

Abstract
In this study, we attempted to understand the mechanism of regulation of the activity and allosteric behavior of the pyruvate kinase M2 enzyme and two of its missense mutations, H391Y and K422R, found in cells from Bloom syndrome patients, prone to develop cancer. Results show that despite the presence of mutations in the intersubunit contact domain, the K422R and H391Y mutant proteins maintained their homotetrameric structure, similar to the wild-type protein, but showed a loss of activity of 75 and 20%, respectively. Interestingly, H391Y showed a 6-fold increase in affinity for its substrate phosphoenolpyruvate and behaved like a non-allosteric protein with compromised cooperative binding. However, the affinity for phosphoenolpyruvate was lost significantly in K422R. Unlike K422R, H391Y showed enhanced thermal stability, stability over a range of pH values, a lesser effect of the allosteric inhibitor Phe, and resistance toward structural alteration upon binding of the activator (fructose 1,6-bisphosphate) and inhibitor (Phe). Both mutants showed a slight shift in the pH optimum from 7.4 to 7.0. Although this study signifies the importance of conserved amino acid residues in long-range communications between the subunits of multimeric proteins, the altered behavior of mutants is suggestive of their probable role in tumor-promoting growth and metabolism in Bloom syndrome patients with defective pyruvate kinase M2.
Pyruvate kinase (PK3; EC 2.7.1.40), a pacemaker of the glycolytic pathway, catalyzes irreversibly the transphosphorylation from P-enolpyruvate to ADP, generating pyruvate and ATP (1, 2). There are four different isozymes (L, R, M1, and M2) in mammalian tissues, which differ in their regulatory properties. These isozymes are allosteric in nature with the exception of the M1 form, present in skeletal muscle and brain (3–7). PKM2 is a ubiquitous prototype enzyme present in all tissues during the embryonic stage and is gradually replaced by other isozymic forms in specific tissues during development. The M2, L, and R isozymes show homotropic cooperative activation with P-enolpyruvate and heterotropic cooperative activation with Fru-1,6-P2 (8–10). The M1 isozyme is regulated by neither P-enolpyruvate nor Fru-1,6-P2 because of its intrinsic active conformation in the R-state (5, 6). Under unfavorable conditions such as hypoxia and lack of glucose supply, the anaerobic tissues and tumor cells rely heavily on PKM2 for ATP production (7). Therefore, stringent control of PK activity is of great importance not only for cell metabolism but also for tumorigenic proliferation.
The M1 and M2 isozymes are produced from a single gene locus by mutually exclusive alternative splicing (11–14). In the human M1 and M2 isozymes, the exon that is exchanged because of alternative splicing encodes 56 amino acids, in which a total of 22 amino acids differ within a length of 45 residues. The residues located in this region form the major intersubunit contact domain (8). The distinguishable kinetic properties of the M1 and M2 isozymes are attributed to these amino acid substitutions. It has been shown by x-ray crystallographic analyses and computer modeling that the corresponding regions of their polypeptides participate directly in the intersubunit contact, which is responsible for the intersubunit communication required for allosteric cooperativity (8, 15).
PK has been largely conserved throughout evolution. The enzyme is usually a homotetramer composed of four identical subunits, and each subunit consists of four domains: the A-, B-, and C-domains and the N-terminal domain. The structure of human PKM2 was recently determined in complex with inhibitors (16). In mammalian cells, PK activity is regulated by two different mechanisms: one at the level of expression and the other through allosteric regulation. The catalytic site usually composes a small part of the enzyme, but allosteric control is transmitted over a long range, thus increasing the number of possible residues involved in regulation. The allosteric transition in PK involves mutual rotations of the A- and C-domains within each subunit and the subunit within the tetramer (14). The residues at the subunit interfaces have the critical function of relaying the allosteric signal from and to the catalytic and regulatory sites. This region also transmits the allosteric signal between P-enolpyruvate- and Fru-1,6-P2-binding sites. Despite the availability of structural details of several PK isozymes, it is difficult to identify the structural elements that play an important role in PK regulation and propagation of the allosteric signals. Although the role of some of the PK residues (positions 340, 389, 398, 401, 402, 408, 423, and 427) has been studied in allosteric regulation (10, 17–19) by in vitro site-directed mutagenesis, the absence of these mutations in any naturally occurring condition presents limitations in attributing a biological role to the introduced changes.
The natural mutations H391Y and K422R (reported previously as K421R) were reported by us for the first time in the PKM2 gene in a Bloom syndrome cell line and in the lymphocytes of an Indian Bloom syndrome patient, respectively (20). The two missense mutations, located in the region of the intersubunit contact domain (Fig. 1, A and B), presented with the biochemical phenotype of down-regulated enzyme activity to different extents (20) and were expected to influence the allosteric nature of the enzyme. The regulatory behavior of allosteric PK has been described by a two-state model that proposes an active (R) and an inactive (T) form of the macromolecule with differential affinity for ligands (15). Upon binding of the substrate or its analogs, the enzyme undergoes a transition from a low activity/low affinity conformation (T state) to a high activity/high affinity conformation (R state). The binding of phenylalanine produces a global structural change and exhibits reduced affinity for substrate P-enolpyruvate in the T state (21–23). Previous studies have demonstrated that each individual domain acts as a rigid body and that, upon transition from the T to the R state, the domain of the functional tetramer modifies its relative orientation by 29°. These movements bring conformational change to the active site, which, upon transition to the T state, undergoes a distortion of the P-enolpyruvate-binding site (24).
FIGURE 1.

A, ribbon diagram of the overall structure of PK showing the positions of the two mutations, H391Y and K422R, along with the active site and Fru-1,6-P2-binding site. B, intersubunit contact domain of PK. The major amino acid residues and side chains at the tetramer interface region are shown.
Because the mutations observed by us previously (20) are located at highly conserved positions not only in different isozymic forms but also across the species (supplemental Fig. S1) and are observed in the genetic background of a syndrome prone to cancer in early age, a study related to the structure-function correlations of these mutations is likely to provide insight into their possible biological importance, especially in the context of recent research highlighting the importance of PKM2 in tumor promotion and growth. In this study, we investigated the role of the two natural missense mutations, after site-directed mutagenesis in the PKM2 gene, in the regulation of allosteric properties as well as their effects on the secondary and tertiary structures in comparison with wild-type PKM2 (PK-WT). An attempt has also been made to understand the effects of these mutations at the interface of the subunits on the signal transmission pathway within the protein.
