

Abstract
We have investigated the possible biochemical basis for enhancements in NO production in endothelial cells that have been correlated with agonist- or shear stress-evoked phosphorylation at Ser-1179. We have found that a phosphomimetic substitution at Ser-1179 doubles maximal synthase activity, partially disinhibits cytochrome c reductase activity, and lowers the EC50(Ca2+) values for calmodulin binding and enzyme activation from the control values of 182 ± 2 and 422 ± 22 nm to 116 ± 2 and 300 ± 10 nm. These are similar to the effects of a phosphomimetic substitution at Ser-617 (Tran, Q. K., Leonard, J., Black, D. J., and Persechini, A. (2008) Biochemistry 47, 7557–7566). Although combining substitutions at Ser-617 and Ser-1179 has no additional effect on maximal synthase activity, cooperativity between the two substitutions completely disinhibits reductase activity and further reduces the EC50(Ca2+) values for calmodulin binding and enzyme activation to 77 ± 2 and 130 ± 5 nm. We have confirmed that specific Akt-catalyzed phosphorylation of Ser-617 and Ser-1179 and phosphomimetic substitutions at these positions have similar functional effects. Changes in the biochemical properties of eNOS produced by combined phosphorylation at Ser-617 and Ser-1179 are predicted to substantially increase synthase activity in cells at a typical basal free Ca2+ concentration of 50–100 nm.
The nitric-oxide synthases catalyze formation of NO and l-citrulline from l-arginine and O2, with NADPH as the electron donor (1). The role of NO generated by endothelial nitricoxide synthase (eNOS)2 in the regulation of smooth muscle tone is well established and was the first of several physiological roles for this small molecule that have so far been identified (2). The nitric-oxide synthases are homodimers of 130–160-kDa subunits. Each subunit contains a reductase and oxygenase domain (1). A significant difference between the reductase domains in eNOS and nNOS and the homologous P450 reductases is the presence of inserts in these synthase isoforms that appear to maintain them in their inactive states (3, 4). A calmodulin (CaM)-binding domain is located in the linker that connects the reductase and oxygenase domains, and the endothelial and neuronal synthases both require Ca2+ and exogenous CaM for activity (5, 6). When CaM is bound, it somehow counteracts the effects of the autoinhibitory insert(s) in the reductase. The high resolution structure for the complex between (Ca2+)4-CaM and the isolated CaM-binding domain from eNOS indicates that the C-ter and N-ter lobes of CaM, which each contain a pair of Ca2+-binding sites, enfold the domain, as has been observed in several other such CaM-peptide complexes (7). Consistent with this structure, investigations of CaM-dependent activation of the neuronal synthase suggest that both CaM lobes must participate (8, 9).
Bovine eNOS can be phosphorylated in endothelial cells at Ser-116, Thr-497, Ser-617, Ser-635, and Ser-1179 (10–12). There are equivalent phosphorylation sites in the human enzyme (10–12). Phosphorylation of the bovine enzyme at Thr-497, which is located in the CaM-binding domain, blocks CaM binding and enzyme activation (7, 11, 13, 14). Ser-116 can be basally phosphorylated in cells (10, 11, 13, 15), and dephosphorylation of this site has been correlated with increased NO production (13, 15). However, it has also been reported that a phosphomimetic substitution at this position has no effect on enzyme activity measured in vitro (13). Ser-1179 is phosphorylated in response to a variety of stimuli, and this has been reliably correlated with enhanced NO production in cells (10, 11). Indeed, NO production is elevated in transgenic endothelium expressing an eNOS mutant containing an S1179D substitution, but not in tissue expressing an S1179A mutant (16). Shear stress or insulin treatment is correlated with Akt-catalyzed phosphorylation of Ser-1179 in endothelial cells, and this is correlated with increased NO production in the absence of extracellular Ca2+ (17–19). Akt-catalyzed phosphorylation or an S1179D substitution has also been correlated with increased synthase activity in cell extracts at low intracellular free [Ca2+] (17). Increased NO production has also been observed in cells expressing an eNOS mutant containing an S617D substitution, and physiological stimuli such as shear-stress, bradykinin, VEGF, and ATP appear to stimulate Akt-catalyzed phosphorylation of Ser-617 and Ser-1179 (12, 13, 20). Although S617D eNOS has been reported to have the same maximum activity in vitro as the wild type enzyme (20), in our hands an S617D substitution increases the maximal CaM-dependent synthase activity of purified mutant enzyme ∼2-fold, partially disinhibits reductase activity, and reduces the EC50(Ca2+) values for CaM binding and enzyme activation (21).
In this report, we describe the effects of a phosphomimetic Asp substitution at Ser-1179 in eNOS on the Ca2+ dependence of CaM binding and CaM-dependent activation of reductase and synthase activities. We also describe the effects on these properties of combining this substitution with one at Ser-617. Finally, we demonstrate that Akt-catalyzed phosphorylation and Asp substitutions at Ser-617 and Ser-1179 have similar functional effects. Our results suggest that phosphorylation of eNOS at Ser-617 and Ser-1179 can substantially increase synthase activity in cells at a typical basal free Ca2+ concentration of 50–100 nm, while single phosphorylations at these sites produce smaller activity increases, and can do so only at higher free Ca2+ concentrations.
EXPERIMENTAL PROCEDURES
Expression and Purification of Proteins—The pCWori vector (22, 23) used for bacterial expression of tagged eNOS has been described in detail previously (21). Phosphomimetic mutations were generated using the QuikChange site-directed mutagenesis kit (Stratagene Inc. La Jolla, CA) according to the manufacturer's instructions. The primers used to generate the S1179D phosphomimetic mutation were: sense gtatacgtaccgaggacttttccctgcag; antisense: ggcctgatccacgtgcacagac. Primers used to add the S617D phosphomimetic mutation so as to produce the double S617/S1179D substitution have been described previously (21). The protein-coding regions in all constructs were verified by performing dye terminator DNA sequencing. Expression and purification of wild type and mutant eNOS and wild type CaM have been described in detail elsewhere (21).
