

Abstract
The small molecular weight G-protein RAB7 is localized to both early and late endosomes and has been shown to be critical for trafficking through the endocytic pathway. The role of RAB7 in the endocytic pathway has been controversial, with some groups reporting that it regulates trafficking from early to late endosomes and others ascribing its role to trafficking between late endosomes and lysosomes. In this study, we use RNA interference to identify the exact step RAB7 regulates in the movement of the epidermal growth factor receptor (EGFR) from the cell surface to the lysosome. In the absence of RAB7, trafficking of the EGF·EGFR complex through the early endosome to the late endosome/multivesicular body (LE/MVB) does not change, but exiting from the LE/MVB is blocked. Ultrastructural analysis reveals that RAB7 is not required for formation of intraluminal vesicles of the LE/MVB, since RAB7-deficient cells have an increased number of enlarged LE/MVBs densely packed with intraluminal vesicles. Biochemical data indicate that the EGFR complex is sequestered in these intraluminal vesicles. Together, these data provide evidence that RAB7 is required for the transfer of cargo from the LE/MVB to the lysosome and for endocytic organelle maintenance.
The endocytic pathway regulates a number of fundamental cellular processes. These include the uptake of nutrients, immune response, intracellular transport, and regulation of cell surface receptor signaling (1). Disruption of normal endocytic trafficking can affect cellular homeostasis and lead to changes in cell physiology that range from hyperproliferation to cell death. Understanding the molecular regulation of endocytic trafficking will provide a better understanding of basic cell biology as well as identify potential molecular targets for diseases characterized by defects in endocytic trafficking.
By following the postinternalization events of cell surface receptors, considerable work has been done to elucidate the molecular details of the endocytic pathway (2). Many cell surface receptors, either constitutively or in response to ligand, use this degradative pathway to regulate receptor and/or ligand levels. Following clathrin-mediated internalization, the endocytic pathway is composed of a series of dynamic stages that progressively shuttle cargo from clathrin-coated vesicles to early endosomes, to late endosomes/multivesicular bodies (LE/MVBs),2 and finally to lysosomes for degradation. Each of these endocytic stages is defined by the morphology and protein composition of the organelle.
Endocytic trafficking is coordinated by a variety of proteins that regulate endosome maturation, movement, fission, and fusion. Primary among these are the small molecular weight G-proteins called RABs (3). Rab proteins are members of the Ras superfamily of GTPases that cycle between GTP-bound active and GDP-bound inactive states. The nucleotide bound state of the RAB determines whether it can interact with downstream effectors. Individual RAB proteins have been shown to act as hubs that regulate distinct trafficking steps temporally and spatially by facilitating vesicle motility, tethering, and fusion (4, 5).
Rab7 localizes to both the early endosome and the LE/MVB and has been shown to be a necessary component of endocytic trafficking and lysosomal degradation (6). However, there is no consensus as to the exact molecular function of RAB7 in the endocytic pathway. Some reports have implicated RAB7 in regulating cargo movement out of early endosomes (7–10), whereas others have reported it to function in the more distal process of lysosomal delivery from LE/MVBs (11, 12). Live cell imaging indicates that RAB7 replaces RAB5 as cargo is trafficked through endocytic compartments (10, 13). However, it remains unclear if the presence of RAB7 indicates that it is immediately functional or if it is positioning itself to be used later in the endocytic pathway. Alternatively, as has been proposed in Caenorhabditis elegans, Rab7 may regulate multiple endocytic steps (14).
Previous attempts to understand the function of RAB7 have relied primarily on overexpression of wild type or mutant RAB7 (11, 12, 15, 16). This approach carries the caveat that high levels of the exogenous protein increase the potential for nonphysiological interactions between an overexpressed RAB and downstream RAB effectors. This concern was highlighted by a recent analysis that showed promiscuity between a variety of RABs and RAB effectors (17). To overcome these issues, we have used the alternative approach of depleting endogenous RAB7 with siRNA and examining EGF·EGFR endocytic trafficking in the absence of RAB7.
In this study, we show that RAB7 is required for lysosomal degradation of the EGF·EGFR complex. Upon dissecting the endocytic pathway of RAB7-deficient cells, we find that cargo can proceed through EEA1 (early endosome antigen 1)-positive early endosomes and into CD63-positive LE/MVB. However, in the absence of RAB7, the EGF·EGFR complex does not exit the LE/MVB and is retained in its intraluminal vesicles. This disrupted trafficking is mirrored by an altered equilibrium between the endocytic organelles, as indicated by the accumulation of enlarged, densely packed LE/MVB and a decrease in the size and number of lysosomes. Based on these data, we have generated a model that RAB7 is dispensable for EGFR endocytic trafficking from the cell surface to the intraluminal vesicles of the LE/MVB but is required for fusion of the LE/MVB and the lysosome.
MATERIALS AND METHODS
Cell Lines—HeLa and tTA-HeLa cells (HeLa cells stably transfected with the tetracycline transactivator (tTA)) were obtained from ATCC (Manassas, VA) and Dr. Sandra Schmid (The Scripps Research Institute, La Jolla, CA), respectively. Cells were maintained in Dulbecco's minimal essential medium (DMEM) containing 5% fetal bovine serum, 100 units/ml penicillin, 100 units/ml streptomycin, and 2 mm glutamine (18). Cell lines were maintained at 37 °C in 5% CO2.