EXPERIMENTAL PROCEDURES
Materials—Culture medium with supplementations (Invitrogen), the QuikChange site-directed mutagenesis kit (Stratagene), restriction enzymes (New England Biolabs), and a sequencing kit (BDT v3.1, Applied Biosystems) were obtained from the indicated manufacturers. Yeast extract, Tryptone, and agar were obtained from Pronadisa (Madrid, Spain). Bradford reagent for protein concentration estimation, chemicals for kinetic assay, and other general chemicals were purchased from Sigma.
Synthesis and Cloning of Wild-type and Mutant PKM2 cDNAs—Lymphocytes isolated from blood samples of normal human subjects (25) were cultured in complete RPMI 1640 medium and stimulated with phytohemagglutinin at a final concentration of 10 μg/ml. Total RNA was purified and reverse-transcribed to cDNA to amplify the PK-WT gene using PK-WT-specific primers. The amplified cDNA (1.6 kb) was cloned into the pGEM-T vector and subcloned into the pGEX-4T1 and pET-28a(+) vectors. The mutant versions of PK were made by site-directed mutagenesis using specific mutagenic primers (supplemental Table SI). The mutant H391Y and K422R constructs were validated for precise incorporation by sequencing.
Expression, Purification, and Size Exclusion Chromatography of Wild-type and Mutant PKM2—For expression of soluble proteins, the Escherichia coli BL21(DE3) strain harboring the appropriate plasmid of wild-type and mutant PKM2 constructs was induced with 0.1 mm isopropyl β-d-thiogalactopyranoside and incubated in a shaker at 20 °C for 10–12 h. The cells were sonicated, and the soluble fraction was passed through a nickelnitrilotriacetic acid column. The bound fraction eluted in buffer containing 250 mm imidazole was further purified to homogeneity using anion exchange DEAE-Sepharose resin. PK activity was measured by NADH/lactate dehydrogenase-coupled assay as described (20). Protein concentration was determined by the Bradford dye-binding assay and by using molar extinction coefficients (30,490 m–1 cm–1 or 0.527 liter·g–1·cm–1) for PKM2 and mutant proteins (26, 27). There was no increase in the evaluated extinction coefficient for the K422R mutant and only a small increase for the H391Y mutant (<3%), which was found to be negligible and within the experimental uncertainty of the measurements. Hence, the reported extinction coefficient for wild-type PK was used for the wild-type as well as the mutant proteins. The tetrameric quaternary state of the purified PK-WT, H391Y, and K422R proteins was confirmed by loading 5.0 mg of purified protein on a Superdex 300 superfine gel permeation column. 0.5-ml fractions were collected at a flow rate of 0.5 ml/min and assayed for total protein concentration and activity.
Kinetic Analysis—Different concentrations of P-enolpyruvate and ADP were used to obtain kinetic parameters for the dependence of ADP and P-enolpyruvate in the presence and absence of the heterotropic activator Fru-1,6-P2. Saturation kinetics were performed using different concentration (0–5 mm) of Fru-1,6-P2 and the allosteric inhibitor Phe (0–2 mm). Initial velocity data were fit to either the Michaelis-Menten equation (Equation 1) or the Hill equation (Equation 2) depending on which model described the experimental data best,
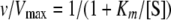 |
(Eq. 1) |
 |
(Eq. 2) |
where v represents the apparent maximal velocity, Vmax is the true maximal velocity under the given experimental conditions, Km is the Michaelis constant (s0.5), [S] is the concentration of substrate, and nH is the Hill number. The effect of phenylalanine on substrate affinity (s0.5) was also measured. The parameters were fit to the Hanes-Woolf graph equation (Equation 3) to calculate the Ki.
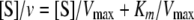 |
(Eq. 3) |
CD and Fluorescence Spectroscopy—CD spectra were recorded in the far-UV region from 200 to 350 nm at 25 °C using a Jasco J-815 spectropolarimeter. The protein concentration used was 0.3 mg/ml in 10 mm phosphate buffer (pH 7.5), 3 mm MgCl2, 100 mm KCl, and 5% glycerol. Data below 200 nm could not be acquired as a result of an increase in the high tension voltage due to solvent absorbing strongly below 200 nm. The percent secondary structure was calculated using the on-line tool SOMCD. The results were expressed as mean residual ellipticity ([θ], degrees cm2/dmol) using Equation 4,
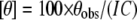 |
(Eq. 4) |
where θobs represents the observed ellipticity in degrees, c is the concentration in moles of residue/liter, and l is the cuvette path length in centimeters. Intrinsic fluorescence measurements were performed using 50 μg/ml wild-type and mutant proteins at 25 °C and pH 7.5 in a Cary Varian Eclipse spectrofluorometer using an excitation wavelength of 295 nm, and the emission intensities were recorded in the range of 310–450 nm. The effect of increasing concentrations of Fru-1,6-P2, P-enolpyruvate, and Phe on the wild-type and mutant proteins was also investigated. The percent fluorescence quenching (Q) was calculated using Equation 5,
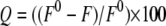 |
(Eq. 5) |
where F and F0 represent the fluorescence intensities in the presence and absence of a ligand, respectively.
pH Dependence and Thermostability Study—The PK-WT, H391Y, and K422R proteins were incubated in reaction buffer at the required pH for 5 min at room temperature, and the reaction was initiated by the addition of lactate dehydrogenase and P-enolpyruvate. The P-enolpyruvate saturation kinetics were also studied over a range of pH to study the effect of pH on Km. Vmax, Km, and Hill number was calculated by fitting the data in Equation 6,
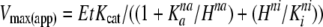 |
(Eq. 6) |
where Vmax(app) is the apparent maximal velocity, EtKcat represents the true maximal velocity, Ka is the concentration for half-maximal activation by H+, na is the number of protons involved in activation, ni is the number of protons involved in inhibition, and H is the concentration of H+. The thermal denaturation of PK-WT and the mutant proteins at a concentration of 0.3 mg/ml at pH 7.5 was monitored by observing changes in ellipticity (CD signal) at 222 nm from 25 to 80 °C. For monitoring thermostability based on activity, the proteins were incubated at a concentration of 0.3 mg/ml at 60 °C for 20 min, and samples were immediately analyzed for biological activity at room temperature after every 5 min. The renaturation profile was studied by cooling the fully denatured (inactive) protein on ice and by checking activity after every 5 min. Similar experiments were also carried out at 37 °C with or without the activator Fru-1,6-P2.