In Vitro Phosphorylation of eNOS—Reactions (250 μl) contained 20 μm each of purified wild type eNOS and CaM in 5 mm Mg2+ acetate, 1 mm ATP, 250 μm Ca2+, 100 mm KCl, and 25 mm Tris, pH 7.5 were incubated for 40 min at 25 °C after addition of the indicated amounts of purified human Akt-1 that was activated by incubation with PDK1 and MAPKAPK2 (Millipore Corp., Billerica, MA). Reactions mixtures were then dialyzed for 2 h at 4 °C against 1 liter of 100 mm KCl, 25 mm Tris, pH 7.5. Final eNOS concentrations in dialysates were determined from optical absorbance spectra as described previously (21). Enzyme prepared in this manner was used in CaM binding and enzyme activity assays. To identify the sites phosphorylated, 20 ng of phosphorylated or control eNOS (incubated in the absence of Akt-1) were subject to SDS-polyacrylamide gel electrophoresis, and phosphorylation was assessed by immunoblotting with phosphospecific antibodies. Enzyme samples analyzed by immunoblotting before and after CaM binding experiments exhibited no detectable change in the levels or pattern of phosphorylation (data not shown). Preactivated human Akt-1 and antibodies against phosphoserines at positions 617, 635, and 1179 in bovine eNOS were purchased from Millipore Corp. Immunoblotting procedures were performed according to the manufacturer's instructions. Bound antibody was detected using an enhanced chemiluminescence kit and high performance chemiluminescence film as detailed by the manufacturer (GE Healthcare, Piscataway, NJ). Densitometric analysis of immunoblots was performed using a flatbed transparency scanner and the Genetools software package (Synoptics Ltd., Cambridge, UK).
Quantitation of eNOS Phosphorylation—Quantitation of Akt-catalyzed phosphorylation of eNOS or S617/1179D eNOS was performed as described in detail elsewhere (24, 25). Briefly, phosphorylation reactions (100 μl) contained 20 μm each of purified wild type or S617/1179D eNOS and CaM in 5 mm Mg2+ acetate, 1 mm [γ-32P]ATP (PerkinElmer Life Sciences, Inc., Boston, MA), 250 μm Ca2+, 100 mm KCl, and 25 mm Tris, pH 7.5, were incubated for 40 min at 25 °C in the presence of the indicated amounts of preactivated Akt-1. Reactions were terminated by adding SDS electrophoresis sample buffer, and 40 pmol of phosphorylated or control eNOS was subject to SDS gel electrophoresis on 8% polyacrylamide gel slabs. Gel slabs were dried, and bands containing 32P were visualized by autoradiography. Because ∼10% of total phosphate incorporation appeared to be in contaminating protein bands (data not shown), the 130-kDa bands corresponding with eNOS were excised from a second gel stained with Coomassie Blue. Gel slices were solubilized and decolorized by incubation with 0.25 ml of 30% H2O2 for 60 min at 90 °C, and the 32P content of each was quantitated by scintillation counting.
Determination of CaM Binding versus [Ca2+] Relationships—Binding of CaM to eNOS as a function of [Ca2+] was determined based on the reduction in the amount of Ca2+-free CaM bound to a fluorescent protein biosensor (BSCaMA) as we have described in detail previously (21). At the CaM and BSCaMA concentrations used, the biosensor is less than 50% saturated with CaM, so there is a direct correspondence between the fractional decrease in the amount of CaM bound to the biosensor and the fractional increase in the amount of CaM bound to eNOS. Because eNOS binds Ca2+-CaM with a Kd value below 1 nm (26), at micromolar protein concentrations and a 1:1 molar ratio of eNOS and CaM fractional binding (FB) of CaM to eNOS is related to the BSCaMA fluorescence emission ratio (r = 525 nm/480 nm) in Equation 1,
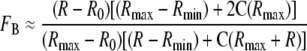 |
(Eq. 1) |
where Rmax and Rmin are the emission ratios for CaM-free and CaM-saturated BSCaMA, and C = Sb/Sf, where Sf and Sb are the fluorescence emission intensities at 480 nm for CaM-free and CaM-saturated BSCaMA. Corresponding free Ca2+ concentrations were derived from concurrent determinations of indo-1 emission ratios (r = 405 nm/485 nm) in the standard manner as detailed previously (21). Measurements of BSCaMA and indo-1 fluorescence were performed using a PTI QM-1 fluorometer (New Brunswick, NJ), with respective excitation wavelengths of 430 and 330 nm. Emission and excitation bandwidths were set to 2.5 nm. Reactions containing 25 mm Tris, pH 7.5, 100 mm KCl, 3 μm indo-1, 5 μm BSCaMA, 0.1 mg/ml BSA, 1.25 mm BAPTA, 3.5 μm eNOS, and 3.5 μm CaM were incubated at 22 °C in a stirred 2-ml quartz cuvette. Measurements of BSCaMA and indo-1 fluorescence were made after incremental additions of CaCl2, which produced negligible increases in the reaction volume.
