

Abstract
Amylin is an endocrine hormone that regulates metabolism. In patients afflicted with type 2 diabetes, amylin is found in fibrillar deposits in the pancreas. Membranes are thought to facilitate the aggregation of amylin, and membrane-bound oligomers may be responsible for the islet β-cell toxicity that develops during type 2 diabetes. To better understand the structural basis for the interactions between amylin and membranes, we determined the NMR structure of human amylin bound to SDS micelles. The first four residues in the structure are constrained to form a hairpin loop by the single disulfide bond in amylin. The last nine residues near the C terminus are unfolded. The core of the structure is an α-helix that runs from about residues 5–28. A distortion or kink near residues 18–22 introduces pliancy in the angle between the N- and C-terminal segments of the α-helix. Mobility, as determined by 15N relaxation experiments, increases from the N to the C terminus and is strongly correlated with the accessibility of the polypeptide to spin probes in the solution phase. The spin probe data suggest that the segment between residues 5 and 17 is positioned within the hydrophobic lipid environment, whereas the amyloidogenic segment between residues 20 and 29 is at the interface between the lipid and solvent. This orientation may direct the aggregation of amylin on membranes, whereas coupling between the two segments may mediate the transition to a toxic structure.
Type 2 diabetes affects over 100 million people worldwide (1) and is thought to cost upward of $130 billion dollars a year to treat in the United States alone (2). The endocrine hormone amylin (also known as islet amyloid polypeptide) appears to have key roles in diabetes pathology (3–5). The normal functions of amylin include the inhibition of glucagon secretion, slowing down the emptying of the stomach, and inducing a feeling of satiety through the actions of the hormone on neurons of the hypothalamus in the brain (5). The effects of amylin are exerted in concert with those of insulin and reduce the level of glucose in the blood (3, 5). Circulating amylin levels increase in a number of pathological conditions, including obesity, syndrome X, pancreatic cancer, and renal failure (3). Amylin levels together with insulin are raised initially in type 2 diabetes but fall as the disease progresses to a stage where the pancreatic islets of Langerhans β-cells that synthesize amylin no longer function (3).
One of the hallmarks of type 2 diabetes, found in 90% of patients, is the formation of extracellular amyloid aggregates composed of amylin (3–5). The amyloid deposits accumulate in the interstitial fluid between islet cells and are usually juxtaposed with the β-cell membranes (3). Aggregates of amylin are toxic when added to cultures of β-cells, so that the amyloid found in situ may be responsible for β-cell death as type 2 diabetes progresses (6, 7). Genetic evidence that amylin is directly involved in pathology includes a familial S20G mutation that leads to early onset of the disease (8) and produces an amylin variant that aggregates more readily (9).
As with all amyloids it is unclear whether fibrillar structures or soluble oligomers are responsible for pathology. A recurrent theme for amyloidogenic proteins is that toxicity appears to be exerted through membrane-bound oligomers that form pores and disrupt ion balance across membranes (4, 10–13). Experimental evidence for such oligomers has been found for the amyloid-β (Aβ)2 peptides (14), which cause Alzheimer disease, and for α-synuclein (αS), the protein involved in Parkinson disease (15), a particular interest of our laboratory. The similar toxic effects exerted by these amyloidogenic molecules may have a common structural and physical basis. Detailed structural models are available for Aβ (16) and αS (17) bound to SDS micelle mimetics of membranes. For amylin there are models of peptide fragments 1–19 (18), 20–29 (19), and 17–29 (20) bound to micelles but as of yet no model of the complete hormone. This turns out to be particularly important as the interplay between structure and dynamics in amylin only comes to light when considering the whole molecule.
Here we report the solution structure of human amylin bound to SDS micelles. We complement the structure with information on dynamics and on the immersion of amylin into micelles.
EXPERIMENTAL PROCEDURES
Materials—Recombinant human amylin (0.5 mg, lot number 718071NAM) and 15N-amylin (1 mg, lot number 70507) were from rPeptide (Bogart, GA). The peptides were expressed in Escherichia coli and differ from human amylin by not having an amidated C terminus. SDS (electrophoresis grade) was from Bio-Rad. D2O, d25-SDS, d4-acetic acid, d6-methanol, MnCl2, and 16-doxyl-stearic acid were from Aldrich.
NMR Sample Preparation—1 mg of 15N-amylin was used for all heteronuclear NMR studies. Sample 1 was prepared by taking up 0.5 mg of lyophilized 15N-amylin powder in a 0.25-ml 90% H2O, 10% D2O solution of 100 mm d25-SDS, 60 mm acetic acid, pH 4.6, to give a final amylin concentration of 0.5 mm. Sample 1 was used for NMR assignments, structure determination and relaxation studies. Sample 2 was prepared by dissolving 0.5 mg of amylin into 0.66 ml of the solution described above to give a final concentration of 0.2 mm. Sample 2 was divided into aliquots and used for the paramagnetic quenching studies with Mn2+ and 16-doxyl-stearate. Sample 3 was prepared by dissolving 0.5 mg of amylin at natural isotope abundance in 99.96% D2O containing 100 mm SDS, and 60 mm d4-acetate, pH 4.2. An internal 2,2-dimethyl-2-silapentanesulfonic acid standard was used for chemical shift referencing.
NMR Spectroscopy—All NMR experiments were done on a 600-MHz Varian Inova spectrometer equipped with a cryogenic probe. Pulse programs were implemented from the Varian ProteinPack. A temperature of 37 °C was used for all experiments. NMR assignments were based on three-dimensional HNHA, HNHB, TOCSY-HSQC (70-ms mixing time), and NOESY-HSQC (100- and 200-ms mixing times) experiments. Spin systems were first grouped by amino acid type using through-bond scalar connectivities. Sequence-specific assignments were then made using dαN and dNN sequential walks (21) using NOESY-HSQC data.
