

Abstract
Deinococcus radiodurans is an extraordinarily radioresistant bacterium that is able to repair hundreds of radiation-induced double-stranded DNA breaks. One of the players in this pathway is an X family DNA polymerase (PolXDr). Deletion of PolXDr has been shown to decrease the rate of repair of double-stranded DNA breaks and increase cell sensitivity to gamma-rays. A 3′→5′ exonuclease activity that stops cutting close to DNA loops has also been demonstrated. The present crystal structure of PolXDr solved at 2.46-Å resolution reveals that PolXDr has a novel extended conformation in stark contrast to the closed “right hand” conformation commonly observed for DNA polymerases. This extended conformation is stabilized by the C-terminal PHP domain, whose putative nuclease active site is obstructed by its interaction with the polymerase domain. The overall conformation and the presence of non standard residues in the active site of the polymerase X domain makes PolXDr the founding member of a novel class of polymerases involved in DNA repair but whose detailed mode of action still remains enigmatic.
DNA replication and repair are functions that are of vital importance for the maintenance of cellular life. These functions are carried out by various DNA replicating engines, most of them acting as multiprotein complexes. Deinococcus radiodurans, a Gram-positive bacterium, is characterized by an extraordinary resistance to ionizing radiation and desiccation. After radiation induced cutting of its 3.28-megabase genome into hundreds of small fragments, it is capable of reassembling it completely (1). Different hypotheses have been suggested to explain this radioresistance. A recently proposed mechanism involves the creation of long linear DNA intermediates by an extended synthesis-dependent strand annealing process, where overlapping chromosomal fragments are used both as primers and as templates for synthesis of complementary single strands (2). Recircularization of chromosomes would be assured by homologous recombination. Although DNA polymerase I is one of the main enzymes involved in this process, it was shown that other proteins affect double strand break repair efficiency in D. radiodurans. One of these is an X family DNA polymerase (PolXDr)5 (3). Cells devoid of PolXDr protein show increased sensitivity to γ-irradiation and a longer delay in the restoration of an intact genome after irradiation. It was therefore proposed that PolXDr has an important role in double strand break repair in D. radiodurans. The contribution of PolXDr may become essential for instance when damage gets too important or, alternatively, it may act in different repair pathways from polymerase I. Indeed, some of the X DNA polymerases, such as Saccharomyces cerevisiae Pol4 and human polymerase λ (4) have been proposed to play important roles in different DNA repair processes, including non-homologous end-joining (5). It was shown that PolXDr also has strong 3′→5′ exonuclease activity that is stimulated by Mn2+ (6). This activity is associated with proofreading mechanisms in other polymerase families and encoded by protein domains or subunits distinct from the polymerase catalytic domain (7). Curiously the exonuclease activity of PolXDr is modulated upon encounter of a stem-loop structure. The combination of both activities leads to the hypothesis that PolXDr might be involved in DNA repair, potentially non-homologous end-joining, by processing damaged DNA or repair intermediates, thus generating substrates for other repair proteins (6). Very recently an orthologue of PolX from Bacillus subtilis was characterized. It was shown that PolXBs is a template-directed DNA polymerase acting on DNA gaps with a downstream 5′ phosphate group, suggesting it may play a role in base excision repair (8).
DNA polymerases all combine a catalytic palm domain, a thumb domain, binding double-stranded DNA, and a finger domain that fixes the incoming nucleotide. The polymerase domain of the X family belongs to the Polβ-like nucleotidyltransferase superfamily, sharing ∼25% amino acid identity with the DNA polymerase domains of Polλ, Pol4, and Polβ. PolXDr has a second domain at the C terminus called PHP, with strong sequence identity with the histidinol phosphatase involved in histidine transport in bacteria. Due to its similarity to histidinol phosphatase and the presence of a trinuclear zinc site, the PolXDr PHP domain is thought to function as phosphoesterase (9). In the context of DNA polymerases, this activity might be responsible for the degradation of pyrophosphate, thus driving the polymerization reaction, or contributes to a nuclease reaction that would be involved in proofreading the newly synthesized strand. The deletion of the PHP domain also had a negative effect on survival of γ-irradiated cells suggesting that this domain possesses a function in DNA repair. Unexpectedly, deletion of the PHP domain destroys structure modulated but not the general 3′→5′ exonuclease activity (6). No activity could be demonstrated for the PHP domain alone.
In this report we present the crystal structure of PolXDr at 2.46-Å resolution. Surprisingly, PolXDr adopts a stretched out conformation instead of the commonly observed closed right hand conformation. In the active site of the polymerase catalytic domain, the two universally conserved aspartates are replaced by two glutamates, whereas the active site of the PHP domain is obstructed by its interaction with the polymerase domain.