Rab7 siRNA—Rab7-specific and control RNA interference were obtained from Dharmacon and previously published (19). Dharmacon Smartpool siRNA was supplied as a mixture of four oligonucleotide duplexes. RAB7 sequences were as follows: duplex 1, sense sequence (5′-CUAGAUAGCUGGAGAGAUGUU-3′) and antisense sequence (5′-CAUCUCUCCAGCUAUCUAGUU-3′); duplex 2, sense sequence (5′-GUACAAAGCCACAAUAGGAUU-3′) and antisense sequence (5′-UCCUAUUGUGGCUUUGUACUU-3′); duplex 3, sense sequence (5′-AAACGGAGGUGGAGCUGUAUU-3′) and antisense sequence (5′-UACAGCUCCACCUCCGUUUUU-3′); duplex 4, sense sequence (5′-CGAAUUUCCUGAACCUAUCUU-3′) and antisense sequence (5′-GAUAGGUUCAGGAAAUUCGUU-3′). siCON sequences were as follows: duplex 1, sense sequence (5′-AUGAACGUGAAUGCUCAAUU-3′) and antisense sequence (5′-UGAGCAUUCACGUUCAUUU-3′); duplex 2, sense sequence (5′-UAAGGCUAUGAAGAGAUACUU-3′) and antisense sequence (5′-AUUCCGAUACUUCUCUAUGUU-3′); duplex 3, sense sequence (5′-AUGUAUUGGCCUGUAUUAGUU-3′) and antisense sequence (5′-CUAAUACAGGCCAAUACAUUU-3′); duplex 4, sense sequence (5′-UGGUUUACAUGUCGACUAAUU-3′) and antisense sequence (5′-UUAGUCGACAUGUAAACCAUU-3′). Cells were plated to ∼50% confluence for transfection the following day. Except where indicated, the cells were transfected with a concentration of 10 nm siRNA duplexes using INTERFERin (Polyplus Transfection) according to the manufacturer's directions. Twenty-four hours post-transfection, the medium was replaced with fresh growth medium. Experiments were carried out 72 h post-transfection, unless otherwise indicated.
Cell Lysate Preparation and Immunoblotting—Cell lysates were generated by washing the cells twice with phosphate-buffered saline (PBS) and solubilizing cells in lysis buffer (150 mm NaCl, 1% Nonidet P-40, 0.5% deoxycholate, 0.1% SDS, 50 mm Tris, pH 8.0, 10 mm sodium pyrophosphate, 100 mm sodium fluoride, and 2 mm phenylmethylsulfonyl fluoride) on ice. Proteins were solubilized by rotating the lysis buffer/cell mix end over end for 10 min at 4 °C; insoluble material was removed by centrifugation for 10 min at 4 °C. The protein concentration of the solubilized protein was assessed by a BCA assay (Pierce), and samples were diluted in SDS-sample buffer. Equivalent amounts of protein (indicated in the figure legends) were separated by SDS-PAGE; transferred to nitrocellulose; and detected using monoclonal antibodies against the hemagglutinin (HA) epitope (12CA5) (Roche Applied Science), RAB7 (Sigma), RAB5 (BD Transduction Laboratories), EEA1 (BD Transduction Laboratories), or a rabbit polyclonal antibody against EGFR (Santa Cruz Biotechnology, Inc., Santa Cruz, CA), followed by probing with a horseradish peroxidase-conjugated goat anti-mouse or goat anti-rabbit secondary antibody. Detected proteins were visualized by enhanced chemiluminescence using a UV Products imaging system or film.
125I-EGF Degradation and Secretion—Cells were incubated for 7.5 min with 1 ng/ml 125I-EGF (catalog number NEX160; PerkinElmer Life Sciences; specific activity 150–200 μCi/μg) at 37 °C in binding buffer (DMEM, 20 mm HEPES, 0.1% bovine serum albumin, pH 7.3). Cells were washed four times in room temperature PBS to remove unbound 125I-EGF. Prewarmed, 37 °C medium was added to the cells, and they were returned to 37 °C for the indicated periods of time. At each time point, the medium was collected. The remaining cells were solubilized in 1% Nonidet P-40, 20 mm Tris, pH 7.4. Cell lysates were incubated with 10% trichloroacetic acid and 1% bovine serum albumin as a carrier protein on ice for 1 h. Intact protein was separated from degraded by centrifugation for 15 min at 14,000 rpm at 4 °C. The radioactivity for all three fractions (secreted, intact, and degraded) was determined by calculating each fraction in a Beckman γ-counter (20).
Rescue of RAB7 Knockdown—tTA-HeLa cells were transfected with RAB7-specific siRNA, as described above. Twenty-four hours after the introduction of RAB7 siRNA, cells were calcium phosphate-transfected with HA-Rab7(canine). This construct was expressed in the pCDF1-MCS2-EF2-copGFP vector (System Biosciences) and is subsequently referred to as pCDF1. pCDF1-HA-Rab7(canine) drives the expression of HA-Rab7(canine) with a cytomegalovirus promoter and green fluorescence protein with a separate constitutive human elongation factor (EF1) promoter. Transfection efficiency was estimated to be 70–80% based on green fluorescence protein expression determined by examining the cells under a fluorescent microscope. Forty-eight hours after the plasmid transfection, cells were serum-starved for 2 h and treated with 10 ng/ml EGF for 0, 15, or 90 min. Cell lysates were prepared and monitored for the amount of EGFR degradation by immunoblot.
In Fig. 2B, following RAB7 knockdown, cells were split at a density of 200,000 cells/35-mm dish in growth medium 24 h following transfection. After the cells recovered for another 24 h, cells were infected with tetracycline-regulatable adenoviruses encoding either a constitutively active or dominant negative mutant of HA-tagged canine Rab7 that have been described previously (11). Twenty-four hours after infection, 125I-EGF degradation was assessed at 60 min, as described above.
FIGURE 2.

Rescue of RAB7 knockdown. A, HeLa cells were transfected with RAB7-specific siRNA or control siRNA. Forty-eight hours after transfection, the cells were calcium phosphate-transfected with wild type, HA-tagged canine Rab7 (canine HA-Rab7). 24 h later, cells were serum-starved for 2 h, followed by stimulation with 10 ng/ml EGF for 0, 15, or 60 min. Cell lysates were prepared, resolved by either 7.5% SDS-PAGE (EGFR) or 12% SDS-PAGE (RAB7, α-tubulin), and immunoblotted (IB) as indicated. B, tTA-HeLa cells were transfected with RAB7-specific siRNA or siCON. Forty-eight hours after transfection, cells were infected with nothing, tetracycline-regulatable adenovirus encoding for the HA-tagged, constitutively active mutant of canine Rab7 (HA-Rab7(Q67L)), or tetracycline-regulatable adenovirus encoding for the HA-tagged, dominant negative mutant of canine Rab7 (HA-Rab7(N125I)). Cells were also infected with HA-Rab7(Q67L) in the presence of tetracycline to inhibit protein expression. After recovery from infection for 24 h, cells were assayed for 125I-EGF degradation following incubation with 125I-EGF for 60 min. Data are plotted as the percentage of intact 125I-EGF (n = 3; mean ± S.E.). *, p < 0.05; **, p < 0.01 (Student's t test) as compared with untransfected and siCON controls. C, cell lysates were prepared from cells treated as in B, resolved by 12% SDS-PAGE, and immunoblotted with an anti-RAB7 antibody for the presence of endogenous Rab7 and exogenous HA-Rab7(Q67L) or HA-Rab7(N125I).