Bioinformatics-based Structural Analysis—The three-dimensional structures of tumor PKM2 (Protein Data Bank code 1t5a), cat muscle PK (code 1pyk), and rabbit muscle PK (code 1pkn) were viewed and compared with each other using Deep-view/Swiss-Pdb Viewer v4.0. Following this, the structure of the mutant version of PK-WT was generated by replacing His391 with Tyr. The H-bond detection threshold used was 0.5–3.5 Å for distances and 120° for angles when hydrogen atoms were present and 1.0 Å and 90° when hydrogen atoms were absent. The stability of mutant proteins was predicted using on-line tool CUPSAT (28).
RESULTS
Overexpression and Solubilization of PK-WT and Mutant Enzymes—Because human PK-WT was cloned downstream of a strong T7 promoter, part of the recombinant protein was trapped in insoluble inclusion bodies. Various conditions such as the concentration of isopropyl β-d-thiogalactopyranoside, the time period, and the temperature of induction were attempted to reduce the inclusion body formation, and success was achieved in obtaining a reasonable amount of active protein (i.e. 3–4 mg of purified protein from 200 ml of culture). All proteins were purified by affinity and ion exchange chromatography to near homogeneity. The purified proteins were used for kinetic and structural studies.
Oligomeric Nature of Mutant Enzymes—The PK-WT, H391Y, and K422R proteins were loaded independently in the gel permeation column and found to be eluted in fractions 15–23 (data not shown). Each of these fractions was analyzed for PK activity, showing the maximal activity in fraction 19. The two mutants maintained their tetrameric form comparable with the wild-type protein. Each fraction was analyzed for PK activity, and both mutants H391Y and K422R showed specific activities reduced by 20 and 75%, respectively.
Alteration in Allosteric Behavior of Mutant Enzymes upon Binding with P-enolpyruvate—The kinetic parameters calculated from the P-enolpyruvate saturation curves for PK-WT and the mutants (supplemental Figs. S2–S4) are displayed in Table 1A. The K422R mutant showed ∼75% reduced maximal activity (Vmax) and a reduced Kcat/Km (efficiency) of 6.31 ± 0.6 s–1 mm–1 compared with 83.7 ± 5s–1 mm–1 for PK-WT. The K422R mutant was observed to be slightly more cooperative, as reflected by an increased Hill coefficient (nH = 2.6 ± 0.07) compared with PK-WT (nH = 2.1 ± 0.08). The affinity of mutant K422R was found to be compromised as shown by an increased [P-enolpyruvate]0.5 of 1.5 ± 0.1 mm compared with PK-WT (0.46 ± 0.01 mm). Interestingly, mutant H391Y exhibited ∼20% reduced maximal velocity in comparison with PK-WT but, unlike mutant K422R, showed a significantly increased Kcat/Km (385.4 ± 9 s–1 mm–1). The H391Y mutant appeared to lose cooperative binding with P-enolpyruvate, as reflected by a reduced Hill coefficient (nH = 1.2 ± 0.08) and increased P-enolpyruvate affinity ([P-enolpyruvate]0.5 = 0.078 ± 0.009 mm) in comparison with PK-WT. Thus, the change of a single amino acid residue resulted in the H391Y mutant being almost non-allosteric, whereas the K422R mutant lost a significant part of its affinity for P-enolpyruvate.
TABLE 1.
Kinetic parameters of recombinant PK-WT and mutant enzymes
|
Protein
|
Vmax
|
[S]0.5
|
nH
|
Kcat/[S]0.5
|
|||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| -Fru-1,6-P2 | +Fru-1,6-P2 | -Fru-1,6-P2 | +Fru-1,6-P2 | -Fru-1,6-P2 | +Fru-1,6-P2 | -Fru-1,6-P2 | +Fru-1,6-P2 | ||||
| units/mg | mm | s-1 mm1 | |||||||||
| A. P-enolpyruvate saturation kineticsa,b,c | |||||||||||
| PK-WT | 724 ± 4 | 810 ± 5 | 0.46 ± 0.01 | 0.047 ± 0.08 | 2.1 ± 0.08 | 1 ± 0.08 | 83.7 ± 5 | 100.79 ± 7 | |||
| K422R | 178 ± 5 | 233 ± 7 | 1.50 ± 0.1 | 0.082 ± 0.02 | 2.6 ± 0.07 | 1 ± 0.07 | 6.31 ± 0.6 | 139.7 ± 4 | |||
| H391Y | 564.2 ± 3 | 852.2 ± 3 | 0.078 ± 0.009 | 0.077 ± 0.05 | 1.2 ± 0.08 | 0.9 ± 0.01 | 385.47 ± 9 | 554.2 ± 9 | |||
| B. ADP saturation kineticsa,c,d | |||||||||||
| PK-WT | 718.3 ± 9 | 812.1 ± 7 | 0.19 ± 0.01 | 0.18 ± 0.04 | 1.1 ± 0.01 | 1 ± 0.01 | 202.6 ± 5 | 263.1 ± 10 | |||
| K422R | 195.2 ± 6 | 240 ± 11 | 0.17 ± 0.02 | 0.11 ± 0.02 | 1.21 ± 0.05 | 1 ± 0.03 | 55.6 ± 5 | 190.9 ± 6 | |||
| H391Y | 555.7 ± 9 | 841.2 ± 9 | 0.312 ± 0.09 | 0.168 ± 0.07 | 1.35 ± 0.03 | 0.95 ± 0.1 | 96.3 ± 8 | 154 ± 6 | |||
Kinetic parameters were obtained by fitting the data to the Hill equation.
Done at 2.5 mm ADP.
Fru-1,6-P2 was used at a final concentration of 2.5 mm wherever required.
Done at 2.5 mm P-enolpyruvate.
Differential Binding of Mutants with ADP—The kinetic parameters calculated from the ADP saturation curves for PK-WT and the mutants (supplemental Figs. S5–S7) are displayed in Table 1B. The K422R mutant showed ∼75% reduced maximal activity but slightly increased affinity for ADP as [ADP]0.5 reduced 0.17 ± 0.02 mm compared with PK-WT (0.19 ± 0.01 mm). The Hill coefficient for K422R (nH = 1.21 ± 0.05) did not show a significant change compared with that for PK-WT (nH = 1.1 ± 0.01). Unlike K422R, H391Y showed an increased [ADP]0.5 (0.31 ± 0.09 mm) and a slight change in cooperativity (nH = 1.35 ± 0.03) compared with PK-WT (nH = 1.1 ± 0.01).