EXPERIMENTAL PROCEDURES
Expression and Purification—PolXDr was expressed as previously described (3). The induced cells were harvested and resuspended in buffer A (20 mm Tris-HCl, pH 7.5, 500 mm NaCl, 10 mm imidazole, and a mixture of anti-proteases (Complete, Roche Applied Science)). After sonication and centrifugation, the supernatant was heated 10 min at 60 °C followed by 10 min cooling on ice. After a second centrifugation, the supernatant was loaded onto a nickel-affinity column (nickel-nitrilotriacetic acid-agarose, Qiagen). The bound proteins were eluted by buffer B (20 mm Tris-HCl, pH 7.5, 150 mm NaCl, 150 mm imidazole). The eluate was loaded onto a 5-ml HiTrap heparin column (Amersham Biosciences) pre-equilibrated with buffer B without imidazole. PolXDr was eluted by a linear gradient from 150 mm to 1 m NaCl on buffer B using anÄKTA Purifier System (Amersham Biosciences). Column fractions were analyzed by SDS-PAGE, and those containing PolXDr were identified and pooled diluted to 50 mm NaCl and concentrated. The yield from 1 liter of culture was 1 mg of PolXDr. The homogeneity and integrity of the protein were finally checked by mass spectrometry (Voyager, PerkinElmer Life Sciences).
Crystallization, Data Collection, Structure Solution, and Refinement—Native protein (4 mg/ml) crystallized at 293 K by the hanging drop, vapor-diffusion method from 1:1 μl drops of protein and precipitant containing 0.1 m NaAc, pH 5, 0.2 m NaCl, 16% 2-methyl-2,4-pentanediol. Plate-like crystals were transferred in the mother liquor containing 30% 2-methyl-2,4-pentanediol and 10 mm HgCl prior to flash-cooling in liquid nitrogen. X-ray diffraction data from a crystal of the native protein was collected on beamline ID23-1 at the European Synchrotron Radiation Facility (Grenoble, France). Data processing was carried out with MOSFLM, XDS, and SCALA (10, 11). The crystals diffracted to a maximum resolution of 2.46 Å and belong to space group P21 with two molecules per asymmetric unit, corresponding to a 50% solvent content. The cell parameters and data collection statistics are reported in Table 1.
TABLE 1.
Data collection and refinement statistics
| Zinc edge | Zinc edge (high resolution) | |
|---|---|---|
| Wavelength (Å) | 1.282 | 1.282 |
| Unit-cell parameters | ||
| a, b, c (Å) | 58.91, 138.5, 67.72 | 58.91, 138.5, 67.72 |
| β (°) | 92.24 | 92.24 |
| Resolution (Å) | 48.393-2.465 | 10-2.465 |
| Total number of reflections | 389,995 (37,516) | 155,376 (15,356) |
| Total of unique reflections | 37,507 (4,456) | 37,234 (4,408) |
| Multiplicity | 10.4 (8.4) | 4.2 (3.2) |
| Rmerge | 13.7 (69.8) | 7.2 (35.1) |
| I/σ(I) | 15.0 (3.2) | 13.3 (3.0) |
| Overall completeness (%) | 95.5 (78.2) | 95.4 (77.7) |
| Reflections (working/test) | 67,975/3,579 | |
| Rcryst/Rfree | 21.2/25.3 | |
| Non-hydrogen atoms | 8,427 | |
| Water molecules | 80 | |
| Bonds (Å) | 0.005 | |
| Angles (°) | 0.773 | |
| Mean B-factor (Å2) protein/zinc/solvent | 19.0/22.1/17.3 | |
| Ramachandran plot (%) | ||
| Most-favored | 91.3 | |
| Allowed | 8.2 | |
| Generously allowed | 0.4 |
The Hyss sub-structure solution module from Phenix was used to find the anomalous scattering sites (12). Refinement and phasing was performed with Sharp (13), solvent flattening with Solomon and resolve (14). An initial model was built in the maps with resolve and manually completed using O. The structure was refined with the refinement module of Phenix (15), using NCS constraints between the two polypeptide chains. The final model contains residues 8–564 and 6–564 for each polypeptide chains respectively, three zinc atoms bound to the PHP domain and one mercury atom bound to C237 in each chain. Coordinates have been deposited (code 2w9m).
Small Angle X-ray Scattering—SAXS data were collected at the Swing beamline at SOLEIL, Saclay. The sample to detector distance was 1.93 m covering the range of scattering vector (q) from 0.007Å–1 to 0.35Å–1 (q = 4π sin θ/λ, with 2θ being the scattering angle and λ = 1.36 Å the wavelength of the x-rays). The detector used was a charge-coupled device camera from Aviex. Twenty-five successive frames of 0.75-s exposure each separated by a 1-s pause were recorded for each protein solution and buffer alike. During exposure, the solution was contained in a quartz capillary 1.5 mm in diameter under vacuum. The solution was continuously pushed into the beam using an automated injection system. In these conditions no radiation damage was detected.
The samples were prepared at concentrations of 1.1, 4.4, and 10.7 mg/ml in 20 mm Tris-HCl, pH 7.5, 150 mm NaCl, 5% (v/v) glycerol. SAXS data were normalized to the intensity of the incident beam, averaged and background-subtracted using the program package PRIMUS (16). The curves recorded at the three concentrations were perfectly identical and therefore free from intermolecular interactions. Intensities were scaled using the scattering by water. The radius of gyration and the intensity at the origin were calculated using the Guinier law over the angular range qRg < 1.3 (17). The pair distribution function P(r) was determined using the indirect Fourier transform approach as implemented in the GNOM program (18). Alternative estimates of the radius of gyration and the intensity at the origin were derived from the calculated P(r).