125I-Transferrin Recycling—Transferrin receptor recycling was monitored using 125I-transferrin (125I-Tfn) (21). Briefly, cells were incubated on ice with 0.05 mg/ml 125I-Tfn (catalog number NEX212; PerkinElmer Life Sciences; specific activity: 0.3–1.0 μCi/μg) in binding buffer (DMEM, 20 mm NaHEPES, 0.1% bovine serum albumin, pH 7.4) for 2 h to achieve equilibrium binding. Free 125I-Tfn was removed with four washes in ice-cold binding buffer. Cells were incubated for the indicated time in 37 °C binding buffer to initiate Tfn receptor endocytic trafficking. At each time point, the medium was collected; cell surface 125I-Tfn was collected with two washes with 0.5 m NaCl, 0.2 m acetic acid, pH 3.5; and the cells were solubilized in 0.1 m NaOH to determine the amount inside the cell. Each fraction was counted in a Beckman γ-counter. As positive control to inhibit Tfn recycling from the early endosome, cells were incubated with 100 μm monensin during the incubation with 125I-Tfn on ice and all subsequent conditions. The percentage of internalized 125I-Tfn was calculated as the amount of intracellular 125I-Tfn divided by the total 125I associated with the cell (medium + cell surface + intracellular). The percentage of secreted 125I-Tfn was calculated as the amount of 125I-Tfn in the medium divided by the total 125I associated with the cell.
Confocal Fluorescence Microscopy—Cells were grown on coverslips, fixed in 4% p-formaldehyde/PBS (PBS, pH 7.4, 0.5 mm CaCl2, 0.5 mm MgCl2), and permeabilized with 0.1% saponin, 5% fetal bovine serum, PBS. After washing, cells were incubated for 1 h with the indicated antibody. The following antibodies were used: EEA1, mouse monoclonal (BD Transduction Laboratories, San Diego, CA); LAMP-2, mouse monoclonal; CD63, mouse monoclonal (University of Iowa Hybridoma Bank, Iowa City, IA). Cells were then incubated 1 h with an Alexa488-conjugated goat anti-mouse secondary antibody (Molecular Probes, Eugene, OR), washed, and mounted on slides with Prolong Gold Antifade with 4′,6-diamidino-2-phenylindole (Molecular Probes, Inc., Eugene, OR). Confocal images were collected using an Olympus confocal FV1000 microscope and analyzed using the Olympus Fluoview software (Olympus America, Inc.). Shown are representative images for each primary antibody scanned with identical confocal settings.
Fluorescent EGF Labeling—Cells were incubated with 10 ng/ml Texas Red EGF (Molecular Probes) for 10 min at 37 °C and washed twice with cold modified PBS (PBS, pH 7.4, 0.5 mm MgCl2, 0.5 mm CaCl2, 5 mm glucose, 0.2% bovine serum albumin), three times with cold citrate buffer (pH 4.6, 25.5 mm citric acid, 24.5 mm sodium citrate, 280 mm sucrose), and re-equilibrated with two PBS washes. Cells were incubated in medium and fixed in 4% paraformaldehyde at the appropriate time point and processed for indirect immunofluorescence.
Colocalization Analysis—Colocalization of Texas Red-EGF and the endosomal marker proteins was quantified as described by Lopez-Alcala et al. (22). To quantify colocalization, a randomly chosen field of cells (5–10 cells/experiment) was selected as a region of interest, and analysis was carried out on three separate planes for each field using ImageJ Software and the Colocalization plug-in (Pierre Bourdoncle, Institut Jacques Monod, Service Imagerie, Paris) to generate a binary image of colocalized pixels from two separate channels. ImageJ was used to automate channel thresholding, and colocalization was established for pixels whose intensities were higher than threshold and for which the ratio of intensity was greater than 50%. Using the “min” operation in the Image Calculator function of ImageJ, the colocalized pixels were converted to the real value of red (Texas Red-EGF) as a 32-bit image. The original Texas Red channel was also converted to a 32-bit image, and the integrated intensity was measured from both images. The data were plotted as the ratio of the integrated intensity from the two images. The ratio of the three planes was averaged for each condition for each experiment. All data represent the average of three separate experiments, with a total of 20–30 cells measured per experiment.
Percoll Gradient Fractionation—Analysis of radioligand distribution was performed as described by Kornilova et al. (23). Briefly, cells were incubated for 15 min with 125I-EGF (1 ng/ml) at 37 °C in binding buffer (DMEM, 20 mm HEPES, 0.1% bovine serum albumin, pH 7.3). Cells were washed four times with PBS and returned to 37 °C in growth medium. After an additional 45 min (60 min total), cells were washed twice with TES (10 mm triethanolamine, 1 mm EDTA, 0.25 m sucrose, pH 7.2) and harvested in 2 ml of TES. Lysates were pipetted up and down 40 times. Lysates were centrifuged for 10 min at 200 × g in a Sorvoll JA25.5 rotor. The supernatant was transferred to a clean tube, and the pellet was resuspended in 2 ml of TES and centrifuged again. The resulting 4 ml of postnuclear supernatant (PNS) was diluted with a 90% Percoll solution in TES to 17% in 11.5 ml. Samples were centrifuged for 25 min at 4 °C at 50,000 × g in a Beckman Ti65 rotor to separate early and late endocytic compartments. Samples were fractionated into 10-drop fractions (∼30 fractions/gradient) from the bottom and measured for radioactivity in a Beckman γ-counter. Every other fraction was immunoblotted for early or late endocytic markers (EEA1 and LAMP-1, respectively). Data are plotted at the percentage of total radioactivity in each tube versus the relative migration of each fraction in the sample. Density was monitored using density marker beads (Amersham Biosciences).