Stereospecific assignments for glycine methylene hydrogens were obtained from an HNHB spectrum as described in the literature (22). Stereospecific assignments for side chain β-methylene hydrogens were made based on qualitative comparisons of the relative sizes of 3JNHβ couplings in HNHB spectra (23) and intraresidue HN-Hβ and Hα-Hβ NOEs in short mixing time NOESY experiments (24). Assignments for micelle-bound amylin have been deposited with the BMRB under accession number 16105.
Structure Determination—A summary of the restraints used to calculate the NMR structures of micelle-bound amylin and of the statistics relating to the qualities of the structures is given in Table 1. Distance restraints were grouped into four ranges (1.8–2.7, 1.8–3.5, 1.8–5.0, and 1.8–5.5 Å) based on the intensities of cross-peaks in the NOESY spectra. Backbone φ angle dihedral restraints of –60 ± 30° were included for 22 residues with 3JHNHα couplings (25) smaller than 6 Hz. Side chain χ1 restraints were included for eight residues based on HNHB coupling data and NOE data from short mixing time NOESY experiments (24).
TABLE 1.
Statistics for the 30 best micelle-bound amylin structuresa

a Values are reported as the means over the final 30 structures ± 1 S.D. These final lowest energy structures had no NOE violations greater than 0.3 Å or dihedral violations greater than 3°.
b The energy was calculated using the X-PLOR (26). Frepel function with van der Waals interactions and atomic radii set to 0.8 times their CHARMM values (64).
c Calculated using the CHARMM empirical energy function (64).
Standard α-helix hydrogen bond restraints were included for 17 residues with 3JHNHα couplings and Hα secondary shifts in the α-helix range. Of the 17 residues, 8 could be identified in one-dimensional 1H NMR spectra as experiencing hydrogen exchange protection for up to 30 min when amylin was dissolved in d25-SDS micelles suspended in D2O (Thr-9, Gln-10, Leu-12, Ala-13, Leu-16, Leu-27, Ile-26, and Ser-20). For the remaining 9 residues, spectral crowding precluded unambiguous information on exchange protection in one-dimensional spectra. We calculated control structures with hydrogen bond restraints included for only the 8 amides unambiguously protected. These structures showed slightly decreased precision (the backbone r.m.s.d. increased from 1.4 to 1.7 Å for residues 6–27 and from 0.4 to 0.5 Å for residues 6–17). Despite the lower precision, there were no systematic changes in structure, and the NMR ensemble calculated with the full complement of hydrogen bonds fell within the breadth of the conformational ensemble calculated with less hydrogen bonds. We feel that the inclusion of all 17 hydrogen bond restraints is justified by the NOE and 3JHNHa coupling data that support hydrogen-bonded α-helical structures for the residues in question, and by the consistency of the hydrogen bonds with preliminary structures calculated without these restraints.
The final NMR structure calculations started from 500 conformations with randomized φ,ψ angles. Structures were calculated with the program X-PLOR (version 3.851) (26) according to a published protocol (27). The 30 lowest energy structures with no distance violations greater than 0.3 Å or dihedral violations greater than 3° were kept for analyses. Coordinates have been deposited with the Protein Data Bank accession code 2KB8.
15N Relaxation Studies of Amylin Dynamics—15N relaxation data were obtained using 1H-15N correlation experiments from the Varian Protein Pack based on published methods (28). Longitudinal relaxation data were obtained from eight spectra collected with T1 relaxation periods of 20, 50, 130, 210, 310, 500, 700, and 1000 ms. To measure time constants for transverse relaxation, eight T2 relaxation periods of 10, 30, 50, 70, 90, 110, 150, and 190 ms were used. Pre-acquisition delays of 2 s were used between transients. T1 and T2 values were determined from nonlinear least squares fits of the intensity decay as a function of relaxation period to Equation 1,
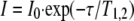 |
(Eq. 1) |
where I is the intensity for relaxation period τ; I0 is the initial amplitude; and T1,2 is the time constant for either T1 or T2 relaxation. Uncertainties in the T1 and T2 values were taken as the standard errors of the fits.
1H-15N NOE values were calculated as the ratio of intensities in an experiment recorded with proton saturation for 4 s to a control experiment where the saturation period was replaced with an equivalent 4-s delay as shown in Equation 2,
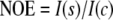 |
(Eq. 2) |
Uncertainties in the NOE values were calculated as shown in Equation 3,
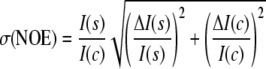 |
(Eq. 3) |
where ΔI(s) and ΔI(c) indicate the root mean square base-line noise in the spectra with and without saturation (29).
S2 order parameters describing the amplitudes of internal motions (30) were calculated with the program TENSOR 2.0 (31) assuming isotropic rotational diffusion (17) for the amylin-micelle complex. To obtain a value for the global correlation time for rotational diffusion, we restricted our analysis to residues 5–17, which had 1H-15N NOE values greater than 0.55, 3JHNHα couplings smaller than 6 Hz, and Hα chemical shifts consistent with stable α-helix structure. Using this subset of residues, we obtained a correlation time of 6.7 ns for global tumbling, which was used to calculate S2 values for individual residues.
Paramagnetic Studies of Amylin Positioning in SDS Micelles—Separate experiments on 0.2 mm amylin in 100 mm SDS were performed with 16-doxyl-stearate and MnCl2. 16-Doxyl-stearate was prepared as a 100 mm stock solution in d6-methanol. The stock solution was used to bring the concentration of sample 2 to 1.85 mm in 16-doxyl-stearate. The aggregation number of SDS micelles is about 60 (32), so that a 100 mm SDS solution corresponds to a micelle concentration of 1.85 mm, or 1 spin probe per micelle. In a second experiment the concentration of 16-doxyl-stearate was raised to 9.2 mm corresponding to five spin probes per micelle. Experiments with paramagnetic Mn2+ were done at concentrations of 0.2 and 1 mm MnCl2, corresponding to ratios of 1:1 and 5:1 of the paramagnetic ion to amylin. 1H-15N HSQC spectra were used to measure paramagnetic quenching. For each experiment a separate control experiment was recorded to obtain cross-peak intensities in the absence of paramagnetic agents.