Scattering patterns from crystal structures were calculated using the program CRYSOL (19). Missing parts in the crystal structure, comprising 11 N-terminal residues and 5 C-terminal residues, were modeled using MODLOOP (20). The closed conformation was built by fitting the fingers and 8-kDa domains in the same orientation as the Polβ protein. To adjust the calculated scattering curve in the experimental one, PolXDr was split into three fragments comprising, respectively, the 11 N-terminal residues, the residues from Asp6 to Arg158, and the 412 C-terminal residues. The relative position of both domains was refined using the rigid-body modeling program SASREF (21), which uses simulated annealing to find an optimal configuration of the domains by fitting the SAXS curve. The connectivity between the two fragments was preserved during refinement by keeping the distance between the Cα atoms of Arg158 and Gln159 and of Pro5 and Asp6 shorter than 4 Å. An ultimate adjustment was performed using the program CRYSOL (19). The goodness of fit was characterized by the χ following parameter,
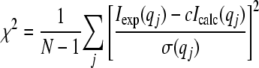 |
(Eq. 1) |
where N is the number of experimental points, c is a scaling factor, and Icalc(qj) and σ(qj) are the calculated intensity and the experimental error at the scattering vector qj, respectively.
RESULTS AND DISCUSSION
Structure Determination and Overall Architecture—The crystals of PolXDr belong to space group P21 with two molecules per asymmetric unit. PolXDr is composed of two distinct parts: the polymerase domain, itself composed of subdomains, and the PHP domain. Sequence homology of the PHP domain of PolX with YcdX from Escherichia coli whose structure is known (PDB code 1M65) suggested the presence of a trinuclear zinc metal binding site. Although no zinc was added to the purification buffers of the recombinant protein, an energy scan at the zinc absorption edge showed the presence of zinc in the crystal, and a 2.46-Å dataset was recorded at the zinc absorption edge. For this crystal, the cryoprotectant solution had been supplemented with 10 mm HgCl2 to aid phasing. Six zinc sites and two mercury sites were found from the anomalous differences. Visual inspection of the zinc heavy atom sites revealed that two sets of three sites had a spatial arrangement similar to the zinc sites in the YcdX PHP domain. Phasing led to poor but interpretable electron density maps, where two models of the PHP domain could be placed in the electron density by superposition of the zinc sites. Models of the subdomains of Polλ were also placed in the density, which provided an initial model used for iterative rebuilding. The structure of PolXDr was fully refined and rebuilt at 2.46-Å resolution.
The two polypeptide chains present in the asymmetric unit superpose perfectly with the exception of the 8-kDa N-terminal sub-domain, which adopts a slightly different orientation in both copies (see below). Apart from this slight difference, the polymerase domain and the PHP domain have the same relative orientations in both polypeptide chains. This is consistent with the idea that PolXDr was trapped in a stable conformation that is not a crystallization artifact.
Structure of the Polymerase Subdomains—The PolXDr polymerase domain shares the modular organization of family X polymerases: an X family-specific 8-kDa domain and the generic polymerase fingers, with the palm and thumb subdomains. Overall the PolXDr structure is most similar to the structure of Polλ (22), although the overall relative orientation of the different domains is dramatically different. Each subdomain will therefore be compared separately before considering the global conformation. These domains will be referred to in the initial notation of Polλ and Polβ, which was later revised for homogeneity with other polymerases.
The thumb domain of PolXDr (Fig. 1, A and B, dark green) is virtually identical to the Polλ domain with the exception of the loop between β7 and β8 (Polλ numbering), which is approximately ten amino acids shorter in PolXDr. The PolXDr palm domain has the β-nucleotide transferase fold, common to all the family X polymerases (23) and related to the prokaryotic replicative PolIII α-subunit (Fig. 1, A and B, light green). Palm domains harbor the carboxylates involved in metal binding and polymerization. The loop between β4 and β5, which is around ten amino acids long in Polλ, Polβ, and the African Swine Fever Virus PolXASFV, is reduced in PolXDr to a three-amino acid β-turn. This difference is important because a longer loop would clash with the fingers subdomain of PolXDr, which has a totally different position relative to the palm domain compared with the other family X DNA polymerases (see further). It is therefore probable that the observed domain configuration is unique to PolXDr.
FIGURE 1.

Structure of PolXDr. A, schematic and surface representation (B) of Polλ in complex with gapped DNA. Domains are colored dark green, light green, light blue, dark blue, for the thumb, palm, fingers, and 8-kDa domains, respectively (PDB code 2BCR). C, schematic and surface representation (D) of PolXDr. The same coloring scheme as in panel A is adopted. The PolXDr PHP domain is colored red, and its linker to the thumb domain is yellow. The orientations of Polλ and PolXDr in views A and B are superposed on the thumb and palm domains. The DNA substrate has been modeled on the structure of PolXDr using the Polλ complex as a template (bottom). Two models were generated: one by superposing with the palm/thumb and the second with the fingers/8-kDa domains. Both are represented on the bottom schematic, clearly showing that the DNA binding sites are on opposite sides of the molecule. E, schematic representation of PolXDr rotated 90° compared with panel B, showing the stretched conformation of the polymerase domain. F, schematic representation of PolXDr palm (green) and fingers (blue) domains with the palm and fingers domain of Polλ superposed on the palm domains. The β4–β5 loop in Polλ is much longer than in PolXDr, which results in a sterical clash with the fingers domain in the stretched conformation.