To further resolve the components of the late endocytic pathway, the high density fractions (fractions 1–9; Rf = 1.0 to 0.7) from the 17% Percoll gradient were pooled and resuspended to 28% Percoll using a 90% Percoll solution in TES (11.5-ml total volume). These samples were centrifuged for 25 min at 4 °C at 50,000 × g in a Beckman Ti65 rotor and fractionated into 10-drop fractions (∼30 fractions/gradient). This assay allows for the separation of late endosomes (ρ = 1.048–1.060 g/ml) and lysosomes (ρ = 1.07–1.11 g/ml) (23).
Enzyme Assay—β-galactosidase was measured using fluorescein-β-d-galctopyranoside as substrate. Equal amounts (100 μl) were taken from each fraction of a 28% Percoll gradient and incubated in 250 μl of 0.1 m sodium acetate buffer, pH 4.4, with 0.1% Triton X-100 and 100 μm substrate (24). The samples were incubated for 3 h at 37 °C, and fluorescent emission was measured with excitation at 480 nm and emission at 520 nm on a FLUOstar OPTIMA (BMG LabTech). The data were plotted as percentage of total β-galactosidase activity minus β-galactosidase activity associated with heat-inactivated PNS (basal) and 28% Percoll. For total basal activity, cells were harvested in TES, and total PNS was measured for β-galactosidase.
Electron Microscopy—For cell compartment quantification, HeLa cells were transfected with RAB7 or siCON siRNA as described above. Seventy-two hours post-transfection, cells were washed 3 × 5 min in serum-free medium at 37 °C. The cells were then treated with 0.4 μg/ml EGF-biotin for 15 min at 37 °C, washed three times in serum-free binding medium, and chased for 45 min with serum-free binding medium at 37 °C similar to previously published methods (25). Cells were fixed in 4% paraformaldehyde, 2% gluteraldehyde in 0.1 m cacodylate buffer at room temperature for 1 h. Cells were postfixed with 1% OsO4 in H2O for 30 min at room temperature, scraped, and pelleted at 1000 × g. Samples were dehydrated in an acetone and propylene oxide series and embedded in Epon Araldite. Ultrathin sections (100 nm) were cut and stained with Sato's Lead and uranyl acetate. Images were collected on a Hitachi H-7600 transmission electron microscope at the Oklahoma Medical Research Foundation Imaging Facility. Individual compartments were measured and quantified using ImageJ (National Institutes of Health). Thirty-five (105 total) individual sections from three independent experiments per condition were counted using images taken at ×15,000 magnification. Total cytosolic area was quantified from images taken at lower magnification (×2000–4500) by subtracting the nuclear area from the total section area.
Data plotted in Fig. 8, D and E, are the “observed size distribution” of LE/MVB and lysosomes. When presenting the results of the analysis of electron micrographs (Fig. 10), we discuss the “true size distribution,” which reflects a correction in the data to account for differences in the positioning of the organelles in the 100-nm electron microscopy section (26).
FIGURE 8.

125I-EGF accumulates in multiple high density fractions in the absence of RAB7. HeLa cells transfected with RAB7 siRNA were pulse-labeled with 125I-EGF for 15 min and chased in DMEM for 45 min at 37 °C. PNS from these cells was resolved over consecutive 17 and 28% Percoll gradients as in Fig. 6B. Fractions from the 28% gradient were collected and assayed for both radioactivity and β-galactosidase activity (β-gal). Data are plotted as a percentage of total enzyme activity (triangle) or radioactivity (square). Data are representative of three independent experiments. The diamonds represent the density bead markers described in the legend to Fig. 6.
FIGURE 10.

Depletion of RAB7 results in the accumulation of enlarged late endocytic compartments. A, size distribution of maximum diameter (nm) for MVBs (siCON, n = 282; RAB7 siRNA, n = 387). B, size distribution (nm) of maximum diameter for lysosomes (siCON, n = 259; RAB7 siRNA, n = 151). C, percentage of total cytosolic area occupied by characteristic late endocytic compartments (mean ± S.E.). D, comparison of the average diameter (nm) of the late endocytic compartments observed in both conditions (mean ± S.E.). Total cytoplasmic area was as follows: siCON, 4914 μm2; RAB7 siRNA, 5000 μm2. Data are representative of 105 sections/condition analyzed from three independent experiments. *, p < 0.05; **, p < 0.01; ***, p < 0.001 (Student's t test). E and F, ILVs were counted from 75 MVBs (25 MVBs/experiment; n = 3) and plotted as the average number of ILVs/MVB (E) or the mean number of ILVs/MVB (F). Data are plotted as the mean ± S.E. ***, p ≤ 0.001 (Student's t test).
The corrected mean diameter (D) of the compartments was calculated using the Fullman equation,
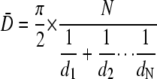 |
(Eq. 1) |
where N is the total number of compartments, and d is the diameter of the individual compartments (27).
Figures were prepared by importing digital images into Adobe Photoshop CS, calibrating scale, and adjusting brightness and contrast as needed. Knockdown of RAB7 and internalization of EGFR were monitored in parallel dishes by Immunoblot and confocal immunofluorescence microscopy, respectively.
Protease Protection Assay—Protease protection assays to detect the percentage of EGFR sorted into intraluminal vesicles were performed using a previously published protocol (28, 29). Cells (2 × 106) were treated with 10 ng/ml EGF (5, 30, or 60 min), washed in PBS, and pelleted at 1500 rpm for 5 min at 4 °C. The cytosol was leaked from the cells by resuspending the cell pellets in 6.5 μg/ml digitonin and incubating for 5 min at room temperature, followed by 30 min on ice. The permeabilized cell suspension was centrifuged at 14,000 rpm for 5 min to pellet the membrane and nuclei. The pellet was resuspended in 300 μl of homogenization buffer (100 mm K2HPO4/KH2PO4, pH 6.7, 5 mm MgCl2, 250 mm sucrose). Equivalent portions (100 μl) from each condition were incubated with H2O, 2.5 ng/ml proteinase K, or 2.5 ng/ml proteinase K with 0.1% Triton X-100 for 10 min at room temperature. The reaction was stopped by adding 20 mm phenylmethanesulfonyl fluoride and 2× preheated SDS sample buffer and boiling at 100 °C for 3 min. The samples were subjected to SDS-PAGE and immunoblotted with an EGFR antibody. EGFR levels were measured using ImageJ software (National Institutes of Health).