Differential Response of Mutants to the Allosteric Activator Fru-1,6-P2—Both PK-WT and the K422R mutant showed an almost 20% increase in maximal activity with the saturating concentration of Fru-1,6-P2 (2.5 mm) (Fig. 2A). The K422R mutant also showed increased affinity for P-enolpyruvate as shown by a decline in the [P-enolpyruvate]0.5 from 1.50 ± 0.1 to 0.082 ± 0.02 mm in the presence of Fru-1,6-P2, somewhat similar to PK-WT, which showed a decline from 0.46 ± 0.01 to 0.047 ± 0.08 mm. There was a loss in cooperativity as the Hill coefficient (nH) reduced to 1 in both cases. Interestingly, for the H391Y mutant, the addition of Fru-1,6-P2 showed a relatively greater increase (∼45%) in Vmax with no significant change in the Hill coefficient and [P-enolpyruvate]0.5 (Table 1A and supplemental Figs. S2–S4). To sum up, both mutants showed a differential sensitivity to Fru-1,6-P2, i.e. a relative increase in affinity in the case of K422R (18-fold in comparison with a 10-fold increase in PK-WT) and an increase in Vmax (852.2 ± 3 units/mg) to bring the activity to almost equivalent to that of PK-WT (810 ± 5 units/mg) in the case of H391Y. Both mutants also showed increased affinity for ADP in the presence of Fru-1,6-P2 in comparison with PK-WT, as the [ADP]0.5 for K422R was reduced to 0.11 ± 0.02 mm from 0.17 ± 0.02 mm, and that of H391Y was reduced to 0.168 ± 0.07 mm from 0.312 ± 0.09 mm (Table 1B and supplemental Figs. S5–S7).
FIGURE 2.

A, Fru-1,6-P2 titration curves. B, phenylalanine titration curves for PK-WT (•) and the K422R (○) and H391Y (▾) mutant proteins. Enzyme activity was assayed at 25 °C and pH 7.5 as described under “Experimental Procedures” with 2.5 mm P-enolpyruvate and 2.5 mm ADP.
Differential Effect of the Allosteric Inhibitor Phe on Mutants—PK-WT exhibited slightly different kinetic behavior in the presence of the allosteric inhibitor Phe in contrast to the K422R and H391Y mutants (Table 2). Upon binding with the allosteric inhibitor, PK-WT showed reduced affinity for the substrate as shown by an increased [P-enolpyruvate]0.5 from 0.46 ± 0.01 to 0.796 ± 0.1 mm. The Ki calculated form the Hanes-Woolf plot (supplemental Figs. S8–S10) for PK-WT was 0.176 ± 0.02 mm. In comparison with PK-WT, the K422R mutant showed almost 3-fold sensitivity for the inhibitor with a Ki of 0.067 ± 0.009 mm. However, the H391Y mutant behaved differentially by showing less of an effect with the inhibitor (Ki = 1.4 ± 0.1 mm) (Table 2). Upon binding to Phe (1 mm), PK-WT and K422R showed losses of 76.5 and 80% activity, respectively, compared with a loss of 23.5% observed in the case of H391Y (Fig. 2B and Table 2).
TABLE 2.
Effect of activator Fru-1,6-P2 and inhibitor Phe on activity of PK-WT and mutant enzymes
|
Protein
|
Fru-1,6-P2 saturation
kinetics,aactivity
increasedb
|
Effect of
Phea
|
|||||
|---|---|---|---|---|---|---|---|
|
Activity
leftc
|
Ki
|
||||||
| -Fru-1,6-P2 | +Fru-1,6-P2 | -Fru-1,6-P2 | +Fru-1,6-P2 | ||||
| % | % | mm | |||||
| PK-WT | 20.5 ± 1 | 23.5 ± 1 | 40.7 ± 2 | 0.176 ± 0.02 | 0.261 ± 0.05 | ||
| K422R | 20 ± 1.5 | 20 ± 2 | 32.1 ± 3.1 | 0.067 ± 0.009 | 0.162 ± 0.04 | ||
| H391Y | 42.5 ± 3 | 76.5 ± 3.3 | 81.12 ± 4 | 1.431 ± 0.1 | 1.78 ± 0.07 | ||
Done at fixed concentrations of 2.5 mm P-enolpyruvate and 2.5 mm ADP.
Done at ≥2.5 mm Fru-1,6-P2.
Done at 1.0 mm Phe.
pH Stability of Mutants—The effects of pH on P-enolpyruvate binding to the active sites of PK-WT and mutants H391Y and K422R are shown in Table 3A. Unbound PK-WT and mutant enzymes showed maximal activity at pH 7.5 (from the Vmax/Km profile) (Fig. 3A), whereas upon binding to P-enolpyruvate (Vmax profile) (Fig. 3B), the pH optimum of PK-WT shifted from 7.5 to 7.4, but that of the mutants showed an abrupt decrease of 0.5 units from 7.5 to 7.0 (Fig. 3, A and B). There was a negligible effect of Fru-1,6-P2 observed over the pH optimum (data not shown). The values obtained from the Vmax/Km profile (Fig. 3A) provided the ionization of free enzyme (PK) and free substrate (P-enolpyruvate). The maximal value obtained from this profile was 1.76 ± 8 min–1 mg–1 for PK-WT, 7.08 ± 6 min–1 mg–1 for H391Y, and 0.946 ± 3 min–1 mg–1 for K422R. The change in the Km versus pH profile (supplemental Fig. S11) revealed the sensitivity of the active site to change in pH. With a deviation of ±1 pH unit from the optimum, the K422R mutant showed a greater loss in affinity compared with PK-WT. However, this loss in affinity was less in the case of the H391Y mutant. The Hill number (nH) versus pH profile also showed alterations, similar to the Vmax/Km profile (supplemental Fig. S12).
TABLE 3.