CD Spectroscopy—Circular dichroism experiments were carried out on an Applied Photophysics II*-180 instrument with samples maintained at a temperature of 37 °C. Far-UV wave-length scans from 280 to 180 nm were collected on 250-μl volume samples held in a 1-mm path length cuvette. The samples contained 35 μm amylin in 60 mm acetic acid, pH 4.6, and varying concentrations of SDS.
RESULTS
Amylin Adopts an α-Helical Structure in the Presence of SDS Micelles—Although human amylin forms fibrils rapidly in water (33), it is soluble and stable for at least 2 months in 100 mm SDS at an acidic pH of 4.6. The 1H-15N HSQC spectrum of amylin under these conditions is shown in Fig. 1.
FIGURE 1.

1H-15N HSQC spectrum of micelle-bound amylin annotated with backbone NMR assignments. Side chain correlations are indicated with the superscript SC. Conditions are as follows: 0.5 mm 15N-amylin, 100 mm SDS, 60 mm acetate, pH 4.6, 37 °C.
Fig. 2A shows CD spectra of amylin with increasing concentrations of SDS at pH 4.6. In the absence of SDS, the CD spectrum of amylin looks roughly like that for an unfolded protein. Weak minima at 208 and 222 nm together with the lack of a pronounced minimum at 195 nm, however, are more indicative of a mixture of random coil and nascent α-helix conformations. The nascent α-helix structure in the absence of SDS probably corresponds to that characterized in detail by NMR for unfolded rat amylin (34). In the presence of 5 mm SDS, there is already significant α-helix structure in the peptide. Under the conditions of our study (37 °C, pH 4.6), the critical micelle concentration of SDS in acetate buffer is about 2 mm (35). As the SDS concentration is raised from 5 to 10 mm, the amount of α-helix structure starts to plateau (Fig. 2A), and there are only small further changes in the CD spectrum between 10 and 100 mm SDS (Fig. 2A). We chose 100 mm SDS for our studies because this value corresponds to a micelle concentration of ∼2 mm (the number of SDS molecules per micelle is expected to be ∼60). Compared with the 0.5 mm amylin concentration, 100 mm SDS ensures an excess of micelles to peptide so that each micelle should have only one molecule of amylin bound. Pulse-field gradient NMR experiments (36) give a diffusion constant for amylin in complex with SDS that within experimental error was the same as that obtained for the complex formed between SDS and Aβ-(1–40), a 40-residue peptide of similar size to the 37-residue amylin (Damylin/DAβ = 0.99 ± 0.06; data not shown). This observation suggests that like Aβ (37), amylin binds to micelles as a monomer at the large 100 mm SDS concentration used for this study.
FIGURE 2.

Data illustrating the α-helix structure of micelle-bound amylin. A, CD spectra of amylin at 0, 5, 10, and 100 mm concentrations of SDS. B, 3JHNHα couplings obtained from a three-dimensional HNHA experiment. C, secondary Hα chemical shifts calculated as the difference between micelle-bound amylin and “random coil” values (38). D, periodicity of HN chemical shifts illustrated as the difference from random coil values (38). E, summary of short-range NOEs.
In addition to work at pH 4.6, we also obtained CD data at pH 10.8 in the presence of SDS (not shown). His-18, the only side chain in amylin that titrates in the physiological range, should lose its positive charge between pH 4.6 and 10.8. The CD spectrum between 200 to 280 nm showed very little difference between the two pH values. In the 180–200 nm range, the spectrum at pH 10.8 lacked the large positive band associated with α-helix structure and was noisier, possibly due to light scattering caused by increased aggregation at pH 10.8.
Fig. 2B shows the sequence profile of 3JHNHα couplings calculated from an HNHA experiment (25). Couplings below 6 Hz indicative of φ dihedral angles in the α-helix range occur for all residues between 5 and 28, except 18 and 22. On average the group of couplings between residues 5 and 17 is smaller than that for residues 23–28.
Fig. 2C shows the difference between random coil values (38) and the Hα chemical shifts of amylin. The secondary shifts for the region between residues 5 and 28 are positive, typical of α-helix structure. The secondary shifts show a periodicity of 3–4 residues, also consistent with α-helix structure (39, 40). Periodicity is also observed for the HN chemical shifts (Fig. 2D), and this type of periodicity has been attributed to α-helix curvature (41, 42). The secondary Hα shifts, which are the most sensitive to secondary structure, appear to be on a gradient decreasing from residues 8 to 28 (Fig. 2C). The data suggest a higher stability for α-helix structure in the N-terminal half of amylin.
Fig. 2E summarizes the short range NOEs observed for amylin. The NOEs are indicative of α-helix structure. There is a break in the pattern near residues 18–22, consistent with a discontinuity in the α-helix structure. Although there is overlap of HN chemical shifts within this segment (Fig. 1) that precludes the detection of HN-HN NOEs, other types of NOEs predicted for an α-helix structure such as dαN(i,i + 3), dαN(i,i + 4), and dαβ(i,i + 3) are weak or missing. Moreover between residues 5 and 28, His-18 and Asn-22 are the only two sites with 3JHNHα values above 6 Hz, consistent with a departure from α-helix structure in this segment. Although not apparent in the semi-quantitative plot, α-helix NOEs are weaker for the 22–28-residue segment compared with the 5–17-residue segment.
NMR Solution Structure of Micelle-bound Amylin—The NMR structure of amylin is shown in Fig. 3. Information on experimental restraints used to calculate the NMR structure ensemble and parameters related to the quality of the structures are given in Table 1.
FIGURE 3.

NMR structure of micelle-bound amylin and sequence conservation relative to the human protein. A, ensemble of the 30 best NMR structures overlaid on the mean Cα, N, and C′ backbone atom coordinates of residues 6–27. B, structures fitted to the backbone mean of residues 6–17. C, structures fitted to the backbone mean of residues 18–27. D, sequences of the 10 closest homologues of human amylin obtained from a BLAST search (46) against the Swiss-Prot data base (47). Residues that differ with respect to the human sequence are shown in inverse type.