RESULTS
Depletion of Rab7 Slows the Rate of EGFR Degradation—To study the role of RAB7 in the endocytic pathway, we monitored trafficking of the EGFR in HeLa cells. HeLa cells express a physiological number of EGFRs (∼50,000 EGFRs/cell) (30), and the endocytic traffic of the EGFR has been well characterized in these cells (31–33). Treatment of HeLa cells with EGF results in almost a complete lysosomal degradation of the EGF·EGFR complex. These features make it an excellent model to study endocytic trafficking.
Human RAB7 siRNA duplexes from Dharmacon (19) were used to attenuate endogenous RAB7 expression in HeLa cells. We first determined the optimal concentration of RAB7 siRNA by transfecting HeLa cells with decreasing concentrations of the siRNA pool (Fig. 1A). We found that 72 h after transfection, 10 nm siRNA complexes abolished all detectable RAB7 by immunoblot, whereas concentrations of <10 nm had low levels of detectable RAB7. In a time course of RAB7 knockdown with 10 nm RAB7 siRNA, there was inhibition of RAB7 expression within 24 h; 72 h post-transfection, the endogenous RAB7 is not detected (Fig. 1B). Parallel dishes of cells were either untransfected or transfected with a pool of nontargeting scrambled siRNA duplexes (siCON) as controls. Expression of the small molecular weight GTPase, RAB5 was unaffected by RAB7-specific siRNA.
FIGURE 1.

Loss of RAB7 slows EGFR degradation. A, HeLa cells were transfected with no siRNA (-), 100 nm siCON, or RAB7 siRNA at a concentration of 100, 25, 10, and 1 nm (lanes 2–5, respectively). Total cell lysate (20 μg) was resolved by 12% SDS-PAGE, transferred to nitrocellulose, and immunoblotted with the indicated antibodies. B, HeLa cells were transfected with no siRNA (-), RAB7 siRNA for 24, 48, and 72 h, or siCON for 72 h, as indicated. Total cell lysate (20 μg) was resolved by 12% SDS-PAGE, transferred to nitrocellulose, and immunoblotted with the indicated antibodies. C and D, HeLa cells were transfected with nothing (untransfected), siCON, or RAB7 siRNA and allowed to recover for 72 h. 125I-EGF degradation and 125I secretion were monitored as described under “Materials and Methods.” Data are plotted as the percentage of intact total radioactivity (B) and the percentage of secreted total radioactivity (C). n = 3; mean ± S.E. **, p < 0.01 (Student's t test) as compared with either siCON-transfected or untransfected samples.
We wanted to determine if knockdown of RAB7 affects endocytic trafficking. Using 125I-EGF as a marker of the EGF·EGFR complex, we monitored 125I-EGF degradation in RAB7 knockdown and control (no siRNA- and siCON-transfected) cells. Following pulse loading the cells with 125I-EGF, we measured the amount of trichloroacetic acid-precipitatable 125I-EGF (intact) and secreted 125I after various times of chase with radioligand-free medium (Fig. 1, C and D). These complementary assays measure radioligand degradation by quantifying the intact ligand and the secreted radiolabeled amino acids following degradation, respectively. We observed that the loss of RAB7 decreased the rate of 125I-EGF degradation and 125I secretion, consistent with our findings using dominant-negative RAB7 (11). We observed that the uptake of 125I-EGF in RAB7 knockdown cells is indistinguishable from control cells (untransfected and siCON-transfected) when analyzed by a paired Student's t test (data not shown). This finding is consistent with the idea that RAB7 is not required for EGFR endocytosis.
Not surprisingly, the inhibition of trafficking was more robust with loss of RAB7 than expression of a dominant negative mutant RAB7 defective in guanine nucleotide binding (11). The weaker phenotype seen with the mutant RAB7 may be due to underlying activity of the endogenous wild type RAB7 and/or the fact that this dominant negative mutant of Rab7 rapidly degrades (11).
To confirm that the change in EGFR degradation was not an off-target effect of the siRNA, we rescued this phenotype by transient expression of an HA-tagged, wild type canine Rab7 (HA-Rab7(canine)). The canine Rab7 differs by 2–3 nucleotides in each of the siRNA-targeted regions of the human RAB7. This difference is sufficient to render the canine Rab7 resistant to the RAB7 siRNA transfection (Fig. 2A, middle). Like radioligand degradation, EGFR degradation is slowed in the absence of RAB7. Introduction of HA-Rab7(canine) restores the ligand-mediated degradation of the EGFR to control levels (Fig. 2A, top).
As a second assay to determine the specificity of RAB7 knockdown, we adenovirally expressed either HA-tagged constitutively active or dominant negative canine Rab7 (HA-Rab7(Q67L) or HA-Rab7(N125I), respectively) in control or RAB7 knockdown cells. Expression of these adenoviruses is tetracycline-regulatable (“tet-off”), which allows us to control for any potential adenoviral effects by examining infected cells in the presence of tetracycline (Fig. 2C). When 125I-EGF degradation was measured after the addition of radioligand, in RAB7 knockdown cells, radioligand degradation was restored by the expression of HA-Rab7(Q67L) (Fig. 2B). Expression of HA-Rab7(N125I) or infection with HA-Rab7(Q67L) in the presence of tetracycline did not restore 125I-EGF degradation (Fig. 2B). Thus, the altered EGF·EGFR trafficking in RAB7 knockdown cells can be differentially restored by the expression of activating and inactivating mutants of Rab7.
The Loss of Rab7 Does Not Affect EGFR Trafficking through Early Endosomes—To interpret the role of RAB7 in endocytic trafficking, we have followed trafficking of the EGF·EGFR complex through the endocytic pathway in the absence of RAB7. We began our analysis by monitoring movement of the ligand-receptor complex through the early endosome, since it has been reported that overexpression of dominant negative RAB7 mutants impairs cargo exit from early endosomes (8, 10).
Rab7 knockdown and control cells were pulse-labeled with fluorescent EGF (Texas Red-EGF) for 10 min, followed by a chase in ligand-free medium. At the indicated time points (10, 30, and 60 min), cells were fixed and stained for EEA1. The extent of colocalization was quantified as described under “Materials and Methods” (22). At all three time points, there was an indistinguishable degree of Texas Red-EGF and EEA1 colocalization (Fig. 3, A and B). Co-localization was highest at the 10 min time point (∼50%) and decreased over time. After 60 min, there was less than 5% colocalization of Texas Red-EGF and EEA1.