Effect of varying pH and temperature on activity of PK-WT and mutant enzymes
|
Protein
|
Optimum pH
|
Slope (na)
|
Slope (ni)
|
||||||
|---|---|---|---|---|---|---|---|---|---|
| -Fru-1,6-P2 | +Fru-1,6-P2 | -Fru-1,6-P2 | +Fru-1,6-P2 | -Fru-1,6-P2 | +Fru-1,6-P2 | ||||
| A. pH-dependent studya,b | |||||||||
| PK-WT | 7.4 ± 0.2 | 7.4 ± 0.2 | 3.68 ± 0.2 | 3.7 ± 0.12 | 0.35 ± 0.12 | 0.34 ± 0.1 | |||
| K422R | 7 ± 0.1 | 7 ± 0.1 | 3.59 ± 0.1 | 3.48 ± 0.1 | 0.91 ± 0.1 | 0.25 ± 0.2 | |||
| H391Y | 7 ± 0.2 | 7 + 0.3 | 1.56 ± 0.1 | 1.5 ± 0.12 | 0.86 ± 0.08 | 0.16 ± 0.1 | |||
|
Protein
|
Incubation at 60 °C, activity lost after 10 min
|
Cooling in ice, activity regained after 15 min
|
Incubation at 37 °C, activity left after 3 h
|
||||||
|
-Fru-1,6-P2
|
+Fru-1,6-P2
|
-Fru-1,6-P2
|
+Fru-1,6-P2
|
-Fru-1,6-P2
|
+Fru-1,6-P2
|
||||
| % | % | % | |||||||
| B. Temperature-dependent studya,b | |||||||||
| PK-WT | 100 | 100 | 0 | 0 | 44 ± 2 | 58 ± 1 | |||
| K422R | 100 | 100 | 0 | 0 | 20 ± 1 | 32 ± 2 | |||
| H391Y | 2 ± 0.01 | 0 | 64 ± 2 | 64 ± 3 | 99 ± 1 | 100 | |||
Assays were done under standard assay conditions as described under “Experimental Procedures.”
Fru-1,6-P2 was used at a final concentration of 2.5 mm wherever required.
FIGURE 3.

A, PK-WT and the mutant enzymes were incubated at different pH values in 50 mm Tris-Cl buffer and assayed under standard conditions as described under “Experimental Procedures.” A graph of log Vmax and pH was plotted to check the pH optimum of the P-enolpyruvate-bound state of the enzymes. B, P-enolpyruvate titration study was done at different pH values in Tris-Cl buffer, and Km values at each individual pH were calculated. The Vmax/Km ratios obtained at different pH values were plotted against pH as shown.
Mutants Have Higher α-Helical Content than PK-WT—The CD spectrum of PK-WT showed a broad negative ellipticity, exhibiting a double minimum at 208 and 222 nm, typical of α-helices (Fig. 4). The K422R and H391Y mutants showed minima at the same wavelength but with a significantly increased α-helical content. Based on SOMCD analysis, PK-WT showed 38% α-helical content, 20% β-sheets, and 42% random coils and turns. This matches well with the values reported from the x-ray crystallography structure (36% α-helical and 19% β-sheets) (16). However, the α-helical content for the H391Y and K422R mutants deduced from their CD spectra was 67 and 97.5%, respectively. The increase in the α-helical content for the K422R and H391Y mutants appears to result from the conversion of a large portion of the random coil structure as observed in the wild-type protein to α-helical structure. Surprisingly, these mutations also resulted in a decrease in the β-sheet content, suggesting that these mutations may affect the folding pathway of the protein and stabilize the α-helical structure more than the β-form. The heterotropic activator Fru-1,6-P2 and the allosteric inhibitor Phe had no effect on the CD spectra even at saturating concentrations (data not shown).
FIGURE 4.

The CD spectra of PK-WT and the mutant proteins were recorded in the far-UV region (from 200 to 250 nm) at 25 °C at a concentration of 0.3 mg/ml in 10 mm phosphate buffer (pH 7.5), 3 mm MgCl2, 100 mm KCl, and 5% glycerol. The mean residual ellipticity (MRE) was plotted for a wavelength from 200 to 250 nm. deg, degrees.
Differential Thermal Stability of Mutants—Thermal denaturation of secondary structure as monitored by CD spectroscopy showed an increased transition temperature (Tm) for the K422R (56.1 °C) and H391Y (55.2 °C) mutants in comparison with PK-WT (51.5 °C) (Fig. 5A). After 10 min of incubation at 60 °C, PK-WT and K422R lost the activity completely, whereas H391Y maintained 98 ± 1% of its total activity (Table 3B). H391Y showed a 40% loss in activity after 15 min of incubation and almost lost the activity completely after 25 min (Fig. 5B). This thermal denaturation was found to be reversible in the case of H391Y because it regained almost 64% of its total activity after cooling in ice for 15 min. This was unlike the results obtained with PK-WT and K422R, which did not regain the activity upon cooling (Fig. 5C). A similar trend was observed at the physiological temperature of 37 °C, at which H391Y did not show any activity loss until 4 h of incubation, whereas PK-WT and K422R showed 44 and 20% activity, respectively, at the end of 3 h (Fig. 5D). The presence of the heterotropic activator Fru-1,6-P2 provided resistance to the change in Tm to some extent (supplemental Fig. S13).
FIGURE 5.

A, loss of ellipticity upon increasing temperature analyzed by CD spectroscopy for PK-WT and the mutant proteins. B, loss of biological activity at 60 °C. C, regain of activity after cooling the denatured proteins on ice. D, activity assay at physiological temperature (37 °C) over a given time period. All assays were done under standard reaction conditions as described under “Experimental Procedures.” mdeg, millidegrees.
Trp Environment Is Significantly Altered in the Mutant Enzyme—Replacement of Lys422 with Arg and of His391 with Tyr resulted in a substantial increase in Trp fluorescence without affecting the emission maximum at 348 nm (Fig. 6A). The addition of P-enolpyruvate led to substantial quenching of the fluorescence intensity of PK-WT. A distinct blue shift of 10 nm from 348 to 338 nm in the emission maxima suggests a large conformational change in the molecule (Fig. 6B). The shift was more prominent in the case of K422R (16 nm). Interestingly H391Y showed no blue shift, suggesting that P-enolpyruvate did not induce any change in H391Y as it did in the structures of PK-WT and K422R (Fig. 6, C and D). In addition, the magnitude of quenching fluorescence intensity upon binding to P-enolpyruvate (5 mm) was also found to be less in H391Y (17%) in comparison with K422R (38%) and PK-WT (50%). A similar pattern was observed for binding to Fru-1,6-P2 (5 mm), where H391Y showed less quenching (10%) in comparison with PK-WT (18%) and K422R (30%) and no red shift as observed in K422R (4 nm) (supplemental Figs. S14–S16). H391Y also showed less quenching (21%) of Trp fluorescence upon binding to the allosteric inhibitor Phe in comparison with PK-WT (73%) and K422R (38.19%) (Fig. 6, E–G).
FIGURE 6.