The finger domain is composed of four helices (F–I) (Fig. 1, A and B, light blue). Helices F and G form a helix-turn-helix motif that binds the primer strand backbone in Polλ. The most noteworthy difference is the I helix that provides the link between the catalytic and the fingers domain. In Polλ, a kink in this helix induces a turn of ∼70 degrees and is sometimes described as two helices (I and I′) (Fig. 1D). In PolXDr, helix I and I′ form a unique, long, and straight helix. The absence of this kink in PolXDr profoundly modifies the standard relative orientation of the fingers and catalytic domains (Fig. 1D), creating a unique configuration of the polymerase domain as a whole (see below).
The 8-kDa domain is composed of 5 helices (A–E) (Fig. 1, A and B, dark blue). In Polλ and Polβ, this domain possesses DNA binding and recognition activity as well as a metal-independent 5′-deoxyribose 5-phosphate-lyase activity. However, despite an overall identical structure, there are important differences in the PolXDr and Polλ 8-kDa domains. In Polλ and Polβ, the 5′-deoxyribose 5-phosphate-lyase uses the Nε of a lysine (Lys312 and Lys72, respectively) as the sole Schiff base nucleophile. This lysine is located in a lysine-rich pocket that contains a 5′-phosphate binding site. In order for the lysine to act as a nucleophile, it must be deprotonated and hence the pKa of its ε-amino group must be lowered. It is believed that the surrounding residues, notably the lysines lining the catalytic pocket, are involved in this process. In PolXDr, the catalytic lysine is replaced by a glutamate (Glu72). The only conserved lysine in PolXDr is Lys33, which corresponds to Arg275 or Lys35 in Polλ and Polβ, respectively, whereas the Polβ conserved lysines Lys60, Lys68, and Lys72 (Lys275, Lys307, and Lys312 in Polλ) are replaced by non-charged residues. In the complex of Polλ (Polβ) with gapped DNA, Lys307 (Lys68) provides the main contribution to 5′-phosphate binding of the downstream primer. Although this conserved lysine is absent in PolXDr, Lys64, and Lys67 are in the vicinity of the hypothetical DNA binding site and could be involved in DNA binding. The conserved tyrosines Tyr279 (Polλ) and Tyr39 (Polβ), which are also implicated in phosphate binding, superpose perfectly with Tyr37 in PolXDr.
As a result, the 8-kDa domain in PolXDr seems to have lost many characteristics of a 5′-phosphate binding site. This questions the existence of 5′-deoxyribose 5-phosphate-lyase activity of PolXDr, which has not yet been documented. In any case, all these differences must contribute to a different DNA binding affinity and selectivity, which could contribute to the selection of special sites on the DNA damaged by irradiation or desiccation.
Structure of the PolXDr PHP Domain—PolXDr has a PHP domain at its C-terminal end (Fig. 1, B and C, red), which is absent in the other family X polymerases such as Polλ and Polβ but present in the replicative polymerase PolIIIα subunits (9, 24, 25). The C-terminal PHP domain of PolXDr is structurally homologous to other members of the PHP family. This domain consists of a (α/β)7 seven-stranded β barrel closed on one side of the β-barrel by a C-terminal helix. The PolXDr PHP domain superposes well with the YcdX protein from E. coli (root mean square deviation of 1.80 Å for 200 residues for 18% sequence identity) and with the PHP domain of the PolIII α-subunit (root mean square deviation of 2.80 Å for 136 residues for 15% sequence identity).
The PHP domain is attached to the thumb domain by a structured 30-amino acid linker, which contains a small helical region (residues 296–326) (Fig. 1, yellow). This linker packs along the α6 and α7 helices of the PHP domain and the N and M helices of the thumb domain through hydrophobic interactions, hydrogen bonds, and salt bridges (Arg299–Asp309, Glu302–Arg534, Arg304–Asp287, and Glu305–Arg190).
Three zinc ions were unambiguously identified in the PolXDr PHP. In contrast to PolXDr, in the structures of YcdX or PolIIIα PHP domains, one or two zincs are missing in the absence of added zinc. This might arise from the fact that the exit of the putative PHP-PolXDr catalytic site is obstructed (see below) preventing the loss the zinc atoms during purification. The three zincs are coordinated by the conserved residues of the PHP family (His332, His334, His364, His428, His456, His518, Asp339, Asp401, and Asp516).