Determination of Enzyme Activity versus [Ca2+] Relationships—The Ca2+ dependence for enzyme activation was determined as described previously (21), under conditions comparable to those used for analyses of CaM binding. Enzyme activities were derived from linear least-squares fits to time courses for the decrease in NADPH fluorescence emission at 460 nm (340-nm excitation) due to its conversion to NADP+. Free Ca2+ concentrations were derived from concurrent measurements of calcium orange fluorescence emission at 570 nm (550 nm excitation) in the standard manner. Reactions containing 25 mm Tris, pH 7.5, 100 mm KCl, 40 μm NADPH, 10 μm tetrahydrobiopterin, 50 μm l-arginine, 0.4 μm calcium orange, 0.1 mg/ml BSA, 1.25 mm BAPTA, 0.7 μm eNOS, and 0.7 μm CaM were incubated at 22 °C in a stirred 2-ml quartz cuvette. In a typical experiment enzyme activity under nominally Ca2+-free conditions was established, an aliquot of a CaCl2 stock solution was added, and the change in calcium orange fluorescence and rate of NADPH oxidation were measured. These measurements were then repeated after adding another aliquot of CaCl2. Because substrate is rapidly consumed, only 1 or 2 sets of measurements were made, after which 1.5 mm CaCl2 was added, and maximum CaM-dependent activity was established for that experiment. Enzyme activities from each set of measurements are expressed as a fraction of the baseline corrected maximum CaM-dependent activity determined for that experiment. Fractional activities derived from measurements at several different intermediate free Ca2+ concentrations were combined to produce a complete Ca2+ titration curve. NADPH, tetrahydrobiopterin, and l-arginine at the concentrations used do not affect the Ca2+ dependences for CaM binding (data not shown).
Analysis of Relationships between CaM Binding or Enzyme Activation and [Ca2+]—EC50(Ca2+) values and cooperativity coefficients were derived from data for fractional CaM binding (FB) or enzyme activation (FA) by non-linear least squares fitting to a simple logistic equation as described previously (21). KD values of 230 and 200 nm were derived for Ca2+ binding to indo-1 and calcium orange by calibrating their responses against those of a fluorescent protein Ca2+ indicator that binds Ca2+ via an EF hand pair (27). Calibration of the indicators in this manner allows the relationships between CaM binding, enzyme activation and [Ca2+] to be quantitatively related according to a previously described sequential model in which formation of the CaM-eNOS complex requires Ca2+ binding to one EF hand pair in CaM, and activation requires binding to both EF hand pairs (21). According to this model, fractional CaM binding, FB, is defined in Equation 2,
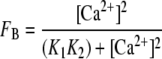 |
(Eq. 2) |
where K1K2 is the product of the Ca2+ binding constants for the pair of Ca2+-binding sites that control binding. Fractional CaM-dependent enzyme activity, FA, is defined in Equation 3,
 |
(Eq. 3) |
where K3K4 is the product of the Ca2+ binding constants for the pair of sites controlling activation. Data sets CaM binding or enzyme activation versus [Ca2+] for native or mutant enzyme were simultaneously fitted to Equations 2 and 3 using the Excel solver to derive the fits shown in Fig. 4, with the K1K2 and K3K4 values listed in Table 2.
FIGURE 4.

Fractional CaM binding (•) and enzyme activation (○) plotted versus [Ca2+] for (A) wild type, (B) S1179D, and (C) S617/1179D eNOS. Binding and activation data were simultaneously fitted to Equations 2 and 3 using the Excel solver add-in to generate the curves shown. The K1K2 and K3K4 values derived from these fits are listed in Table 2.
TABLE 2.
Parameters derived by simultaneously fitting CaM binding and enzyme activation versus [Ca2+] data to an ordered model
In this model CaM binding occurs when one pair of Ca2+-binding sites in CaM is occupied and activation occurs when the second pair is occupied (Eqs. 2 and 3). K1K2 and K3K4 are the products of the dissociation constants for first and second pair of Ca2+-binding sites. Standard errors were derived from the variance-covariance matrices for the fits.
| eNOS | K1K2 | K3K4 |
|---|---|---|
| nm2 | nm2 | |
| Wild typea | 28712 ± 2810 | 142582 ± 12330 |
| S617Da | 11832 ± 1165 | 54196 ± 4742 |
| S1179D | 15687 ± 2806 | 72168 ± 11595 |
| S617/1179D | 6183 ± 501 | 12325 ± 1014 |
Values taken from Tran et al. (21).
RESULTS
Enzyme Activities—The maximum CaM-dependent synthase activities of the S1179D and S617/1179D eNOS mutants are both ∼2-fold higher than the wild type activity (Fig. 1). In the absence of CaM, the wild type and mutant enzymes have activities that are ≤ 15% of the maximum CaM-dependent values. The activity of the S617/1179D mutant appears to be slightly higher than the wild type activity under these conditions (Fig. 1). As seen in Fig. 2A in the absence of CaM the NADPH oxidase activities of the wild type and mutant enzymes are ≤ 25% of the maximum CaM-dependent values. The activities of both mutant proteins are significantly higher than the wild type activity under these conditions, and the activity of the double mutant is also significantly higher than the activity of the single mutant. In the presence of CaM, NADPH oxidase activities determined for the mutant enzymes are ∼2-fold higher than the wild type activity. As previously reported by Gao et al. (28), these activity measurements are consistent with an NADPH:NO ratio of ∼2, higher than the value of 1.5 expected based on the catalytic mechanism. This may reflect uncoupled reductase activity or chemical oxidation of NADPH. Despite this discrepancy, the relative NADPH oxidase and synthase activities of the mutant and wild type enzymes are well correlated (Figs. 1 and 2A), supporting our use of the former to determine activity versus [Ca2+] relationships.
FIGURE 1.

Nitric-oxide synthase activities of WT eNOS and the indicated mutants determined in the presence and absence of saturating concentrations of Ca2+ and CaM. Error bars are standard deviations for the means of six independent determinations made using a minimum of two different enzyme preparations. Nitric oxide production was monitored by measuring the conversion of l-arginine to l-citrulline as described previously (21). The specific activity determined for wild type eNOS under these conditions is 82.4 ± 3.3 nmol/min-mg. Reactions contained 50 mm Tris, pH 7.5, 10 μm BH4, 100 μm DTT, 50 μm NADPH, 51 μm l-[3H]arginine, 100 mm KCl, 0.1 mg/ml BSA, 250 μm CaCl2, and 125 nm eNOS, plus or minus 1.3 μm CaM. Values indicated by an asterisk are significantly higher (p < 0.005) than the corresponding wild type value.