At the N terminus, residues 1-4 are constrained to a hairpin topology by a disulfide bond between Cys-2 and Cys-7. The Cys-7 part of the disulfide is in the α-helix and is ordered, giving a nonaveraged side chain χ1 of –60°. The Cys-2 residue is disordered. The last 8 residues between Thr-30 and Tyr-37 are also disordered. The core of the structure consists of an α-helix running from residues 5 to 28. Most of the members of the NMR ensemble have a kink or bend in the α-helix between residues 18 and 22. When the NMR ensemble is superposed on the mean backbone coordinates of the entire α-helix (residues 6–27), the resulting root mean square deviation (r.m.s.d.) of 1.4 Å is rather large (Fig. 3A and Table 1). Consequently, we did a systematic search for residue ranges that would improve the r.m.s.d., and we found that the precision of the structure improves if the segments 6–17 (Fig. 3B) and 18–27 (Fig. 3C) are considered separately. Leaving out segment 18–22 did not improve the fit significantly compared with that obtained for residues 6–27. This indicates that the poor r.m.s.d. when the whole helix is considered is because of an uncoupling of the relative orientations of the 6–17- and 18–27-residue segments rather than disorder in the middle of the helix. We used the MolMol program (43) to calculate a value of 30 ± 18° for the inter-helical angle between segments 6–17 and 18–27.
The organization of the micelle-bound amylin structure into subdomains is reminiscent of that seen with αS and the Alzheimer Aβ-(1–40) peptide. In the presence of SDS micelles αS folds into an α-helix hairpin structure (17, 44). The Aβ-(1–40) peptide when bound to SDS micelles adopts an α-helical structure with a kink between residues 26 and 28 (16). The two helical segments 15–24 and 28–36 flanking the kink superpose well individually but have different orientations relative to each other with an inter-helical angle of 48 ± 15° (16). Similarities have been noted in the neurotoxic effects of amylin and Aβ, which may extend to mechanistic similarities (45).
Of the two α-helix segments, residues 6–17 gave a more precise structure than residues 18–27 (Fig. 3, B and C, and Table 1). This is consistent with the magnitudes of the 3JHNHα couplings (Fig. 2B) and Hα secondary shifts (Fig. 2C), which suggest the α-helix structure is more stable in the 6–17-residue segment. As described below, micelle-bound amylin shows a gradient of backbone dynamics, with the 6–17-residue segment corresponding to the least flexible part of the polypeptide chain. The structural precision and dynamics of micelle-bound amylin correlate with the amino acid sequence conservation in amylin homologues. Fig. 3D shows the sequences of the 10 closest homologues of human amylin identified in a BLAST search (46) against the Swiss-Prot data base (47). The N-terminal half of amylin, which has the highest avidity for membranes (48), shows the highest sequence conservation. The C-terminal part of amylin, which in human amylin is the most amyloidogenic (48, 49), has the lowest sequence conservation.
Dynamic Properties of Micelle-bound Amylin—Fig. 4 shows 15N relaxation data for micelle-bound amylin. The experimental relaxation parameters T1 (Fig. 4A), T2 (Fig. 4B), and the 1H-15N NOE (Fig. 4C) suggest a gradient of increasing flexibility from the N to the C terminus of amylin.
FIGURE 4.

Dynamics of micelle-bound amylin. A, 15N T1 values. B, 15N T2 values. C, 1H-15N NOEs. D, S2 order parameters calculated (31) from 15N relaxation data using an isotropic model of rotational diffusion for the amylinmicelle complex. Errors in S2 values were calculated with the TENSOR program (31) using 100 Monte Carlo simulations to probe the uncertainty in internal mobility because of errors in the experimental relaxation data.
Calculations to obtain a correlation time for global rotational diffusion were done with the program TENSOR 2.0 (31) using data for the least flexible segment between residues 5 and 17. The calculations assuming isotropic tumbling (17) gave a global correlation time of 6.7 ns. For comparison, free SDS micelles have a correlation time of ∼5.5 ns at 35 °C (50), and the correlation time of a complex formed between SDS micelles and a 22-residue peptide similar in size to the 37-residue amylin was 6.6 ns (51). Although the agreement observed is good, we note that the correlation time of SDS micelles depends on a number of variables, including detergent concentration, ionic strength, and the molecules complexed to the micelle (52). Nevertheless, the 6.7-ns rotational correlation time is most consistent with amylin binding to the SDS micelles as a monomer.
The TENSOR 2.0 program (31) was used to calculate S2 order parameters, which describe the amplitudes of internal motions on the fast ps to ns time scale. The data are shown in Fig. 4D and suggest a subdivision of amylin dynamics into three subdomains. The segment running from residues 4 to 18 has the lowest mobility with all S2 values greater than 0.8. Residues 19–30 have S2 values between 0.7 and 0.5. Finally, the last 7 residues have S2 values smaller than 0.4. Only two residues Asn-3 and Thr-4 required R2ex exchange broadening terms (>2 Hz) to account for the experimental relaxation data.
Positioning of Amylin in the SDS Micelle—Fig. 5 shows the results of paramagnetic quenching experiments used to define the interactions of amylin with SDS micelles. Paramagnetic Mn2+ provides a probe of surface accessibility to bulk solution. At a 1:1 ratio of Mn2+ to amylin, the C-terminal half of the protein is already substantially broadened (Fig. 5A). The 30–37-residue segment, which is unstructured, experiences the most severe quenching. The 21–29-residue segment, although α-helical, experiences more broadening than the 5–17-residue segment.
FIGURE 5.

Spin probe studies of amylin positioning in SDS micelles. A, effects of MnCl2 on 1H-15N HSQC cross-peak intensities. B, effects of 16-doxyl-stearate on cross-peak intensities. C, control 1H-15N HSQC of micelle-bound amylin showing intensities in the absence of paramagnetic spin probes. D, spectrum in the presence of 1.0 mm MnCl2. The correlations that persist are labeled. Correlations from side chains are indicated with the superscript SC.