Domain Organization—In the structures of Polβ and Polλ, domains are arranged similarly to a closed right hand, which envelops both DNA and the incoming nucleotides (26, 27). Substrate binding is often accompanied by structural rearrangements of the polymerase subdomains. The structure of human Polβ, solved in complex with gapped DNA in the presence or absence of dNTP, has revealed an open and closed conformation of the enzyme. The 8-kDa domain shows significant movement during DNA binding, while the conformational change upon dNTP binding involves a slight reorientation of the thumb subdomain (28, 29). On the contrary, the structure of the Polλ family X Polymerase has shown that it does not undergo large subdomain movements during catalysis (22). The configuration of the PolXDr domains is radically different from Polλ and Polβ in the open or closed conformation. The finger, palm, and thumb domains in PolXDr do not create the familiar closed right hand but, on the contrary, adopt a completely extended conformation (Fig. 1C). This suggests that the thumb and palm domain on one hand and the fingers and 8-kDa domains on the other have moved as “rigid” bodies. It is interesting to note that the PolXDr conformation does not result from a simple opening of the Polλ “hand” as classically seen in the small movement of Polλ upon DNA binding: the finger and 8-kDa N-domains in PolXDr are swung out by 90° compared with the Polλ conformation around a hinge region centered on Arg158, situated in the αI–αJ loop between the palm and fingers domain (Fig. 1D). This implies that the putative DNA binding region of the 8-kDa domain and of the palm catalytic site are on opposite sides of the protein surface (Fig. 1B). The PHP domain binds across the length of the polymerase domain and makes interactions with all four polymerase domains. The PHP domain might be involved in stabilizing the observed stretched conformation of PolXDr by binding at the interface between palm and fingers (Fig. 1, B and C). However, the fingers and 8-kDa domain can be modeled in the same conformation, because they adopt in Polβ without severely clashing with the PHP domain. There is no steric hindrance preventing the polymerase to flip into a closed conformation, which would also free the entrance of the PHP domain active site. In the PolIII α-subunit the PHP domain wedges between the thumb and palm domain, but these interactions do not involve the putative active site region (25). The function of the PolIII PHP domain is also unknown.
Thermal parameter analysis of the structure using the TLS Motion Determination server indicated that the 8-kDa domain was the most obvious candidate for defining a TLS group for refinement (see Fig. 3A). This confirms the hypothesis that the 8-kDa domain is slightly flexible in one copy of PolX in the crystal structure. Further division of the structure in TLS groups identified the fingers, palm, thumb, and PHP domains. However, inclusion of up to five TLS domains for the modeling of atomic displacement parameters during refinement did not yield any improvement in the refinement statistics.
FIGURE 3.

Conformation of PolXDr in solution by SAXS and crystallo-graphic B-factors. A, representation of the crystallographic B-factors of PolX against the protein sequence. B, comparison of the experimental curve (black dots) with the curves calculated from the crystal structure (green), from a model of the closed conformation (blue), and from a model (red) in which the fingers and 8-kDa domain (light and dark blue) positions were optimized for best fit to the experimental data. The models used for calculating the curves are shown in surface representation with a dashed line indicating the direction of the fingers/8-kDa domain orientation. The inset represents the Guinier plot from experimental data.
The linker and PHP domain make extensive interactions with all other subdomains. This interaction buries in total 2000 Å2 and involves 7 salt bridges and 7 hydrogen bonds. Surprisingly the main interaction site is situated at the top of the PHP β-barrel, which coincides with the putative catalytic site containing the trinuclear zinc. The cavity of the PHP domain containing the zinc ions is occluded by the hinge between helix E (8 kDa) and F (fingers). The PHP β5α5 loop makes extensive contacts in the groove formed between the 8-kDa and fingers domains (Fig. 1C). This loop could act as a latch that locks the 8-kDa/fingers domain in the stretched configuration.
As a result of these tight interactions, the access to the putative active site of the PHP domain is completely blocked. It cannot be excluded that binding of an appropriate substrate could provoke a substantial conformational change which would expose the PHP active site.
Catalytic Mechanism—It was shown for Polβ that the binding of the incoming nucleotide closes the active site, bringing into place the catalytic groups. These changes are characterized by rotations of thumb helices and closing in of the 8-kDa domain. Polλ, however, is in a closed state before nucleotide binding, but dNTP binding shifts the DNA template strand in the active site creating crucial interactions with the minor groove of DNA. In view of the radically different relative configuration of the PolXDr domains compared with other family X members, it remains an open question whether the same mechanism is at work here. It is interesting to note that the African swine fever PolXASFV altogether lacks the finger and 8-kDa domains but is a genuine polymerase (30). Therefore the role of the positioning or even the presence of the individual subdomains on polymerase activities has not definitely been settled.
Structural analysis and sequence analysis strongly suggest that the phosphoryl transfer reaction of all polymerases is catalyzed by a two metal ion mechanism (31). The structure of Polβ in complex with gapped DNA and dideoxy-CTP showed that the two metal ions are bound by three carboxylates of the palm domain (32). Superposition of the PolIIIα and Polβ palm domains aligns all three catalytic aspartates. Strikingly, superposition of these palm domains with the PolXDr palm domain reveals the replacement of two of these metal binding aspartates (for instance Asp429 and Asp490 in Polλ numbering) by two glutamate residues Glu199 and Glu234 (Fig. 2A). The third aspartate (Asp427 in Polλ numbering), which bridges the two catalytic metal ions is absent in PolXDr. No other obvious residue is present in the vicinity to complete the coordination of the metal ions in PolXDr. In the polymerases of family C and X, the loop between strands β1 and β2 is part of the incoming nucleotide binding pocket and contains a conserved glycine-serine/glycine motif (Gly179 and Ser180 in Polβ) involved in hydrogen bonding to the β and γ phosphates of the nucleotide. In PolXDr, the glycine is conserved (Gly186), but the serine/glycine is replaced by an aspartate (Asp187). The presence of an aspartate in this position is surprising and probably leads to important changes in the binding mode of the incoming nucleotide. However, the two Polβ arginines Arg149 and Arg183, which are involved in binding the phosphates of the incoming nucleotide, are conserved in PolXDr (Arg160 and Arg190), whereas the Gly190, which is hydrogen bonded to the γ-phosphate is replaced by Arg196. The positively charged residues that bind the 3′-phosphate in the primer strand in Polλ, Polβ, and PolIII (Arg254, Arg488, and Lys616, respectively) are replaced in PolXDr by a proline (Pro232) but possibly Arg196 could take over this role.