FIGURE 2.

A, NADPH oxidase activities of WT eNOS and the indicated mutants determined in the presence and absence of saturating concentrations of Ca2+ and CaM. The reaction buffer contained 50 mm Tris, pH 7.5, 100 mm KCl, 0.1 mg/ml BSA, 10 μm BH4, 100 μm DTT, 50 μm NADPH, 250 μm CaCl2, 50 μm l-arginine, and 125 nm eNOS, plus or minus 1.3 μm CaM. B, cytochrome c reductase activities of WT eNOS and the indicated mutants determined in the presence and absence of saturating concentrations of Ca2+ and CaM. The reaction buffer contained 50 mm Tris, pH 7.5, 100 mm KCl, 0.1 mg/ml BSA, 50 μm NADPH, 100 μm cytochrome c, 250 μm CaCl2, and 125 nm enzyme, plus or minus 1.3 μm CaM. In both panels, error bars are standard deviations for the mean of six independent determinations made using a minimum of two different enzyme preparations. Minus CaM values indicated by daggers are significantly higher (p < 0.005) than the wild type value, and those indicated by asterisks are significantly higher than the value for S1179D eNOS. Plus CaM values indicated by asterisks are significantly higher (p < 0.005) than the corresponding wild type value. The small difference between the reductase activities of the S617/1179D mutant measured in the presence and absence of CaM presented in panel B is not significant (p = 0.11).
To investigate the effects of phosphomimetic substitutions on the reductase component of eNOS activity, cytochrome c reductase assays were performed (Fig. 2B). As seen with both synthase and NADPH oxidase activities, maximum CaM-dependent reductase activity is increased ∼2-fold by the S1179D or S617/1179D substitution (Fig. 2B). In the absence of CaM, the reductase activity of the S1179D mutant is significantly higher than the wild type enzyme activity, but the double substitution completely disinhibits reductase activity, eliminating its dependence on Ca2+ and CaM (Fig. 2B).
In summary, the S1179D substitution increases synthase and reductase activities ∼2-fold in the presence of CaM and partially disinhibits reductase activity, lowering its dependence on CaM. These effects are essentially identical to those produced by an S617D substitution (21). The double S617/1179D substitution has essentially identical effects on these enzyme activities in the presence of CaM. However, cooperativity between the two substitutions completely disinhibits reductase activity and slightly elevates CaM-independent synthase activity.
Relationships among CaM Binding, Enzyme Activation and [Ca2+]—Given stoichiometric amounts of eNOS and CaM at concentrations at least 1000-fold higher than the apparent Kd for the CaM-enzyme complex (26), the dependence of the complex on [Ca2+] is determined by its intrinsic Ca2+ binding properties, or more precisely, the Ca2+ binding properties of CaM bound to eNOS (21). The intrinsic EC50(Ca2+) values determined under these conditions allow the Ca2+ dependence of different CaM-enzyme complexes to be compared quantitatively. Fits of data for fractional CaM binding (FB) versus [Ca2+] to a logistic equation are presented in Fig. 3A. The EC50(Ca2+) values determined for wild type, S1179D and S617/1179D eNOS are 182 ± 2, 116 ± 2, and 77 ± 2 nm (Table 1). Data for fractional NADPH oxidase activity versus [Ca2+] were determined under conditions similar to those used for binding assays, except for the presence of 10 μm BH4, 40 μm NADPH, and 50 μm l-arginine, which do not significantly affect EC50(Ca2+) values for CaM binding (21). Fits of these data to a logistic equation are presented in Fig. 3B. EC50(Ca2+) values of 422 ± 23, 300 ± 10 and 130 ± 5 nm were derived for wild type and S1179D and S617/1179D eNOS (Table 1). As seen in Table 1, the EC50(Ca2+) values for enzyme activation and CaM binding determined for the S1179D mutant are essentially identical to those reported previously for the S617D mutant (21).
FIGURE 3.

A, dependence of fractional CaM binding on [Ca2+] for wild type (▴), S1179D (○), and S617/1179D (•) eNOS. The reaction buffer contained 25 mm Tris, pH 7.5, 100 mm KCl, 3 μm indo-1, 5 μm BSCaMA, 0.1 mg/ml BSA, 1.25 mm BAPTA, and 3.5 μm eNOS and CaM. B, dependence of fractional enzyme activation on Ca2+ for wild type (▴), S1179D (○), and S617/1179D (•) eNOS. The reaction buffer contained 25 mm Tris, pH 7.5, 100 mm KCl, 40 μm NADPH, 10 μm tetrahydrobiopterin, 50 μm l-arginine, 0.1 mg/ml BSA, 1.25 mm BAPTA, 0.7 μm eNOS, and CaM and 0.4 μm calcium orange. The curves in both panels correspond with nonlinear least-squares fits to a logistic binding equation. The parameters derived from these fits are presented in Table 1. These data sets are representative of experiments performed with a minimum of two different enzyme preparations.
TABLE 1.