The intrinsic tryptophan fluorescence of the proteins (50 μg/ml) was measured upon excitation at 295 nm and by scanning emission in the range of 310–450 nm. A, emission spectra of PK-WT and the K422R and H391Y mutant proteins. B–D, P-enolpyruvate titration of PK-WT, K422R, and H391Y, respectively, showing quenching with increasing concentrations of P-enolpyruvate (PEP). E–G, PK-WT, H391Y, and K422R, respectively, showing quenching with increasing concentrations of the inhibitor phenylalanine. au, absorbance units.
Mutations in the Intersubunit Contact Domain Can Affect the Local and Distant Atomic Interactions—Structure analysis of the H391Y model revealed the possibility of the formation of a unique H-bond 2.98 Å in length by the replaced tyrosine residue with the backbone of Glu386, connecting the A- and C-domains of the protein (Fig. 7, A and B). The H391Y mutant was observed to be more thermostable with an average ΔΔG of 2 kcal/mol using the on-line tool CUPSAT, whereas a reduced thermostability with an average ΔΔG of –2 kcal/mol was observed for the K422R mutant. It was observed that His391 is buried in the structure and surrounded by many hydrophobic residues such as Leu392, Pro446, Tyr390, and Phe395 (Fig. 8A) and that replacement of any amino acid that is more hydrophobic than His results in a more stable molecule. Lys422 in the K422R mutant was observed to be involved in making a H-bond with the Ala419 backbone to align the position of Glu418 within the monomer to make a stable H-bond with Arg399 of the neighboring monomer (Fig. 8B).
FIGURE 7.

A, the wild-type human PKM2 structure shows His391 (red) of the A-chain and Glu386 (blue) of the C-chain. B, a unique H-bond 2.98 Å in length was predicted between the replaced tyrosine residue (red) and the backbone of Glu386 (blue) connecting the A- and C-domains of the protein. It is hypothesized that formation of a new hydrogen bond connecting the A- and C-domains could restrict the movements along the hinges of the A- and C-chains and cause compromised dynamics in the molecule.
FIGURE 8.

A, His391 in human PKM2 is surrounded by hydrophobic residues such as Leu392, Pro446, Tyr390, and Phe395, which make a local hydrophobic cluster. B, Lys422 (blue) with its protruding side chain is found to interact with Ala419 (turquoise) to align the position of Glu418 (orange) of the same monomer to make a stable H-bond with Arg399 (red) of the neighboring monomer.
DISCUSSION
The altered metabolic phenotypes associated with tumorigenesis have recently been associated with differential regulation of a glycolytic pathway isozyme, PKM2. However, it is not known if any genetic variation in PKM2 could result in differential regulation of the enzyme and a possible facilitation of tumor promotion in a natural syndromic condition prone to cancers. Our observation of two missense mutations in the intersubunit contact domain of PKM2 in Bloom syndrome patients (20), who are prone to spontaneous cancers in early age (29), paved the way to initiate a study to investigate systematically the structural and functional implications of such mutations, which may be correlated with a biological outcome. This study was therefore designed to investigate the differential phenotypic effect of the two natural missense mutations to understand the regulation of the allosteric properties and their effects on the secondary and tertiary structures of the mutant PKM2 proteins to unravel the structure-function correlations of these mutations and to understand the complex allosteric behavior.
Structural Implications of Mutations—The missense mutation H391Y, located within helices 13 and 14, constituting a helix-loop-helix region (residues 390–422) in the C-terminal domain of the intersubunit region, showed a massive loss in cooperativity. An almost 6-fold increase in substrate affinity shifted the sigmoidal curve to a typical hyperbolic curve to attain non-allosteric behavior (supplemental Fig. S4 and Table SI). Investigation of the compromised cooperativity of this mutant using bioinformatics tools revealed that a H-bond could form between the replaced tyrosine residue and the backbone of Glu386, connecting the A- and C-domains of the protein (Fig. 7, A and B). It has been observed that a similar H-bond is present in the structure of the non-allosteric form of PK in cat muscle (30) and rabbit muscle (31) and is absent in allosteric proteins such as human PKM2 (16). These findings are consistent with our earlier molecular study (20). It has been suggested that the high affinity R state requires rotation of all the subunits by these flexible hinges, the H-bonds (32). The probable existence of this H-bond could thus restrict the free rotation, which possibly hinders the allosteric structural transition and results in a compact, rigid, non-allosteric molecule (20). The structural and thermal stability acquired by the replacement of His391 with Tyr (H391Y) has also been reported previously in an α-helixrich protein, glucoamylase (33). Furthermore, His391 is surrounded by hydrophobic residues such as Leu392, Pro446, Tyr390, and Phe395 (Fig. 8A), resulting in a local hydrophobic cluster. Introduction of the relatively more hydrophobic tyrosine is expected to contribute more in stabilizing the hydrophobic environment by making the protein structure more compact by providing extra stability as observed in the case of glucoamylase (33).
Helices 13 and 14, representing the helix-loop helix region (residues 390–422) in the C-terminal domain of PK, are also known to adopt at least two different conformations, T (tensed) and R (relaxed) (21, 34), where Lys421 has been reported to maintain the R state conformation by interacting with Glu409 of the neighboring monomer in rabbit muscle PK (21). In allosteric proteins such as human PKM2 (24), Lys422 with its protruding side chain is found to interact with the Ala419 backbone to align the position of Glu418 within the monomer to make a stable H-bond with Arg399 of the neighboring monomer (Fig. 8B). Introduction of a more basic residue like Arg in the K422R mutant could disrupt the proper alignment of these residues and shift the structure more toward the T state by disturbing the allosteric signal transduction. Another PK mutant (Y443F) of rabbit muscle and kidney (35) was reported to exhibit almost identical kinetic properties, similar to those of the K422R mutant in this study, showing reduced maximal activity, increased [P-enolpyruvate]0.5, and increased response to the allosteric effectors (Fru-1,6-P2 and Phe) probably because of a break in the chain of interactions from the interface to the active site (Lys421–Glu409–Tyr443), shifting the enzyme more toward the inactive T state. It is possible that mutating the evolutionarily conserved residues Tyr443 and Lys421 could lead to interruption of the molecular linkage between Lys421 and the active site. In our study, the P-enolpyruvate saturation curve of the K422R mutant was more sigmoidal, indicating a shift toward the T state, suggesting it to be an essential residue in the intersubunit communications and in stabilizing the R state conformation.