FIGURE 2.

Comparison of the active sites of PolXDr and Polβ. Stereo representation of PolXDr (A) and Polβ (B) active sites. Polβ is in complex with incoming nucleotide and DNA. The DNA, fingers, and 8-kDa domains have been omitted for clarity. The incoming nucleotide is in sticks. The two magnesium atoms bound to the two catalytic aspartates are shown as yellow spheres.
Clearly, because the polymerase activity of PolXDr is strongly stimulated by Mn2+, we expected a participation of this metal in the reaction mechanism. The supposed metal binding site in the palm, however, deviates from what is observed in other members of the family X polymerases. This suggests that the active site may be organized differently in PolXDr and/or that a variant of the canonical two-metal catalytic step is operating in PolXDr. Complexes with nucleotides and metals are needed to settle these questions.
Exonuclease Activity—Many DNA polymerases contain a 3′→5′ exonuclease activity that is part of a proofreading mechanism (Polγ,-δ, and -ε) while it is lacking in others (Polα and -β and the eukaryotic polymerases of the Y family). In some polymerases, this exonuclease is embedded in a separate domain (Polδ and -ε) while in others it is associated to a separate subunit of the replicative complex (e.g. E. coli PolIII). It has recently been shown that the PHP domain of Thermus thermophilus replicase has 3′→5′ exonuclease activity that may act as an additional proofreading exonuclease (33). Surprisingly, the strong Mn2+-dependent 3′→5′ exonuclease activity of PolXDr is located in the polymerase domain, because deletion of the PHP domain does not abolish canonical 3′→5′ exonuclease activity. The stretched conformation of PolXDr further shows that the putative active site of the PHP domain is obstructed by its interaction with the polymerase domain. Therefore the PHP domain cannot be active as a nuclease in this conformation of the full-length protein. The only known biochemical effect of deletion of the PHP domain is to reduce the structure modulated nuclease activity of PolXDr. Because the PHP seems to participate in stabilizing the stretched conformation of PolXDr, it is tempting to speculate that the stretched conformation is involved in the stem-loop-specific nuclease activity of PolXDr.
Mutations of two conserved glycines 104 and 106 does not affect the 3′→5′ exonuclease activity on homopolymeric DNA but leads to a complete loss of the pausing site on stem-loop oligonucleotides (6). These glycines are part of the conserved GXG motif shown to interact with the DNA phosphate backbone in Polβ (27). These glycines are situated at the entrance of the fingers and the interaction with DNA requires the cusp configuration (where they are located at ∼15-Å distance from the catalytic carboxylates). In the stretched PolXDr configuration, this glycine motif is far removed (31 Å) from the palm catalytic carboxylates, and it is hard to imagine how it could interact with stem loop DNA without a drastic rearrangement of the domains.
We have tested whether the conserved glutamates of the palm domain of Polymerase active site might be important for exonuclease activity. The E199A, E234A, and E253A mutants exhibited the same structure modulated exonuclease activity as the wild-type enzyme, indicating that the polymerase active site is not involved (results not shown). It was recently shown that PolXBs contains a 3′→5′ exonuclease activity that resides in its PHP domain (34). Mutation of two evolutionary conserved histidines (His339 and His441) belonging to a motif predicted to bind putative catalytic zinc ions abolished the 3′→5′ exonuclease activity. The equivalent histidines of PolXDr (His353 and His355) are direct ligands of the zinc cluster, and mutagenesis results of PolXBs seem therefore compatible with the PolXDr structure. As mentioned the zinc cluster is totally inaccessible in our present PolXDr structure, which probably represents an inactive conformation as far as the exonuclease activity concerns. The PHP domain in PolXDr makes two direct salt bridges with the β5–β6 loop of the palm (Glu193–Arg500 and Arg190–Glu305, Fig. 2). The homologous position to Arg190 in Polβ binds the incoming nucleotide. This offers a possibility to couple polymerase and exonuclease activities in PolXDr. The PHP domain may change its position upon encounter of mismatches in the active site, thereby liberating its active site zinc-cluster.