At a 5:1 ratio of Mn2+ to amylin, most of the backbone HN signals from residues after Val-17 are barely visible. The 2–16-residue segment survives even at this higher concentration of Mn2+, indicating it is immersed in the micelle (Fig. 5, C and D). Interestingly, the 2–16-residue segment also shows at least partial protection of side chain amide protons. Each of the side chains of Asn-14, Arg-11, Gln-10, and Asn-3 has at least one HN proton inaccessible (Fig. 5D).
In addition to Mn2+ we also looked at paramagnetic quenching with 16-doxyl-stearate (Fig. 5B). This lipid has a spin label near the ω-end of the fatty acid chain, so the paramagnetic probe should be incorporated near the hydrophobic center of the micelle. In contrast to Mn2+, 16-doxyl probe caused little specific quenching, except perhaps for residues 2, 5, and the glycine at position 33. These data suggest that although amylin is immersed below the surface of the micelle, it does not traverse to its center.
DISCUSSION
In this study we report NMR assignments, the solution structure, and dynamics of amylin when bound to SDS micelles. Although SDS micelles are not physiological, their relatively small size makes them amenable to high resolution structure studies by NMR. Indeed, the most detailed structural models currently available for the amyloidogenic proteins Aβ (16) and α-synuclein (17) in membrane-like environments come from work with SDS micelles. We envision that the micelle-bound structure of amylin reported in this work will pave the way for studies with more realistic membrane mimetics like bilayers or vesicles.
Aspects of the amylin NMR structure related to its dynamic and micelle-binding properties are highlighted in Fig. 6. Fig. 6A shows a ribbon representation of the NMR structure color-coded according to the S2 order parameters derived from 15N relaxation data (Fig. 4). Micelle-bound amylin folds into an α-helix running from about residue 5 to 28. The structure is not uniformly stable but shows a ramp of increasing flexibility toward the C terminus (color coded from blue for rigid, to red for flexible). Residues 5–17 form the most rigid part of the α-helix, and disorder increases from residues 18 to 28. Residues 1–4 are constrained to form a hairpin turn by the Cys-7–Cys-2 disulfide bond, and residues ∼30–37 are unfolded. We note that in contrast to the recombinant peptide studied here, amylin in vivo undergoes post-translational processing that results in an amidated C terminus. Amidation is required for amylin activity (3), presumably due to the interactions of the hormone with its receptors. Because the last 8 residues in the micelle-bound amylin structure are disordered, we think it is unlikely that amidation has a role in the interaction of the peptide with membranes.
FIGURE 6.

Properties of the micelle-bound amylin structure. A, ribbon diagram of the NMR structure color-coded according to S2 order parameters (data from Fig. 4D). The color ramp ranges from blue (large S2, rigid) to red (small S2 flexible). Residues without data are colored gray. The side chains of Cys-2 and Cys-7 are shown in yellow to illustrate the disulfide bond. B, stereo pair (cross-eyed) showing the precision of side chain conformations for residues 6–16 is similar for the hydrophobic (brown) and hydrophilic (violet) faces of the helix. C, schematic diagram illustrating the structure of micelle-bound amylin. The ensemble of 30 NMR structures were superposed on the mean backbone coordinates of residues 6–17, and the ribbon diagrams were color-coded according to spin probe accessibility with 1.0 mm MnCl2 (data from Fig. 5A). The color ramp ranges from blue for backbone amides protected from the spin probe to red for amides that experience large NMR peak intensity losses in the presence of 1 mm MnCl2. The gray ellipsoid illustrates an SDS micelle. The micelle and peptide are not drawn to scale, and we do not have quantitative information on the distance the peptide penetrates into the micelle. The schematic is simply to illustrate that based on spin probe data the N-terminal half of the peptide is immersed into the micelle, and that immersion correlates with the dynamic properties of the peptide (B).
The α-helix of amylin has a kink or discontinuity near residues 18–22. We have noted that in the micelle-bound states of other amyloidogenic proteins, kinks or distortions from α-helix conformations correlate with the locations of turns in the fibrillar β-sheet structures (53). Thus in micelle-bound Aβ the first 15 residues are disordered, and a kink in the α-helix occurs in the 26–28-residue region (16). The first 15 residues are also disordered in Aβ fibrils, and a reverse turn occurs between residues 26 and 30 (54). The micelle-bound state of α-synuclein consists of a broken helix with an interruption of α-helical conformations near residues 42–44 (17). This location roughly corresponds to a turn between strands β1 and β2 in the fibril structure, and more generally regions of α-synuclein with small S2 order parameters (17) in the micelle-bound structure (30–37, 65–70, 83–89) appear to correlate with gaps between β-strands in a recent model of α-synuclein fibrils (55). For amylin, residues 12–17, 22–27, and 30–37 are thought to make up the strands of β-sheet in amyloid fibrils of the peptide (56). The strands correspond to the dynamic subdomains of micelle-bound amylin identified in the current work, and the discontinuity in the α-helix structure near residues 18–22 corresponds to a turn in the putative fibril structure. These observations suggest that intrinsic sequence turn propensities may have roles in determining β-sheet topology in the fibrils.
The location of α-helix structure in micelle-bound amylin is roughly consistent with previous studies. A very recent NMR structure of a 1–19-residue fragment of human amylin in dodecylphosphocholine micelles identified α-helix structure between residues 7 and 17 (18). The difference from our structure at the C terminus is probably due to end fraying effects in the fragment. A second 20–29-residue fragment in SDS micelles adopted a structure consisting of a series of reverse turns (19). A third 17–29-residue fragment showed NMR and CD evidence for weakly stable α-helix structure (20). Fragment studies pose the problem that results on structure as well as dynamics can depend on the location and size of the fragments. A model for intact amylin in large unilamellar vesicles is available from EPR studies (48). This work identified α-helix structure running from residues 9 to 22. The helix starts later than position 5 in our structure. This may result from the substitution of the cysteines at positions 2 and 7 with alanines for the EPR studies. Although the helix ends at position 22 in the EPR study rather than 28 in this work, the data are consistent with our dynamic structure of amylin. The EPR experiments measure mobility and accessibility of the spin labels introduced into amylin (48). Although residues 23–28 are in an α-helix conformation according to the NMR data (Fig. 2, B–D), both mobility (Fig. 4D) and solvent accessibility (Fig. 5A) are increased in this region compared with the 5–17-residue segment.