Functional Implications of Mutant Proteins—Experiments under different pH conditions provided further support to observations related to the differential behavior of the two mutants. With increasing pH, the slope of the log Vmax versus pH profile (rounding to the nearest whole integer, na and ni) represented four activating protons and no inhibiting proton for PK-WT in the P-enolpyruvate-bound stage (Fig. 3B and Table 3A), whereas detection of an inhibiting proton in the case of the mutant protein under similar pH conditions defined the weak interaction with P-enolpyruvate and accounted for the reduced activity. The log Vmax/Km versus pH profile indicated that a significant amount of the H391Y mutant enzyme was left unutilized in comparison with PK-WT and the K422R mutant within the studied pH range (Fig. 3A). In the case of the H391Y mutant, upon binding to P-enolpyruvate, no blue shift and lesser quenching of Trp fluorescence (Fig. 6D) indicated the absence of structural perturbations in comparison with PK-WT and the K422R mutant. The loss of structural dynamics, required for allosteric signal transduction from one subunit to another, reflected how the non-allosteric nature of the H391Y protein was acquired due a single base change. This finding is important in the context of an earlier observation of a non-allosteric form of PK acquiring an allosteric character by a single base change (36).
The binding of Fru-1,6-P2 did not affect the log Vmax versus pH profile for PK-WT, whereas it hindered the interaction of the single inhibiting proton in the case of both mutants (Table 3A), indicating that the mutant proteins become more responsive toward activator as observed in kinetic experiments. Less quenching and almost no red shift observed in the Trp emission peak in the case of the H391Y mutant enzyme indicated an absence of structural transition that may occur upon binding to Fru-1,6-P2 compared with PK-WT and the K422R mutant enzyme (supplemental Figs. S14–S16). This also indicated the absence of the phenomenon of ligand-induced domain closure occurring in allosteric proteins such as human PKM2, which has been shown earlier by Trp482-based fluorescence quenching (16). Upon binding of the allosteric inhibitor Phe, the H391Y mutant enzyme resisted the structural perturbations and hence the effect on its activity (Fig. 6F), showing less quenching of fluorescence intensity. whereas the K422R mutant enzyme showed significantly higher quenching, also reflected by greater loss in activity as shown by kinetic assay. The fluorescence studies thus provided an insight in the direct correlation between structural perturbations and the activity profile of the mutant and wild-type enzymes.
The mutants of human PKM2 studied showed differential thermal stability over a period of time, whereas thermal denaturation studies using CD spectroscopy indicated almost 4–5 °C increases in the Tm of the mutant enzymes in comparison with PK-WT (Fig. 5A). However, enhanced thermostability of activity was evident only for the H391Y mutant, and the K422R mutant was as sensitive to temperature as PK-WT under similar conditions (Fig. 5B). We hypothesize that the increased transition temperature of K422R as observed in the secondary structure melting profile was due to the higher α-helical content (97.5%) observed by CD spectroscopy compared with the H391Y mutant (67%) and PK-WT (38%) enzymes (Fig. 4). Having higher α-helical content is likely to provide more structural stability to the K422R mutant enzyme, as observed in our study. In addition, the reversible thermal denaturation of H391Y also indicated its structural rigidity in comparison with PK-WT and K422R (Fig. 5C and Table 3B).
Over the years, the importance of the lowered activity of PKM2 in cells by dissociation of PKM2 to the dimeric form has been proposed to promote cell growth under stressed or pathological conditions such as cancer. This allows the accumulation of glycolytic intermediates (phosphometabolites) for nucleic acid synthesis (4, 36, 38–42). Recent observations suggest that subunit dissociation is not a mandatory condition for lowering the activity of PKM2, which possibly occurs after binding with phosphotyrosine, leading to promotion of the cell growth (19, 43). The studies of the mutant enzymes of PKM2 provide an alternative insight into the processes of differential down-regulation of the enzyme activity and altered allostericity, providing a step toward the creation of a stressful situation in a cell to facilitate tumor progression. We propose that because altered allostericity and down-regulated activity together as well as alone could potentially modulate glycolysis, the K422R mutant with its significantly down-regulated activity and the H391Y mutant with its modulated allosteric behavior and partially lost activity might help in tumor progression. The mutants thus provide a possible explanation for the early onset of tumor development in Bloom syndrome patients. In addition, these natural mutations have helped to understand the key regulation of allosteric behavior by the evolutionarily conserved sparse networks of amino acid interactions, which represent structural motifs for allosteric communication in proteins (37). The understanding obtained in this work on the structural, functional, and allosteric nature of the mutant proteins could pave the way for correcting the structural and functional aspects by using small molecule therapeutics.
Supplementary Material
Acknowledgments
We thank Ponnusamy Kalaiarasan (NCAHG, SLS, JNU, New Delhi) for help in the bioinformatics work.
The work performed in the National Centre of Applied Human Genetics was supported by the University Grant Commission (New Delhi, India).
The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. S1–16 and Table SI.
Footnotes
The abbreviations used are: PK, pyruvate kinase; PK-WT, wild-type pyruvate kinase M2.