Conformation of PolXDr in Solution—To study the extent of the flexibility and to establish whether or not the crystal structure corresponds to the conformation in solution, we have investigated its structure by SAXS measurements. The radius of gyration value derived from the pair distribution function is 27.9 ± 0.1 Å, close to the value 27.8 ± 0.1 Å obtained from the Guinier law. The maximal extension of the protein is estimated from the pair distribution function: Dmax = 88 ± 3 Å. These values of Rg and Dmax are very close to the values corresponding to the crystal structure (27.5 Å and 91 Å, respectively). Fig. 3B shows that the calculated scattering curve from the crystal structure does not fit the experimental curve (the best adjustment gives χ = 5.2). Thus the protein in solution adopts a conformation different from the crystal structure. We have also tried to adjust the experimental curve to the curve calculated from a model of PolXDr in the closed conformation (see “Experimental Procedures” for modeling details), but this conformation does account neither for the data in solution (χ = 11.3). The orientations of the different PolXDr domains were therefore adjusted by rigid-body refinement (see “Experimental Procedures” for details). Optimizing the orientation of the 8-kDa domain could not account for the experimental data (not shown). However, optimized models where both the fingers and 8-kDa domains were both reoriented (as rigid bodies around the link between Arg158 and Gln159 (Fig. 1)) gave good fits to the experimental data with typical value of χ = 1.3 (Fig. 3). Several independent runs starting from either the crystal structure or the closed conformation converged to very similar models where the first domain has turned of ∼20° from the twisted conformation (Figs. 1D and 3). The results demonstrate that the conformation of PolXDr is different from the crystal structure but is close to the twisted conformation. One can expect that the structure is dynamic, and the model shown in the Fig. 3 must be regarded as an “average” rather than as a unique conformation adopted by the protein.
CONCLUSION
The extended conformation observed in our crystal structure raises interesting questions as to the function of PolXDr. At first sight, our observations suggest that the stretched conformation does not represent a (near) catalytically competent state of the enzyme and that, for polymerase activity, the enzyme must undergo drastic structural rearrangements to adopt the canonical Polβ cusp-like arrangement. The present PolXDr configuration for which the fingers and 8-kDa domains are far removed from the palm catalytic domain could be compared with the polymerase of the African swine fever virus (PolXASFV), an X family polymerase, which consists uniquely of a thumb and palm domain. PolXASFV binds gapped DNA as efficiently as does Polβ (35). The lack of the fingers and 8-kDa domain in this polymerase is thought to contribute to the low fidelity of nucleotide insertion. The crystal structure and conformation in solution of PolXDr enrich the structural diversity of family X polymerases with a new twisted conformation and suggest that polymerases can explore a much greater conformational space than was previously thought. The twisted conformation of PolXDr might present specific advantages in the repair of some specific subsets of DNA lesions. Alternatively, PolXDr might adopt different conformations, for example in the presence of specific structures in damaged DNA, which might expose either polymerase or exonuclease activities. The presence of non-standard residues in the active site might indicate that proteins with polymerase activities might have been overlooked in sequence data base based on search against the canonical motifs.
Acknowledgments
We thank I. Shevelev and M. Blasius for participating in the initial work. We thank J. Perez and G. David (SOLEIL synchrotron) for help with the SAXS data collection.
The atomic coordinates and structure factors (code 2w9m) have been deposited in the Protein Data Bank, Research Collaboratory for Structural Bioinformatics, Rutgers University, New Brunswick, NJ (http://www.rcsb.org/).
Footnotes
The abbreviations used are: PolXDr, X family DNA polymerase; Pol, polymerase; SAXS, small angle x-ray scattering; PHP, polymerase and histidinol phosphatase; TLS, translation/libration/screw.
References
- 1.Blasius, M., Hubscher, U., and Sommer, S. (2008) Crit. Rev. Biochem. Mol. Biol. 43 221–238 [DOI] [PubMed] [Google Scholar]
