelman D, Lichtarge O. ET viewer: an application for predicting and visualizing functional sites in protein structures. Bioinformatics. 2006;22:2049–50. doi: 10.1093/bioinformatics/btl285. [DOI] [PubMed] [Google Scholar]
- 12.Mihalek I, Res I, Lichtarge O. Evolutionary trace report_maker: a new type of service for comparative analysis of proteins. Bioinformatics. 2006;22:1656–7. doi: 10.1093/bioinformatics/btl157. [DOI] [PubMed] [Google Scholar]
- 13.Mihalek I, Res I, Yao H, Lichtarge O. Combining inference from evolution and geometric probability in protein structure evaluation. J Mol Biol. 2003;331:263–79. doi: 10.1016/s0022-2836(03)00663-6. [DOI] [PubMed] [Google Scholar]
- 14.Geva A, Lassere TB, Lichtarge O, Pollitt SK, Baranski TJ. Genetic Mapping of the Human C5a Receptor. Identification Of Transmembrane Amino Acids Critical For Receptor Function. J Biol Chem. 2000;275:35393–35401. doi: 10.1074/jbc.M005602200. [DOI] [PubMed] [Google Scholar]
- 15.Lichtarge O, Yamamoto KR, Cohen FE. Identification of functional surfaces of the zinc binding domains of intracellular receptors. J Mol Biol. 1997;274:325–327. doi: 10.1006/jmbi.1997.1395. [DOI] [PubMed] [Google Scholar]
- 16.Sowa ME, He W, Wensel TG, Lichtarge O. A regulator of G protein signaling interaction surface linked to effector specificity. Proc Natl Acad Sci U S A. 2000;97:1483–1488. doi: 10.1073/pnas.030409597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Landgraf R, Xenarios I, Eisenberg D. Three-dimensional cluster analysis identifies interfaces and functional residue clusters in proteins. J Mol Biol. 2001;307:1487–1502. doi: 10.1006/jmbi.2001.4540. [DOI] [PubMed] [Google Scholar]
- 18.Armon A, Graur D, Ben-Tal N. ConSurf: an algorithmic tool for the identification of functional regions in proteins by surface mapping of phylogenetic information. J Mol Biol. 2001;307:447–463. doi: 10.1006/jmbi.2000.4474. [DOI] [PubMed] [Google Scholar]
- 19.Hannenhalli SS, Russell RB. Analysis and prediction of functional subtypes from protein sequence alignments. J Mol Biol. 2000;303:61–76. doi: 10.1006/jmbi.2000.4036. [DOI] [PubMed] [Google Scholar]
- 20.Yang Z, Nielsen R. Codon-substitution models for detecting molecular adaptation at individual sites along specific lineages. Mol Biol Evol. 2002;19:908–917. doi: 10.1093/oxfordjournals.molbev.a004148. [DOI] [PubMed] [Google Scholar]
- 21.Franchini LF, Elgoyhen AB. Adaptive evolution in mammalian proteins involved in cochlear outer hair cell electromotility. Mol Phylogenet Evol. 2006;41:622–635. doi: 10.1016/j.ympev.2006.05.042. [DOI] [PubMed] [Google Scholar]