EC50(Ca2+) values and Hill coefficients (n) derived from logistic fits of CaM binding and enzyme activation versus [Ca2+] data for wild-type eNOS and the indicated mutants
These parameters correspond with the fits to a logistic equation seen in Fig. 3. Standard errors were derived from the variance-covariance matrices for the fits. Values indicated by a dagger were determined in f-tests to be significantly (p < 0.001) smaller than the corresponding values derived for wild-type eNOS. Values indicated by an asterisk were determined in f-tests to be significantly smaller than the corresponding values derived for S1179D eNOS.
|
eNOS
|
Binding
|
Activation
|
||
|---|---|---|---|---|
| EC50(Ca2+) | n | EC50(Ca2+) | n | |
| nm | nm | |||
| Wild typea | 182 ± 2 | 2.25 ± 0.04 | 422 ± 23 | 2.06 ± 0.21 |
| S617Da | 109 ± 2† | 2.06 ± 0.05 | 258 ± 11† | 2.39 ± 0.27 |
| S1179D | 116 ± 2† | 2.0 ± 0.04 | 300 ± 10† | 2.95 ± 0.32 |
| S617/1179D | 77 ± 2* | 2.1 ± 0.04 | 130 ± 5* | 2.90 ± 0.24 |
Values are taken from Tran et al. (21).
The difference that we have observed between the Ca2+ dependence for CaM binding and enzyme activation suggests a sequential process in which Ca2+ binding to one pair of sites in CaM controls formation of the CaM-enzyme complex, and subsequent binding to the other pair controls enzyme activation (21). The CaM binding and enzyme activation versus [Ca2+] data for each protein were therefore fit simultaneously to Equations 2 and 3 to derive K1K2 and K3K4 values. No attempt was made to derive individual Ca2+ binding constants, which are poorly determined because Ca2+ binding to each pair of sites is highly cooperative. In any event, the products of the two constant are sufficient for our purposes. The fitted curves are presented in Fig. 4, and the parameters derived are listed in Table 2. As seen in the figure, a sequential model for CaM binding and enzyme activation provides a good fit to the data, including the property that enzyme activity exhibits a steeper dependence on [Ca2+] than CaM binding, with the difference in steepness becoming larger as the binding and activation curves approach one another (Fig. 4). Thus, as we have noted previously (21), increases in the Hill coefficients for eNOS activation associated with phosphomimetic substitutions (see Table 1) are fully accounted for by a sequential model for CaM binding and activation.
In summary, the S1179D substitution reduces the EC50(Ca2+) values for CaM binding and enzyme activation ∼35%, essentially identical to the effect of an S617D substitution (21). Combined S617D and S1179D substitutions appear to act cooperatively, reducing EC50(Ca2+) values twice as much as either does alone. The Ca2+ dependence determined for CaM binding and enzyme activation are consistent with a sequential process in which binding of Ca2+ to one of the two pairs of Ca2+-binding sites in CaM controls formation of the CaM-enzyme complex, and binding to the other then produces enzyme activation.
Comparison of the Functional Effects of Phosphorylations and Phosphomimetic Substitutions at Ser-617 and Ser-1179—To confirm that results obtained using Asp substitutions at Ser-617 and Ser-1179 are analogous to those produced by bona fide phosphorylations, we functionally characterized enzyme specifically phosphorylated at these positions by incubation with preactivated Akt-1. Akt-catalyzed incorporation of phosphate into wild type eNOS or the S617/1179D mutant was quantitated in electrophoretically purified enzyme (see “Experimental Procedures”). As seen in Fig. 5A, Akt catalyzed a dose-dependent incorporation of phosphate into eNOS, with ∼1.8 mol of phosphate incorporated/mol enzyme at a kinase concentration of 500 nm. There is negligible incorporation into the S617/1179D mutant under these conditions, demonstrating that Ser-617 and Ser-1179 are specifically phosphorylated (Fig. 5A). Phosphoimmunoblots confirm that at a kinase concentration of ∼500 nm Ser-617 and Ser-1179 are both phosphorylated, and there is negligible phosphorylation of Ser-635 (Fig. 5B, inset). These observations suggest that Akt-1 catalyzes phosphorylation of Ser-617 and Ser-1179 with similar efficiency. However, a method for quantitating doubly and singly phosphorylated enzyme produced at submaximal kinase concentrations would be required to properly evaluate the phosphorylation mechanism. We therefore chose to characterize only the highly phosphorylated protein produced after incubation with 500 nm Akt-1.
FIGURE 5.

Akt-catalyzed phosphorylation of Ser-617 and Ser-1179 and its effect on the [Ca2+] dependence of CaM binding. A, 32P incorporation into electrophoretically purified eNOS measured after incubation for 40 min with 1 mm [γ-32P]ATP and the indicated concentrations of preactivated Akt-1. Error bars are standard deviations for means of three replicates. Corresponding 32P autoradiography and Coomassie Blue staining of eNOS protein bands are shown below the main panel. B, CaM binding versus [Ca2+] relationships determined for eNOS incubated in the presence (•) and absence (▴) of 500 nm preactivated Akt-1. The solid lines correspond with nonlinear least-squares fits of these data to a logistic binding equation. Respective EC50(Ca2+) values of 165 ± 2 and 62 ± 2 nm were derived from these fits. Assay conditions are given in the legend to Fig. 3.
Fractional CaM binding versus [Ca2+] relationships determined for eNOS after incubation in the presence and absence of 500 nm Akt-1 are presented in Fig. 5B. Respective EC50(Ca2+) values of 165 ± 2 and 62 ± 2 nm were derived for control and phosphorylated enzyme from the logistic fits shown. These are similar to values of 182 ± 2 and 77 ± 2 nm derived for the untreated wild type enzyme and the S617/1179D mutant protein (see Table 1). We also determined the NADPH oxidase and reductase activities of control and maximally phosphorylated eNOS in the presence and absence of Ca2+-CaM (Fig. 6). As seen in Fig. 6A, phosphorylation doubles maximal Ca2+-CaM-dependent oxidase activity and slightly increases CaM independent activity. Similarly, as seen in Fig. 6B, phosphorylation doubles maximal reductase activity and eliminates its dependence on CaM. These effects are not significantly different from the effects of the S617/1179D double mutation (Fig. 2, A and B).
FIGURE 6.