FIGURE 3.

Loss of RAB7 does not affect EGFR progression through EEA1 positive early endosomes. A, HeLa cells were transfected with nothing (No siRNA), RAB7 siRNA, or siCON and grown on coverslips. Seventy-two hours post-transfection, the cells were serum-starved for 2 h and pulse-labeled with Texas Red-EGF (TR-EGF) (Molecular Probes) as described under “Materials and Methods.” At the indicated time points, the cells were fixed and processed for confocal immunofluorescence microscopy using an anti-EEA1 mouse monoclonal antibody (Transduction Laboratories). Images were collected with an Olympus FV500 confocal microscope and processed using the FV500 software. Insets are higher resolution images of the boxed region. Images are single planes and representative of three independent experiments. Size bar, 10 μm. Inset size bar, 1 μm. B, endosomes positive for both EEA1 and Texas Red EGF were counted and plotted as a percentage of total EEA1-positive endosomes. Data are plotted as the mean ± S.E. (n = 3). Three consecutive planes (0.20-μm steps) from three separate fields containing 5–15 cells were analyzed from each experiment for each condition. *, p < 0.05 (Student's t test). Control (C) or RAB7 siRNA-transfected cells (D) were pulse-labeled with 125I-EGF for 15 min at 37 °C and chased with DMEM for an additional 45 min at 37 °C. Postnuclear supernatant from these cells was separated over a 17% Percoll gradient. Fractions were collected and assayed for associated radioactivity. Data are plotted as the percentage of total radioactivity found in each fraction. An immunoblot was performed on every other fraction using LAMP-1 to mark late endosomes/lysosomes and EEA1 to mark early endosomes. Diamonds on the x axis represent migration of density beads (∼Rf0.22 = 1.040 g/ml; ∼Rf0.8 = 1.069 g/ml; ∼Rf0.85 = 1.080 g/ml; ∼Rf0.9 = 1.109 g/ml). WT, wild type.
The conclusion of this single cell assay is supported by a complementary biochemical approach. Control and RAB7 knockdown cells were pulse-labeled for 15 min with 125I-EGF, washed, and chased for an additional 45 min in serum-free medium. PNSs were separated on 17% Percoll gradients to resolve early and late endocytic compartments (23). Gradient fractions were assayed for the presence of radioactivity. In RAB7 knockdown cells, 125I accumulated mainly in fractions of a density characteristic of late (Rf = 0.8–1.0) endocytic vesicles (Fig. 3D). As predicted by the 125I-EGF degradation experiment shown in Fig. 1B, control cells had little 125I accumulation, although there were small peaks corresponding to the predicted locations of early (Rf = 0.0–0.2) and late (Rf = 0.8–1.0) endocytic vesicles (Fig. 3C). Although ∼10% of the total radioactivity accumulated in light fractions of control cells, this amount of radioactivity represents only a fraction of what was observed in RAB7 siRNA-treated cells. Together, the immunofluorescent and biochemical data indicate that RAB7 is not required for trafficking into or out of the early endosome.
Knockdown of Rab7 Does Not Affect Transferrin Receptor Recycling—To determine whether RAB7 was required for recycling from the early endosome, rather than progression to LE/MVB, we analyzed the endocytic trafficking of an established marker of recycling, the transferrin receptor. Briefly, cells were incubated with 125I-Tfn on ice for 2 h to achieve steady-state binding to the receptor. After washing away unbound 125I-Tfn, endocytosis was initiated by transitioning the cells to 37 °C by incubating them in prewarmed medium. At various times following the addition of medium, the amount of internalized 125I-Tfn and secreted 125I-Tfn was analyzed. The kinetics of 125I-Tfn uptake and recycling in RAB7 knockdown cells was indistinguishable from controls (untransfected and siCON-transfected cells) (Fig. 4, A and B). As positive control for inhibited recycling, cells were incubated with the early endosomal proton transport inhibitor monensin (21). Treatment with monensin prevents recycling from the early endosome and causes the accumulation of 125I-Tfn within the cell. These data indicate that RAB7 is dispensible for 125I-Tfn recycling and support the notion that it functions downstream of the early endosome.
FIGURE 4.

Knockdown of RAB7 does not affect transferrin recycling through the early endosome. HeLa cells were transfected with nothing or siCON or RAB7 siRNA. After recovery, cells were analyzed for 125I-Tfn recycling, as described under “Materials and Methods.” As a positive control to inhibit 125I-Tfn recycling, cells were incubated with 100 μm monensin. A, time course of internalized 125I-Tfn; B, time course of secreted 125I-Tfn. Data are plotted as the mean ± S.E. (n = 3). *, p < 0.05; **, p < 0.01 (Student's t test).
Loss of RAB7 Results in the Accumulation of EGFR in Late Endocytic Compartments—Because the previous data indicate that the EGFR exits EEA1-positive compartments, we wanted to determine if the immediate downstream trafficking to late endocytic structures required RAB7. We monitored trafficking of fluorescently labeled EGF into compartments immunofluorescently stained with markers of the LE/MVB and lysosomes.
CD63 (melanoma 1 antigen) was used as an LE/MVB marker because it has been shown that the EGFR traffics through a subpopulation of MVBs positive for this protein en route to lysosomes (34). Very little Texas Red-EGF is detected in CD63-positive compartments at 10 min (Fig. 5, A and B). At 30 and 60 min poststimulation, Texas Red-EGF colocalizes with CD63, and the amount of colocalization is indistinguishable in RAB7 knockdown and control cells (Fig. 5, A and B). However, at 90 min poststimulation, the total amount of Texas Red-EGF colocalizing with CD63 is reduced in control cells (from ∼40% colocalization to ∼20%). In contrast, in RAB7-deficient cells, 90 min after the introduction of Texas Red-EGF, there is an increase in the amount of Texas Red-EGF that colocalizes with CD63 (from ∼40 to ∼65%). One explanation for this increased colocalization is that Texas Red-EGF is able to enter, but not exit, the CD63-positive compartment.
FIGURE 5.