As noted for micelle-bound α-synuclein (17), protein backbone dynamics are highly correlated with the degree of immersion of individual sites into the micelle. Indeed, conformational restriction of segments of the polypeptide chain bound to micelles is a common theme in peripherally bound proteins (57). Although amylin binds roughly parallel to the micelle surface, the degree of immersion varies from site to site based on accessibility to paramagnetic Mn2+. The S2 order parameters of amylin (Fig. 4D) are linearly correlated with the quenching data obtained at 0.2 mm Mn2+, with an R2 of 0.73. This indicates that 73% of the variance in S2 values can be explained by the variance in solvent exposure of amylin. The strong correlation probably arises because the lower dielectric constant of the micelle environment should favor hydrogen bonding of polar groups in the polypeptide backbone (4). Greater immersion in the micelle would thus increase the stability of hydrogen-bonded α-helix structure and decrease backbone mobility.
Interestingly, our paramagnetic quenching data with Mn2+ show that some of the NH-containing side chains in amylin are at least partially protected from solvent by the micelle. A stereopair illustrating the side chain conformations of residues 6–17 is shown in Fig. 6B. The brown side chains in Fig. 6B are hydrophobic and should point toward the interior of the micelle, whereas the violet hydrophilic side chains should be oriented toward the solvent (48). Fig. 6B shows that the precision of the side chains in the NMR ensemble is similar for the two faces of the amphipathic α-helix, which seems consistent with the paramagnetic quenching data that suggest hydrophilic residues in the 5–17-residue region are partially immersed in the micelle. Consistently, EPR insertion depth measurements indicate that the center of the stable α-helix of amylin is immersed 6–9 Å below the lipid headgroups, with the immersion depth decreasing from the N to the C terminus of the helix. Monolayer experiments are also consistent with an insertion of the N-terminal part of amylin into lipid membranes (58).
A highly schematic model illustrating our results on the binding of amylin to SDS micelles is shown in Fig. 6C. The gray ellipsoid in Fig. 6C represents an SDS micelle. The ribbon diagrams in Fig. 6C illustrating the NMR ensemble are color-coded according to spin probe accessibility, with blue representing inaccessible and red representing accessible. The segment between residues 5 and 17 is immersed just below the surface of the micelle, stabilizing α-helical structure and dampening the dynamics of this region. The Cys-70–Cys-2 disulfide directs residues 1–4 to form a hairpin turn that presumably brings Lys-1 near the surface of the micelle to avoid burying a positively charged group in a hydrophobic environment. The N-terminal part of amylin thus acts as a “hook” anchoring the hormone to the micelle.
Although amylin can bind to phospholipids as a monomer (58), we are unaware of such an interaction in the normal function of amylin. At the same time amylin needs to bind to transmembrane receptors (59) and to cross the blood-brain barrier (60) so the hydrophobic character of the N-terminal part of the hormone could facilitate those functions.
At the end of the 5–17-residue segment, residues 18–28 show a gradient of diminishing protection from paramagnetic quenching and increased backbone dynamics. Presumably these residues are located at the interface between the micelle and solvent. Although residues 18–28 are in α-helical conformations, the axis of the helix shows considerable variance relative to the 5–17-residue segment with an average inter-helix angle of 30°. The ability of the N terminus to insert into membranes could facilitate the assembly of amylin into toxic aggregates. Anchoring in the membrane would work to increase the effective concentration of amylin and would reduce the entropic cost of self-association by orienting molecules within the plane of the membrane lipid bilayers (4). The dynamic character of the 22–29-residue segment would allow induced fit association, and its placement at the interface between lipids and solvent may nucleate aggregation. Under the conditions of the present studies, amylin is very stable, but we have an excess of SDS micelles to peptide. When the peptide predominates, with different types of lipids or under slightly different solution conditions, membranes could catalyze amylin aggregation (61–63).
The gradient in dynamics, the correlation of the helices when coordinates are fitted over the entire 5–28-residue region, and the presence of a few short range NOEs that span the 18–22-residue region suggest that although there is flexibility in the relative orientation of the α-helical segments 5–17 and 18–28, their structures remain coupled. This is important because although our work provides a model for amylin peripherally associated to membranes, a toxic pore composed of oligomeric amylin would have to traverse the membrane lipid bilayers. Presumably, aggregation of the amyloidogenic 20–29-residue segment results in the transmission of a conformational switch to the 5–17-residue segment that allows it to traverse the membrane bilayer. Structural coupling between the amyloidogenic and membrane-immersed segments could thus facilitate the transition to a toxic state when membrane-bound amylin oligomerizes. Our NMR characterization of the micelle-bound monomer paves the way to studies under different sample conditions that could provide structural information on the elusive membrane-bound aggregates and on the mechanisms by which they exert their toxicity.
Acknowledgments
We thank Andrew Mehta for help with spin probe experiments and Carol Teschke for making the CD instrument available to us and teaching us how to use it. Arlene Albert, Debra Kendall, and Nathan Alder are acknowledged for useful discussions about micelle-bound proteins.
This work was supported, in whole or in part, by National Institutes of Health Grant 1S10RR016760 from NCRR (NMR instrumentation). This work was also supported by a Roger C. Duvoisin, M.D., research grant from the American Parkinson Disease Association.
The atomic coordinates and structure factors (code 2KB8) have been deposited in the Protein Data Bank, Research Collaboratory for Structural Bioinformatics, Rutgers University, New Brunswick, NJ (http://www.rcsb.org/).
Footnotes
The abbreviations used are: Aβ, amyloid-β; αS, α-synuclein; r.m.s.d., root mean square deviation; NOE, nuclear Overhauser effect; NOESY, nuclear Overhauser effect spectroscopy; HSQC, heteronuclear single quantum coherence; TOCSY, total correlation spectroscopy.