The effects of Akt-catalyzed phosphorylation of Ser-617 and Ser-1179 on NADPH oxidase (A) and cytochrome c reductase (B) activities. These activities were measured in the presence and absence of Ca2+ using eNOS incubated in the presence or absence of 500 nm Akt-1. NADPH oxidase reactions contained 50 mm Tris, pH 7.5, 100 mm KCl, 0.1 mg/ml BSA, 10 μm BH4, 100 μm DTT, 50 μm NADPH, 1.3 μm CaM, 50 μm l-arginine, and 125 nm eNOS, plus or minus 250 μm CaCl2. Cytochrome c reductase reactions contained 50 mm Tris, pH 7.5, 100 mm KCl, 0.1 mg/ml BSA, 50 μm NADPH, 100 μm cytochrome c, 1.3 μm CaM, and 125 nm enzyme, plus or minus 250 μm CaCl2. Error bars are standard deviations for the means of six independent determinations made using a minimum of two different enzyme preparations. Values indicated by asterisks are significantly higher (p < 0.005) than the corresponding wild type value.
In summary, we have demonstrated that Ser-617 and Ser-1179 are specifically phosphorylated by Akt-1 in vitro, and have confirmed the phosphorylations and phosphomimetic Asp substitutions at these positions have similar effects on the functional properties of eNOS.
Predicted Effects of Phosphorylation on the Relationship between Synthase Activity and [Ca2+]—Based on these results we can predict the activity ratios, or fold-increases in activity, that are produced by combined phosphorylation of Ser-617 and Ser-1179, and by a single phosphorylation at Ser-1179, which appears to be representative of either singly phosphorylated species. As seen in Fig. 7A, phosphorylation at both positions is predicted to increase synthase activity ∼150- and ∼50-fold at Ca2+ concentrations of 50 and 100 nm. A single phosphorylation is predicted to increase synthase activity by much smaller factors of ∼7 and ∼6 at these Ca2+ concentrations. As the Ca2+ concentration is increased the activity ratios for doubly and singly phosphorylated enzyme both approach a value of 2, corresponding with the effect of phosphorylation on maximal CaM-dependent activity. Above a Ca2+ concentration of ∼500 nm, this effect appears to be the primary contributor to phosphorylation-dependent increases in enzyme activity.
FIGURE 7.

A, predicted activity ratios (fold-increases) in eNOS activity produced by double and single phosphorylations of eNOS plotted versus [Ca2+]. Calculations were performed using Equation 3 and the K1K2 and K3K4 values for S617/1179D (solid line), S1179D (dashed line), and wild type eNOS listed in Table 2, The predicted effect of phosphorylation on maximum CaM-dependent activity was accounted for by a factor of 2. Activity ratios for singly phosphorylated enzyme are also plotted on an expanded scale in the inset. B, predicted normalized fractional activities for doubly, singly, and nonphosphorylated eNOS plotted versus [Ca2+]. Fractional activities were calculated using Equation 3 and the K1K2 and K3K4 values for S617/1179D (solid line), S1179D (dashed line), and wild type (dotted line) eNOS listed in Table 2. These values were normalized with respect to the maximal activity of the non-phosphorylated enzyme. The predicted effect of phosphorylation on maximum CaM-dependent activity was accounted for by a factor of 2, so in this plot the maximum values for the non-phosphorylated and phosphorylated proteins are, respectively, 1 and 2.
While activity ratios provide a revealing look at the predicted effects of phosphorylation, it is important to also consider the corresponding absolute enzyme activities. The predicted relationships between fractional enzyme activity and [Ca2+] for singly, doubly, and nonphosphorylated enzyme are presented in Fig. 7B. For comparative purposes the fractional activities given in this figure have been “normalized” to the maximum activity for the nonphosphorylated enzyme, so the maximum for this enzyme species is 1, and the maximum given for the phosphorylated species is 2. As seen in the figure, the predicted normalized fractional activities for the doubly phosphorylated enzyme are ∼0.2 and ∼0.9 at Ca2+ concentrations of 50 and 100 nm. Thus, at a Ca2+ concentration of 100 nm this enzyme species is predicted to have an activity equivalent to ∼90% of the maximum activity of non-phosphorylated eNOS. The predicted normalized fractional activities for singly phosphorylated enzyme at Ca2+ concentrations of 50 and 100 nm are almost insignificant, with values of ∼0.01 and ∼0.1.
In summary, combined phosphorylation at Ser-617 and Ser-1179 is predicted to produce substantial increases in eNOS activity at a typical basal Ca2+ concentration of 50–100 nm. In contrast, single phosphorylations at Ser-617 or Ser-1179 are predicted to produce smaller activity increases and to do so only at significantly higher Ca2+ concentrations.
DISCUSSION
Biochemical Implications—The effects of the S1179D substitution on EC50(Ca2+) values that we report here are similar to those produced by an S617D substitution (21). The effects of the S1179D substitution on synthase and reductase activity that we report are similar to those reported by McCabe et al. (29). These investigators did not report an effect of this substitution on the EC50(Ca2+) value for enzyme activation, but they did note that it decreased the apparent rate at which the CaM-enzyme complex inactivates after addition of a Ca2+ chelator (29). Combined S617D and S1179D substitutions appear to act cooperatively on EC50(Ca2+) values and reductase activity, but the double substitution produces the same maximum CaM-dependent synthase and reductase activities as a single substitution. The disconnect between the effects of the double and single substitutions on reductase activity and synthase activity is consistent with a model in which CaM activates the reductase and oxygenase domains via somewhat different mechanisms. Activation of the reductase appears to involve reversing the effects of at least two autoinhibitory elements (4, 30), while activation of the oxygenase appears to involve a facilitation of electron transfer from the reductase to the active site heme (31).