Loss of RAB7 results in the accumulation of EGF in CD63-positive endosomes. A, HeLa cells were transfected with nothing (No siRNA), RAB7 siRNA, or siCON and pulse-labeled with Texas Red-EGF (TR-EGF) (red) for the indicated times. Cells were fixed and processed for indirect immunofluorescence staining with antibody against CD63 (multivesicular bodies) (green). Nuclei were stained with 4′,6-diamidino-2-phenylindole (blue). Size bar, 10 μm. Images were collected with an Olympus FV1000 confocal microscope and processed using the FV1000 software. Insets are higher resolution images of the boxed region. Top, Texas Red signal; middle, CD63 signal; bottom, merged image. Inset size bar, 1 μm. Shown are single planes that are representative of 3–6 independent experiments. B, endosomes positive for both CD63 and Texas Red EGF were counted and plotted as a percentage of total CD63-positive endosomes. Data are plotted as the mean ± S.E. (n = 3, 10 min; n = 6, 30, 60, and 90 min). Three consecutive planes from three separate fields containing 5–15 cells were analyzed from each experiment for each condition. ***, p ≤ 0.001 (Student's t test).
As a second approach to determine where knockdown of RAB7 was causing a defect in endocytic trafficking, we monitored Texas Red-EGF movement into compartments positive for the lysosomal marker, LAMP-2 (lysosome-associated membrane protein 2). In the absence of RAB7, only a small fraction of Texas Red-EGF colocalized with LAMP-2 (Fig. 6, A and B). Control cells exhibit a time dependent increase in co-localization of Texas Red-EGF and LAMP-2. However, in RAB7 knockdown cells, even 90 min post-stimulation, there was never more than 10% of the total Texas Red-EGF colocalized with LAMP-2. Despite a decrease in the total Texas Red-EGF in control cells, there is an increase in the percentage of fluorescent ligand in the lysosome. Taken with the CD63 indirect immunofluorescence data, our findings support the idea that RAB7 is not necessary for entry into the LE/MVB but is required for subsequent events in endocytic trafficking.
FIGURE 6.

The EGFR does not reach LAMP-2-positive lysosomes in the absence of RAB7. A, HeLa cells were transfected with nothing (No siRNA), RAB7 siRNA, or siCON and pulse-labeled with Texas Red EGF (red) for the indicated times. Cells were fixed and processed for indirect immunofluorescence staining with antibody against LAMP-2 (lysosomes) (green). Nuclei were stained with 4′,6-diamidino-2-phenylindole (blue). Size bar, 10 μm. Images were collected with an Olympus FV1000 confocal microscope and processed using the FV1000 software. Insets are higher resolution images of the boxed region. Top, Texas Red signal; middle, CD63 signal; bottom, merged image. Inset size bar, 1 μm. Shown are single planes that are representative of three independent experiments. B, endosomes positive for both LAMP-2 and Texas Red EGF were counted and plotted as a percentage of total endosomes positive for LAMP-2 only. Data are plotted as the mean ± S.E. (n = 3). Three consecutive planes from three separate fields containing 5–15 cells were analyzed from each experiment for each condition. *, p < 0.05 (Student's t test).
Lysosomal Enzyme Activity Is Decreased and Redistributed in the Absence of Rab7—Rab7 has been reported to regulate biogenesis and maintenance of the lysosome (35). Thus, the absence of a functional lysosome could explain the failure of Texas Red-EGF to exit the LE/MVB. To determine if there is a functional lysosome, we examined β-galactosidase activity. We observed that RAB7 knockdown cells with and without EGF stimulation had on average a 23% decrease in total β-galactosidase activity (Fig. 7A). Although this change is statistically significant, it is unlikely to account for the almost complete inhibition of 125I-EGF degradation in RAB7-deficient cells.
FIGURE 7.

Total lysosomal enzyme activity is decreased and endocytic compartment density is altered in the absence of RAB7. A, HeLa cells were transfected with RAB7 siRNA or siCON. Seventy-two hours post-transfection, cells were stimulated without (0′ EGF) or with EGF (60′ EGF) for 60 min. PNS was collected from an equal number of cells and assayed for total β-galactosidase activity. Enzyme activity was normalized to protein levels. Data were normalized to enzyme activity from unstimulated siCON-treated cells. Data are shown as the mean ± S.E. from five independent experiments. *, p < 0.05 (Student's t test). siCON-transfected (B) and RAB7 siRNA-transfected HeLa cells (C) were stimulated for 60 min with EGF. PNS was separated over a 17% Percoll gradient, and the late endosomal fractions (Rf 1.0-0.7) were then further resolved over a 28% Percoll gradient. Fractions from the 28% gradient were assayed for β-galactosidase activity, and each fraction was plotted as a percentage of total activity. Data are representative of four independent experiments. Diamonds on the x axis represent migration of density beads (Rf0.2 = 1.040 g/ml; Rf0.48 = 1.055 g/ml; Rf0.74 = 1.069 g/ml; Rf0.79 = 1.080 g/ml; Rf0.85 = 1.109 g/ml). Two consecutive fractions from the first 26 fractions of the 28% Percoll gradient were combined, resuspended in 2× Sample Buffer, resolved by 7.5% SDS-PAGE, and immunoblotted (IB) for the presence of CD63 (insets).
An alternative possibility is that the β-galactosidase is not localized properly in the absence of RAB7. We monitored β-galactosidase activity in PNS that had been separated over a 28% Percoll gradient that resolves late endocytic compartments. Quite strikingly, when RAB7 levels were attenuated, the β-galactosidase activity accumulated in more dense fractions as compared with control cells (Fig. 7, B and C). In control cells, the β-galactosidase activity is found in intermediate fractions (Rf = 0.2–0.5) that have a density (1.048–1.060 g/ml) characteristic of both late endosomes (23) and hybrid vesicles arising from an MVB-lysosome fusion (36). In RAB7 knockdown cells, β-galactosidase activity accumulated in more dense fractions (Rf = 0.6–1.0) characteristic of lysosomes (1.07–1.11 g/ml) (23).
We also probed the fractions for CD63. In control cells, CD63 co-migrates with the peak of β-galactosidase activity and sediments at a density characteristic of hybrid vesicles (36). In contrast, in RAB7 knockdown cells, CD63 distributes diffusely across the gradient. (Fig. 7, B and C). Together with the shift in β-galactosidase migration, the redistribution of CD63 indicates an alteration in the profile of late endocytic compartments in the absence of RAB7.