- 2.Zahradka, K., Slade, D., Bailone, A., Sommer, S., Averbeck, D., Petranovic, M., Lindner, A. B., and Radman, M. (2006) Nature 443 569–573 [DOI] [PubMed] [Google Scholar]
- 3.Lecointe, F., Shevelev, I. V., Bailone, A., Sommer, S., and Hubscher, U. (2004) Mol. Microbiol. 53 1721–1730 [DOI] [PubMed] [Google Scholar]
- 4.Hubscher, U., Maga, G., and Spadari, S. (2002) Annu. Rev. Biochem. 71 133–163 [DOI] [PubMed] [Google Scholar]
- 5.Ramadan, K., Shevelev, I. V., Maga, G., and Hubscher, U. (2004) J. Mol. Biol. 339 395–404 [DOI] [PubMed] [Google Scholar]
- 6.Blasius, M., Shevelev, I., Jolivet, E., Sommer, S., and Hubscher, U. (2006) Mol. Microbiol. 60 165–176 [DOI] [PubMed] [Google Scholar]
- 7.Shevelev, I. V., and Hubscher, U. (2002) Nat. Rev. Mol. Cell Biol. 3 364–376 [DOI] [PubMed] [Google Scholar]
- 8.Banos, B., Lazaro, J. M., Villar, L., Salas, M., and de Vega, M. (2008) J. Mol. Biol. 384 1019–1028 [DOI] [PubMed] [Google Scholar]
- 9.Aravind, L., and Koonin, E. V. (1998) Nucleic Acids Res. 26 3746–3752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Leslie, A. (1992) Joint CCP4 and EACMB Newsletter Protein Crystallography, Daresbury Laboratory, Warrington, UK
- 11.Kabsch, W. (1993) J. Appl. Crystallogr. 26 795–800 [Google Scholar]
- 12.Adams, P. D., Grosse-Kunstleve, R. W., Hung, L. W., Ioerger, T. R., McCoy, A. J., Moriarty, N. W., Read, R. J., Sacchettini, J. C., Sauter, N. K., and Terwilliger, T. C. (2002) Acta Crystallogr. Sect. D Biol. Crystallogr. 58 1948–1954 [DOI] [PubMed] [Google Scholar]
- 13.Bricogne, G., Vonrhein, C., Flensburg, C., Schiltz, M., and Paciorek, W. (2003) Acta Crystallogr. Sect. D Biol. Crystallogr. 59 2023–2030 [DOI] [PubMed] [Google Scholar]
- 14.Terwilliger, T. (2004) J. Synchrotron Radiat. 11 49–52 [DOI] [PubMed] [Google Scholar]
- 15.Adams, P. D., Gopal, K., Grosse-Kunstleve, R. W., Hung, L. W., Ioerger, T. R., McCoy, A. J., Moriarty, N. W., Pai, R. K., Read, R. J., Romo, T. D., Sacchettini, J. C., Sauter, N. K., Storoni, L. C., and Terwilliger, T. C. (2004) J. Synchrotron Radiat. 11 53–55 [DOI] [PubMed] [Google Scholar]
- 16.Konarev, P. V., Volkov, V. V., Sokolova, A. V., Koch, M. H. J., and Svergun, D. I. (2003) J. Appl. Crystallogr. 36 1277–1282 [Google Scholar]
- 17.Guinier, A., and Fournet, G. (1955) Small Angle Scattering of X-Rays, Wiley, New York
- 18.Svergun, D. I. (1992) J. Appl. Crystallogr. 25 495–503 [Google Scholar]
- 19.Svergun, D. I., Barberato, C., and Koch, M. H. (1995) J. Appl. Crystallogr. 28 768–773 [Google Scholar]
- 20.Fiser, A., and Sali, A. (2003) Bioinformatics 19 2500–2501 [DOI] [PubMed] [Google Scholar]
- 21.Petoukhov, M. V., and Svergun, D. I. (2005) Biophys. J. 89 1237–1250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Garcia-Diaz, M., Bebenek, K., Gao, G., Pedersen, L. C., London, R. E., and Kunkel, T. A. (2005) DNA Repair 4 1358–1367 [DOI] [PubMed] [Google Scholar]
- 23.Sawaya, M. R., Pelletier, H., Kumar, A., Wilson, S. H., and Kraut, J. (1994) Science 264 1930–1935 [DOI] [PubMed] [Google Scholar]
- 24.Bailey, S., Wing, R. A., and Steitz, T. A. (2006) Cell 126 893–904 [DOI] [PubMed] [Google Scholar]
- 25.Lamers, M. H., Georgescu, R. E., Lee, S. G., O'Donnell, M., and Kuriyan, J. (2006) Cell 126 881–892 [DOI] [PubMed] [Google Scholar]
- 26.Garcia-Diaz, M., Bebenek, K., Krahn, J. M., Kunkel, T. A., and Pedersen, L. C. (2005) Nat. Struct. Mol. Biol. 12 97–98 [DOI] [PubMed] [Google Scholar]
- 27.Pelletier, H., Sawaya, M. R., Wolfle, W., Wilson, S. H., and Kraut, J. (1996) Biochemistry 35 12742–12761 [DOI] [PubMed] [Google Scholar]
- 28.Sawaya, M. R., Prasad, R., Wilson, S. H., Kraut, J., and Pelletier, H. (1997) Biochemistry 36 11205–11215 [DOI] [PubMed] [Google Scholar]
- 29.Beard, W. A., and Wilson, S. H. (2000) Mutat. Res. 460 231–244 [DOI] [PubMed] [Google Scholar]
- 30.Maciejewski, M. W., Shin, R., Pan, B., Marintchev, A., Denninger, A., Mullen, M. A., Chen, K., Gryk, M. R., and Mullen, G. P. (2001) Nat. Struct. Biol. 8 936–941 [DOI] [PubMed] [Google Scholar]
- 31.Steitz, T. A. (1999) J. Biol. Chem. 274 17395–17398 [DOI] [PubMed] [Google Scholar]
- 32.Beard, W. A., Osheroff, W. P., Prasad, R., Sawaya, M. R., Jaju, M., Wood, T. G., Kraut, J., Kunkel, T. A., and Wilson, S. H. (1996) J. Biol. Chem. 271 12141–12144 [DOI] [PubMed] [Google Scholar]
- 33.Stano, N. M., Chen, J., and McHenry, C. S. (2006) Nat. Struct. Mol. Biol. 13 458–459 [DOI] [PubMed] [Google Scholar]
- 34.Banos, B., Lazaro, J. M., Villar, L., Salas, M., and de Vega, M. (2008) Nucleic Acids Res. 36 5736–5749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Showalter, A. K., Byeon, I. J., Su, M. I., and Tsai, M. D. (2001) Nat. Struct. Biol. 8 942–946 [DOI] [PubMed] [Google Scholar]