References
- 1.Amos, A. F., McCarty, D. J., and Zimmet, P. (1997) Diabetes Med. 14 Suppl. 5, 1–85 [PubMed] [Google Scholar]
- 2.Hogan, P., Dall, T., and Nikolov, P. (2003) Diabetes Care 26 917–932 [DOI] [PubMed] [Google Scholar]
- 3.Cooper, G. J. (1994) Endocr. Rev. 15 163–201 [DOI] [PubMed] [Google Scholar]
- 4.Jayasinghe, S. A., and Langen, R. (2007) Biochim. Biophys. Acta 1768 2002–2009 [DOI] [PubMed] [Google Scholar]
- 5.Zdrojewicz, Z., and Belowska-Bién, K. (2006) Diabetologia Doœswiadczalna i Kliniczna 6 169–172 [Google Scholar]
- 6.Janson, J., Ashley, R. H., Harrison, D., McIntyre, S., and Butler, P. C. (1999) Diabetes 48 491–498 [DOI] [PubMed] [Google Scholar]
- 7.Lorenzo, A., Razzaboni, B., Weir, G. C., and Yankner, B. A. (1994) Nature 368 756–760 [DOI] [PubMed] [Google Scholar]
- 8.Sakagashira, S., Sanke, T., Hanabusa, T., Shimomura, H., Ohagi, S., Kumagaye, K. Y., Nakajima, K., and Nanjo, K. (1996) Diabetes 45 1279–1281 [DOI] [PubMed] [Google Scholar]
- 9.Ma, Z., Westermark, G. T., Sakagashira, S., Sanke, T., Gustavsson, A., Sakamoto, H., Engstrom, U., Nanjo, K., and Westermark, P. (2001) Amyloid 8 242–249 [DOI] [PubMed] [Google Scholar]
- 10.Chiti, F., and Dobson, C. M. (2006) Annu. Rev. Biochem. 75 333–366 [DOI] [PubMed] [Google Scholar]
- 11.Ferreira, S. T., Vieira, M. N., and De Felice, F. G. (2007) IUBMB Life 59 332–345 [DOI] [PubMed] [Google Scholar]
- 12.Lansbury, P. T., and Lashuel, H. A. (2006) Nature 443 774–779 [DOI] [PubMed] [Google Scholar]
- 13.Ross, C. A., and Poirier, M. A. (2004) Nat. Med. 10 S10–S17 [DOI] [PubMed] [Google Scholar]
- 14.Chimon, S., Shaibat, M. A., Jones, C. R., Calero, D. C., Aizezi, B., and Ishii, Y. (2007) Nat. Struct. Mol. Biol. 14 1157–1164 [DOI] [PubMed] [Google Scholar]
- 15.Lashuel, H. A., Hartley, D., Petre, B. M., Walz, T., and Lansbury, P. T., Jr. (2002) Nature 418 291. [DOI] [PubMed] [Google Scholar]
- 16.Coles, M., Bicknell, W., Watson, A. A., Fairlie, D. P., and Craik, D. J. (1998) Biochemistry 37 11064–11077 [DOI] [PubMed] [Google Scholar]
- 17.Ulmer, T. S., and Bax, A. (2005) J. Biol. Chem. 280 43179–43187 [DOI] [PubMed] [Google Scholar]
- 18.Nanga, R. P., Brender, J. R., Xu, J., Veglia, G., and Ramamoorthy, A. (2008) Biochemistry 47 12689–12697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mascioni, A., Porcelli, F., Ilangovan, U., Ramamoorthy, A., and Veglia, G. (2003) Biopolymers 69 29–41 [DOI] [PubMed] [Google Scholar]
- 20.Pappalardo, G., Milardi, D., Magri, A., Attanasio, F., Impellizzeri, G., La Rosa, C., Grasso, D., and Rizzarelli, E. (2007) Chemistry 13 10204–10215 [DOI] [PubMed] [Google Scholar]
- 21.Wuthrich, K. (2003) Angew. Chem. Int. Ed. Engl. 42 3340–3363 [DOI] [PubMed] [Google Scholar]
- 22.Düx, P., Whitehead, B., Boelens, R., Kaptein, R., and Vuister, G. W. (1997) J. Biomol. NMR 10 301–306 [DOI] [PubMed] [Google Scholar]
- 23.Archer, S. J., Ikura, M., Torchia, D. A., and Bax, A. (1991) J. Magn. Res. 95 636–641 [Google Scholar]
- 24.Case, D. A., Dyson, H. J., and Wright, P. E. (1994) Methods Enzymol. 239 392–416 [DOI] [PubMed] [Google Scholar]
- 25.Vuister, G. W., and Bax, A. (1993) J. Am. Chem. Soc. 115 7772–7777 [Google Scholar]
- 26.Brunger, A. T. (1992) A System for X-ray Crystallography and NMR. X-PLOR, Version 3.1, Yale University Press, New Haven, CT
- 27.Gronenborn, A. M., Filpula, D. R., Essig, N. Z., Achari, A., Whitlow, M., Wingfield, P. T., and Clore, G. M. (1991) Science 253 657–661 [DOI] [PubMed] [Google Scholar]
- 28.Farrow, N. A., Zhang, O., Forman-Kay, J. D., and Kay, L. E. (1995) Biochemistry 34 868–878 [DOI] [PubMed] [Google Scholar]
- 29.Alexandrescu, A. T., and Shortle, D. (1994) J. Mol. Biol. 242 527–546 [DOI] [PubMed] [Google Scholar]
- 30.Lipari, G., and Szabo, A. G. (1982) J. Am. Chem. Soc. 104 4546–4559 [Google Scholar]
- 31.Dosset, P., Hus, J. C., Blackledge, M., and Marion, D. (2000) J. Biomol. NMR 16 23–28 [DOI] [PubMed] [Google Scholar]
- 32.Lindberg, M., Jarvet, J., Langel, U., and Graslund, A. (2001) Biochemistry 40 3141–3149 [DOI] [PubMed] [Google Scholar]
- 33.Abedini, A., and Raleigh, D. P. (2005) Biochemistry 44 16284–16291 [DOI] [PubMed] [Google Scholar]