References
- 1.Kayne, F. J., and Price, N. C. (1973) Arch. Biochem. Biophys. 159 292–296 [DOI] [PubMed] [Google Scholar]
- 2.Kuo, D. J., O'Connell, E. L., and Rose, I. A. (1979) J. Am. Chem. Soc. 101 5025–5030 [Google Scholar]
- 3.Valentini, G., Chiarelli, L., Fortin, R., Speranza, M. L., Galizzi, A., and Mattevi, A. (2000) J. Biol. Chem. 275 18145–18152 [DOI] [PubMed] [Google Scholar]
- 4.Mazurek, S., Boschek, C. B., and Eigenbrodt, E. (1997) J. Bioenerg. Biomembr. 29 315–330 [DOI] [PubMed] [Google Scholar]
- 5.Imamura, K., and Tanaka, T. (1982) Methods Enzymol. 90 150–165 [DOI] [PubMed] [Google Scholar]
- 6.Cardenas, J. M. (1982) Methods Enzymol. 90 140–149 [DOI] [PubMed] [Google Scholar]
- 7.Cardenas, J. M., Dyson, R. D., and Strandholm, J. J. (1975) in Isozymes: Molecular Structure (Markert, C. L., ed), Vol. 1, pp. 523–541, Academic Press, New York [Google Scholar]
- 8.Muirhead, H., Clayden, D. A., Barford, D., Lorimer, C. G., Fothergill-Gilmore, L. A., Schiltz, E., and Schmitt, W. (1986) EMBO J. 5 475–481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Boles, E., Schulte, F., Miosga, T., Freidel, K., Schlüter, E., and Zimmermann, F. K. (1997) J. Bacteriol. 179 2987–2993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Demina, A., Varughese, K. I., Barbot, J., Forman, L., and Beutler, E. (1998) Blood 92 647–652 [PubMed] [Google Scholar]
- 11.Noguchi, T., Yamada, K., Inoue, H., Matsuda, T., and Tanaka, T. (1987) J. Biol. Chem. 262 14366–14371 [PubMed] [Google Scholar]
- 12.Noguchi, T., Inoue, H., and Tanaka, T. (1986) J. Biol. Chem. 261 13807–13812 [PubMed] [Google Scholar]
- 13.Takenaka, M., Noguchi, T., Inou, H., Yamada, K., Matsuda, T., and Tanaka, T. (1989) J. Biol. Chem. 264 2363–2367 [PubMed] [Google Scholar]
- 14.Takenaka, M., Noguchi, T., Sadahiro, S., Hirai, H., Yamada, K., Matsuda, T., Imai, E., and Tanaka, T. (1991) Eur. J. Biochem. 198 101–106 [DOI] [PubMed] [Google Scholar]
- 15.Muirhead, H. (1990) Biochem. Soc. Trans. 8 193–196 [DOI] [PubMed] [Google Scholar]
- 16.Dombrauckas, J. D., Santarsiero, B. D., and Mesecar, A. D. (2005) Biochemistry 44 9417–9429 [DOI] [PubMed] [Google Scholar]
- 17.Friesen, R. H. E., and Lee, J. C. (1998) J. Biol. Chem. 273 14772–14779 [DOI] [PubMed] [Google Scholar]
- 18.Ikeda, Y., and Noguchi, T. (1998) J. Biol. Chem. 273 12227–12233 [DOI] [PubMed] [Google Scholar]
- 19.Christofk, H. R., Vander Heiden, M. G., Wu, N., Asara, J. M., and Cantley, L. C. (2008) Nature 452 181–186 [DOI] [PubMed] [Google Scholar]
- 20.Anitha, M., Kaur, G., Baquer, N. Z., and Bamezai, R. (2004) DNA Cell Biol. 23 442–449 [DOI] [PubMed] [Google Scholar]
- 21.Wooll, O. J., Friesen, R. H. E., White, M. A., Watowich, S. J., Fox, R. O., Lee J. C., and Czerwinski, E. W. (2001) J. Mol. Biol. 312 525–540 [DOI] [PubMed] [Google Scholar]
- 22.Monod, J., Wyman, J., and Changeux, J. P. (1965) J. Mol. Biol. 12 88–118 [DOI] [PubMed] [Google Scholar]
- 23.Consler, T. G., Woodard, S. H., and Lee, J. C. (1989) Biochemistry 28 8756–8764 [DOI] [PubMed] [Google Scholar]
- 24.Mattevi, A., Valentini, G., Rizzi, M., Speranza, M. L., Bolognesi, M., and Coda, A. (1995) Structure (Lond.) 3 729–741 [DOI] [PubMed] [Google Scholar]
- 25.Boyum, A. (1968) Scand. J. Clin. Lab. Investig. Suppl. 97 9–29 [PubMed] [Google Scholar]
- 26.Bradford, M. M. (1976) Anal. Biochem. 72 248–254 [DOI] [PubMed] [Google Scholar]
- 27.Pace, C. N., Vajdos, F., Fee, L., Grimsley, G., and Gray, T. (1995) Protein Sci. 4 2411–2423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Parthiban, V., Gromiha, M. M., and Schomburg, D. (2006) Nucleic. Acids Res. 34 239–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Reddy, B. S., Kochhar, A. M., Anitha, M., and Bamezai, R. (2000) Int. J. Dermatol. 39 760–763 [DOI] [PubMed] [Google Scholar]
- 30.Stuart, D. I., Levine, M., Muirhead, H., and Stammers, D. K. (1979) J. Mol. Biol. 134 109–142 [DOI] [PubMed] [Google Scholar]
- 31.Larsen, T. M., Laughlin, L. T., Holden, H. M., Rayment, I., and Reed, G. H. (1994) Biochemistry 33 6301–6309 [DOI] [PubMed] [Google Scholar]
- 32.Van Solinge, W., Kraaijengagen, R. J., Rijkson, G., Van Wijk, R., Stoffer, B. B., Gajhede, M., and Nielsen, F. C. (1997) Blood 90 4987–4995 [PubMed] [Google Scholar]
- 33.Wang, Y., Fuchs, E., Silva, R. D., Daniel, A. M., Seibel, J., and Ford, C. (2006) Starch/Stärke 58 501–508 [Google Scholar]
- 34.Viappiani, C. (2004) Proc. Natl. Acad. Sci. U. S. A. 101 144–149 [Google Scholar]
- 35.Friesen, R. H. E., Castellani, R. J., Lee, J. C., and Braun, W. (1998) Biochemistry 37 15226–15276 [DOI] [PubMed] [Google Scholar]
- 36.Ikeda, Y., Tanaka, T., and Noguchi, T. (1997) J. Biol. Chem. 272 20495–20501 [DOI] [PubMed] [Google Scholar]
- 37.Suel, G. M., Lockless, S. W., Wall, M. A., and Ranganathan, R. (2002) Nat. Struct. Biol. 10 59–69 [DOI] [PubMed] [Google Scholar]
- 38.Mazurek, S., Grimm, H., Boschek, C. B., Vaupel, P., and Eigenbrodt, E. (2002) Br. J. Nutr. 87 S23–S29 [PubMed] [Google Scholar]
- 39.Eigenbrodt, E., Reinacher, M., Scheefers-Borchel, U., Scheefers, H., and Friis, R. (1992) Crit. Rev. Oncog. 3 91–115 [PubMed] [Google Scholar]
- 40.Zwerschke, W., Mazured, S., Massimi, P., Eigenbrodt, E., and Jansen-Durr, P. (1999) Proc. Natl. Acad. Sci. U. S. A. 96 1291–1296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mazurek, S., Boschek, C. B., Hugo, F., and Eigenbrodt, E. (2005) Semin. Cancer Biol. 15 300–308 [DOI] [PubMed] [Google Scholar]
- 42.Kato, H., Fukuda, T., Parkison, C., McPhie, P., and Cheng, S. (1989) Proc. Natl. Acad. Sci. U. S. A. 86 7861–7865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Christofk, H. R., Vander Heiden, M. G., Harris, M. H., Ramanathan, A., Gerszten, R. E., Wei, R., Fleming, M. D., Schreiber, S. L., and Cantley, L. C. (2008) Nature 452 230–233 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.