Previous investigations of interactions between neuronal NOS and CaM fragments corresponding with the N-ter and C-ter lobes of the protein suggest that the C-ter lobe binds prior to the N-ter lobe and drives a relatively weak association with the N-ter lobe that is required for enzyme activation (8). This is consistent with the different dependence of CaM binding and eNOS activation on [Ca2+], as these fit a sequential model (Equation 3), and further suggests that the EC50(Ca2+) values for CaM binding and enzyme activation correspond with Ca2+ binding to the C-ter and N-ter CaM lobes. We have previously proposed that a CaM mutant in which the N-ter lobe has been replaced by a copy of the C-ter lobe may be unable to activate the neuronal synthase because the mutant N-ter lobe is weakly associated with the CaM-binding domain in the enzyme (32). Roman et al. (33) have recently demonstrated that this mutant CaM partially activates a mutant neuronal synthase whose reductase activity has been disinhibited by removal of an autoinhibitory domain. Perhaps the energetic consequences of disinhibition increase association with the N-ter lobe in the mutant CaM enough to produce partial activation. This raises the possibility that phosphorylation acts through a similar mechanism, increasing maximal CaM-dependent synthase activity by increasing the degree to which the N-ter lobe in wild type CaM is associated with the CaM-binding domain in the enzyme.
Physiological Implications—Stimulation of endothelial cells by shear stress or insulin is correlated with Akt-catalyzed phosphorylation of Ser-1179 and increased NO production in the absence of extracellular Ca2+ (17–19). Akt-catalyzed phosphorylation or an S1179D substitution has also been correlated with increased synthase activity at low [Ca2+] in cell extracts (17). Based on these observations, it has been proposed that Akt-dependent phosphorylation of eNOS at Ser-1179 activates eNOS activity through a Ca2+-independent mechanism (17). However, it has recently become clear that physiological stimuli such as shear stress, bradykinin, VEGF, and ATP stimulate phosphorylation of Ser-617 in addition to Ser-1179 (12, 20). This raises the possibility that some of the effects attributed to phosphorylation of Ser-1179 may also require phosphorylation of Ser-617. Our results demonstrate that Ser-617 and Ser-1179 are both specifically phosphorylated by Akt-1 in vitro and also suggest that phosphorylation at both sites is required to produce significant enyzme activation at basal Ca2+ concentrations. As we have noted previously (21), the micromolar concentrations of CaM and eNOS we have used for determinations of EC50(Ca2+) values for CaM binding and enzyme activation are similar to estimates of their concentrations in endothelial cells. The EC50(Ca2+) values determined at these protein concentrations correspond with intrinsic Ca2+ dependence (21) and should be applicable to eNOS in the cell. Our data clearly do not support a Ca2+-independent mechanism for enzyme activation, but rather suggest that phosphorylation of both Ser-617 and Ser-1179 reduces the EC50(Ca2+) value for enzyme activation enough to substantially activate CaM-dependent synthase activity at a typical basal Ca2+ concentration of 50–100 nm. Our results also suggest that a single phosphorylation at Ser-617 or Ser-1179 can enhance synthase activity during stimulus-evoked increases in the intracellular free Ca2+ concentration.
To quantitatively relate the observed phosphorylation-dependent biochemical changes in eNOS to cellular responses it is necessary to determine the population of singly and doubly phosphorylated enzyme, and the free Ca2+ concentrations experienced by these population(s). Spatial heterogeneity in the cell makes such determinations challenging. For example, cytoplasmic [Ca2+] signals produced in response to shear stress can be largely confined to perinuclear and subplasma membrane regions (34). Furthermore, the degree and pattern of eNOS phosphorylation appear to vary with subcellular location (35), and this difficulty is compounded by the fact that physiological changes in the phosphorylation status of eNOS involve several sites in the enzyme and an assortment of kinases and phosphatases (10–12). Interpretation of correlations between phosphorylation and changes in NO production in the cell are further complicated by the presence of additional regulatory factors such as HSP90 and caveolin (10–12). Nevertheless, our results suggest that phosphorylation-dependent changes in the biochemical properties of eNOS could account for Akt-mediated increases in NO production in endothelial cells, particularly at basal free Ca2+ concentrations.
This work was supported, in whole or in part, by National Institutes of Health Grant GM074887 (to A. P.).
Footnotes
The abbreviations used are: eNOS, endothelial nitric-oxide synthase; FP, fractional phosphorylation; FB, fractional binding; FA, fractional activity; CaM, calmodulin; BSCaMA, fluorescent biosensor containing a CaM binding sequence based on the IQ domain in neuromodulin, BAPTA, 1,2-bis(2-aminophenoxy)ethane-N,N,N′N′-tetraacetic acid; BH4, tetrahydrobiopterin; BSA, bovine serum albumin; WT, wild type; DTT, dithiothreitol.