- 34.Williamson, J. A., and Miranker, A. D. (2007) Protein Sci. 16 110–117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Paul, B. C., and Ismail, K. (1993) Bull. Chem. Soc. Jpn. 66 703–708 [Google Scholar]
- 36.Nesmelova, I. V., Idiyatullin, D., and Mayo, K. H. (2004) J. Magn. Reson. 166 129–133 [DOI] [PubMed] [Google Scholar]
- 37.Jarvet, J., Danielsson, J., Damberg, P., Oleszczuk, M., and Graslund, A. (2007) J. Biomol. NMR 39 63–72 [DOI] [PubMed] [Google Scholar]
- 38.Wishart, D. S., Bigam, C. G., Holm, A., Hodges, R. S., and Sykes, B. D. (1995) J. Biomol. NMR 5 67–81 [DOI] [PubMed] [Google Scholar]
- 39.Jimenez, M. A., Blanco, F. J., Rico, M., Santoro, J., Herranz, J., and Nieto, J. L. (1992) Eur. J. Biochem. 207 39–49 [DOI] [PubMed] [Google Scholar]
- 40.Wiltscheck, R., Kammerer, R. A., Dames, S. A., Schulthess, T., Blommers, M. J., Engel, J., and Alexandrescu, A. T. (1997) Protein Sci. 6 1734–1745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.McLeish, M. J., Nielsen, K. J., Najbar, L. V., Wade, J. D., Lin, F., Doughty, M. B., and Craik, D. J. (1994) Biochemistry 33 11174–11183 [DOI] [PubMed] [Google Scholar]
- 42.Zhou, N. E., Zhu, B.-Y., Sykes, B. D., and Hodges, R. S. (1992) J. Am. Chem. Soc. 114 4320–4326 [Google Scholar]
- 43.Koradi, R., Billeter, M., and Wuthrich, K. (1996) J. Mol. Graph. 14 51–55 [DOI] [PubMed] [Google Scholar]
- 44.Chandra, S., Chen, X., Rizo, J., Jahn, R., and Sudhof, T. C. (2003) J. Biol. Chem. 278 15313–15318 [DOI] [PubMed] [Google Scholar]
- 45.Lim, Y. A., Ittner, L. M., Lim, Y. L., and Gotz, J. (2008) FEBS Lett. 582 2188–2194 [DOI] [PubMed] [Google Scholar]
- 46.Altschul, S. F., Madden, T. L., Schaffer, A. A., Zhang, J., Zhang, Z., Miller, W., and Lipman, D. J. (1997) Nucleic Acids Res. 25 3389–3402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bairoch, A., Boeckmann, B., Ferro, S., and Gasteiger, E. (2004) Brief. Bioinform. 5 39–55 [DOI] [PubMed] [Google Scholar]
- 48.Apostolidou, M., Jayasinghe, S. A., and Langen, R. (2008) J. Biol. Chem. 283 17205–17210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Moriarty, D. F., and Raleigh, D. P. (1999) Biochemistry 38 1811–1818 [DOI] [PubMed] [Google Scholar]
- 50.Lycknert, K., Rundlf, T., and Widmalm, G. (2002) J. Phys. Chem. 106 5275–5280 [Google Scholar]
- 51.Jarvet, J., Zdunek, J., Damberg, P., and Graslund, A. (1997) Biochemistry 36 8153–8163 [DOI] [PubMed] [Google Scholar]
- 52.Gangabadage, C. S., Najda, A., Bogdan, D., Wijmenga, S. S., and Tessari, M. (2008) J. Phys. Chem. 112 4242–4245 [DOI] [PubMed] [Google Scholar]
- 53.Alexandrescu, A. T., and Croke, R. L. (2008) in Protein Misfolding (O'Doherty, C. B., and Byrne, A. C., eds) Nova Science Publishers, Hauppauge, NY
- 54.Luhrs, T., Ritter, C., Adrian, M., Riek-Loher, D., Bohrmann, B., Dobeli, H., Schubert, D., and Riek, R. (2005) Proc. Natl. Acad. Sci. U. S. A. 102 17342–17347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Vilar, M., Chou, H. T., Luhrs, T., Maji, S. K., Riek-Loher, D., Verel, R., Manning, G., Stahlberg, H., and Riek, R. (2008) Proc. Natl. Acad. Sci. U. S. A. 105 8637–8642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kajava, A. V., Aebi, U., and Steven, A. C. (2005) J. Mol. Biol. 348 247–252 [DOI] [PubMed] [Google Scholar]
- 57.Gierasch, L. M., Lacy, J. E., Thompson, K. F., Rockwell, A. L., and Watnick, P. I. (1982) Biophys. J. 37 275–284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Engel, M. F., Yigittop, H., Elgersma, R. C., Rijkers, D. T., Liskamp, R. M., de Kruijff, B., Hoppener, J. W., and Antoinette Killian, J. (2006) J. Mol. Biol. 356 783–789 [DOI] [PubMed] [Google Scholar]
- 59.Young, A. (2005) Adv. Pharmacol. 52 47–65 [DOI] [PubMed] [Google Scholar]
- 60.Young, A. (2005) Adv. Pharmacol. 52 67–77 [DOI] [PubMed] [Google Scholar]
- 61.Domanov, Y. A., and Kinnunen, P. K. (2008) J. Mol. Biol. 376 42–54 [DOI] [PubMed] [Google Scholar]
- 62.Jayasinghe, S. A., and Langen, R. (2005) Biochemistry 44 12113–12119 [DOI] [PubMed] [Google Scholar]
- 63.Knight, J. D., and Miranker, A. D. (2004) J. Mol. Biol. 341 1175–1187 [DOI] [PubMed] [Google Scholar]
- 64.Brooks, B. R., Bruccoleri, R. D., Olafson, B. D., States, D. J., Swaminathan, S., and Karplus, M. (1983) J. Comp. Chem. 4 187–193 [Google Scholar]