References
- 1.Masters, B. S. S., McMillan, K., Sheta, E. A., Nishimura, J. S., Roman, L. J., and Martasek, P. (1996) FASEB J. 10 552–558 [DOI] [PubMed] [Google Scholar]
- 2.Moncada, S., Palmer, R. M. J., and Higgs, E. A. (1991) Pharmacol. Rev. 43 109–142 [PubMed] [Google Scholar]
- 3.Daff, S. (2003) Biochem. Soc. Trans. 31 502–505 [DOI] [PubMed] [Google Scholar]
- 4.Nishida, C. R., and Ortiz de Montellano, P. R. (1999) J. Biol. Chem. 274 14692–14698 [DOI] [PubMed] [Google Scholar]
- 5.Venema, R. C., Sayegh, H. S., Kent, J. D., and Harrison, D. G. (1996) J. Biol. Chem. 271 6435–6440 [DOI] [PubMed] [Google Scholar]
- 6.Zhang, M., and Vogel, H. J. (1994) J. Biol. Chem. 269 981–985 [PubMed] [Google Scholar]
- 7.Aoyagi, M., Arvai, A. S., Tainer, J. A., and Getzoff, E. D. (2003) EMBO J. 22 766–775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Persechini, A., McMillan, K., and Leakey, P. (1994) J. Biol. Chem. 269 16148–16154 [PubMed] [Google Scholar]
- 9.Persechini, A., Stemmer, P. M., and Ohashi, I. (1996) J. Biol. Chem. 271 32217–32225 [DOI] [PubMed] [Google Scholar]
- 10.Boo, Y. C., and Jo, H. (2003) Am. J. Physiol. Cell Physiol. 285 C499–C508 [DOI] [PubMed] [Google Scholar]
- 11.Fleming, I., and Busse, R. (2003) Am. J. Physiol.-Reg. Integr. Comp. Physiol. 284 R1–R12 [DOI] [PubMed] [Google Scholar]
- 12.Mount, P. F., Kemp, B. E., and Power, D. A. (2007) J. Mol. Cell. Cardiol. 42 271–279 [DOI] [PubMed] [Google Scholar]
- 13.Bauer, P. M., Fulton, D., Boo, Y. C., Sorescu, G. P., Kemp, B. E., Jo, H., and Sessa, W. C. (2003) J. Biol. Chem. 278 14841–14849 [DOI] [PubMed] [Google Scholar]
- 14.Matsubara, M., Hayashi, N., Jing, T., and Titani, K. (2003) J. Biochem. 133 773–781 [DOI] [PubMed] [Google Scholar]
- 15.Kou, R. Q., Greif, D., and Michel, T. (2002) J. Biol. Chem. 277 29669–29673 [DOI] [PubMed] [Google Scholar]
- 16.Scotland, R. S., Morales-Ruiz, M., Chen, Y., Yu, J., Rudic, R. D., Fulton, D., Gratton, J. P., and Sessa, W. C. (2002) Circ. Res. 90 904–910 [DOI] [PubMed] [Google Scholar]
- 17.Dimmeler, S., Fleming, I., Fisslthaler, B., Hermann, C., Busse, R., and Zeiher, A. M. (1999) Nature 399 601–605 [DOI] [PubMed] [Google Scholar]
- 18.Fulton, D., Gratton, J. P., McCabe, T. J., Fontana, J., Fujio, Y., Walsh, K., Franke, T. F., Papapetropoulos, A., and Sessa, W. C. (1999) Nature 399 597–601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Montagnani, M., Chen, H., Barr, V. A., and Quon, M. J. (2001) J. Biol. Chem. 276 30392–30398 [DOI] [PubMed] [Google Scholar]
- 20.Michell, B. J., Harris, M. B., Chen, Z. P., Ju, H., Venema, V. J., Blackstone, M. A., Huang, W., Venema, R. C., and Kemp, B. E. (2002) J. Biol. Chem. 277 42344–42351 [DOI] [PubMed] [Google Scholar]
- 21.Tran, Q. K., Leonard, J., Black, D. J., and Persechini, A. (2008) Biochemistry 47 7557–7566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.McMillan, K., and Masters, B. S. (1995) Biochemistry 34 3686–3693 [DOI] [PubMed] [Google Scholar]
- 23.Roman, L. J., Sheta, E. A., Martasek, P., Gross, S. S., Liu, Q., and Masters, B. S. (1995) Proc. Natl. Acad. Sci. U. S. A. 92 8428–8432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.King, M. M., Fitzgerald, T. J., and Carlson, G. M. (1983) J. Biol. Chem. 258 9925–9930 [PubMed] [Google Scholar]
- 25.Nadeau, O. W., Anderson, D. W., Yang, Q., Artigues, A., Paschall, J. E., Wyckoff, G. J., McClintock, J. L., and Carlson, G. M. (2007) J. Mol. Biol. 365 1429–1445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tran, Q. K., Black, D. J., and Persechini, A. (2005) Cell Calcium 37 541–553 [DOI] [PubMed] [Google Scholar]
- 27.Persechini, A., Lynch, J. A., and Romoser, V. A. (1997) Cell Calcium 22 209–216 [DOI] [PubMed] [Google Scholar]
- 28.Gao, Y. T., Panda, S. P., Roman, L. J., Martasek, P., Ishimura, Y., and Masters, B. S. S. (2007) J. Biol. Chem. 282 7921–7929 [DOI] [PubMed] [Google Scholar]
- 29.McCabe, T. J., Fulton, D., Roman, L. J., and Sessa, W. C. (2000) J. Biol. Chem. 275 6123–6128 [DOI] [PubMed] [Google Scholar]
- 30.Lane, P., and Gross, S. S. (2002) J. Biol. Chem. 277 19087–19094 [DOI] [PubMed] [Google Scholar]
- 31.Panda, K., Ghosh, S., and Stuehr, D. J. (2001) J. Biol. Chem. 276 23349–23356 [DOI] [PubMed] [Google Scholar]
- 32.Persechini, A., Gansz, K. J., and Paresi, R. J. (1996) Biochemistry 35 224–228 [DOI] [PubMed] [Google Scholar]
- 33.Roman, L. J., and Masters, B. S. (2006) J. Biol. Chem. 281 23111–23118 [DOI] [PubMed] [Google Scholar]
- 34.Geiger, R. V., Berk, B. C., Alexander, R. W., and Nerem, R. M. (1992) Am. J. Physiol. 262 C1411–C1417 [DOI] [PubMed] [Google Scholar]
- 35.Fulton, D., Fontana, J., Sowa, G., Gratton, J. P., Lin, M., Li, K. X., Michell, B., Kemp, B. E., Rodman, D., and Sessa, W. C. (2002) J. Biol. Chem. 277 4277–4284 [DOI] [PubMed] [Google Scholar]