To determine where cargo distributes relative to β-galactosidase activity, we repeated the fractionation experiment in RAB7 knockdown cells that had been incubated with 125I-EGF for 60 min. Following fractionation of the gradients, we monitored both the distribution of radioactivity and β-galactosidase activity on 28% Percoll gradients. Cells that had been transfected with siCON were not used in these experiments, because, as shown in Fig. 3B, they do not have significant accumulation of radioactivity following incubation with 125I-EGF. There were three major peaks of 125I activity. Two of the 125I fractions co-sedimented with the major peak (density: 1.069-1.109 g/ml; Rf = 0.9–0.7) and minor peak (density: ∼1.050 g/ml; Rf ∼0.3) of β-galactosidase activity. The third peak of 125I does not contain β-galactosidase activity (Fig. 8).
Despite the observation that a substantial portion of the total cellular radioactivity (∼60%) does not colocalize with the major peak of β-galactosidase activity, the dense β-galactosidase/125I-positive fractions cannot be ignored. One possibility is that the dense 125I and β-galactosidase peaks represent two distinct vesicle populations. This may arise because the 125I-containing fraction represents accumulation in an aberrant compartment of high density. This possibility is consistent with the increased density at which CD63 migrates (Fig. 7B). An alternative explanation is that, despite the presence of β-galactosidase, a nonfunctional lysosome accumulates in the dense fraction. This possibility is not favored based on the immunofluorescence data from Fig. 5 that indicate that there is little colocalization of Texas Red-EGF with the lysosomal marker LAMP-2 after 60 min of ligand incubation.
Rab7 Is Not Necessary for Maturation of Multivesicular Bodies, but Depletion of Rab7 Results in the Accumulation of Enlarged LE/MVB Compartments—The altered distribution of CD63 on Percoll gradients led us to investigate whether the absence of RAB7 affects endocytic organelle formation and/or maintenance. We used transmission electron microscopy to examine the ultrastructural morphology of late endocytic compartments in both RAB7 siRNA- and siCON-transfected cells. MVBs have been classified as compartments of >200 nm containing intraluminal vesicles (ILVs) but lacking a lamellar morphology (34). Analysis of cells with depleted RAB7 revealed enlarged MVBs packed with intraluminal vesicles and in some cases containing electron-dense content (Fig. 9, B, D, and E). A similar MVB morphology has been reported in cells lacking the endosomal sorting complex required for transport (ESCRT)-III protein, CHMP5 (29). From these electron micrographs, we measured the diameter of the LE/MVB and lysosomes from siCON and RAB7 knockdown conditions (Fig. 10, A and B). The mean observed diameter of MVBs in RAB7 knockdown cells was ∼50% larger than in control cells (Fig. 10D). Since these observed diameters reflect the profile of organelles in 100-nm electron microscopy sections, we corrected for variation in apparent vesicle size due to the limitations of analyzing spheres in thin electron microscopy sections using the Fullman Equation (see “Materials and Methods”). The overall size of both siCON control and RAB7 knockdown cells increased (480 and 671 nm, respectively), but the relative difference in diameter did not decrease significantly. The increase in LE/MVB size was reflected in the observation that the LE/MVB comprised 2.5–3.0 times more of the cytosol in RAB7 knockdown cells than in siCON-treated cells (Fig. 10C).
FIGURE 9.

Rab7 is not required for the biogenesis of LE/MVBs or ILV formation, but loss of RAB7 alters LE/MVB size and morphology. HeLa cells were transfected with siCON or RAB7 siRNA. After 72 h of recovery, cells were stimulated for 60 min with EGF and processed for electron microscopy. Shown are representative electron micrographs of sections from siCON-treated (A) and RAB7 siRNA-treated cells (B). Endocytic compartments are marked as follows: LE/MVBs (large arrowheads), clustered lysosomes (small arrowhead), or aberrant compartments containing intraluminal vesicles (arrows). Scale bars, 2 μm. C–G, representative LE/MVBs and lysosomes are shown for each condition. Scale bar, 500 nm.
Lysosomes were also observed in both conditions (Fig. 9, A and B), and there was no significant difference in their observed size with or without RAB7 (Fig. 10D). The same held true when the observed measurements were corrected using the Fullman equation. Under both conditions, lysosomes comprised comparable percentages of the cytosol (Fig. 10C).
Next, we counted the number of ILVs per MVB. The mean number of ILVs/MVB in RAB7 knockdown cells is twice what is seen in the siCON control cells (Fig. 10E). However, the number of ILVs per unit MVB area did not change (Fig. 10F). These data indicated that the enlarged LE/MVBs in RAB7 knockdown cells have an increased number of ILVs but no change in ILV size. These data confirm that formation of ILVs is not regulated by RAB7 and that the increased size of LE/MVB is correlated with an increased number of ILVs. Further, our findings are consistent with a role for RAB7 in the transport of ILVs out of the LE/MVB.
The EGFR Enters ILVs of the LE/MVB in the Absence of RAB7—We next wanted to know whether the EGFR that was retained in the CD63-positive LE/MVB was localized to the limiting membrane or in ILVs. To answer this question, we performed a proteinase K protection assay (28, 29, 37). This assay is predicated on the fact that proteinase treatment of LE/MVBs will cleave EGFRs localized to the limiting membrane, whereas those receptors in intraluminal vesicles will be resistant to protease treatment. Following stimulation with EGF, cells were permeabilized, and membrane fractions were treated with proteinase K in the absence or presence of Triton X-100. No proteinase K treatment at 5 min is a reference for the total amount of EGFR. Triton X-100 solubilizes the LE/MVB membranes and is a positive control for total receptor cleavage. We monitored the rate of EGFR degradation as a control, indicating that RAB7 activity had been ablated. Consistent with previous experiments, knockdown of RAB7 slows the rate of ligand-mediated EGFR degradation (Fig. 11B).
FIGURE 11.

EGFR is sequestered in intraluminal vesicles in the absence of RAB7. A, HeLa cells were transfected with RAB7 siRNA or siCON. After 72 h of recovery, cells were stimulated with EGF for 5, 30, or 60 min. Cells were digitonin-permeabilized, and membranes were isolated and incubated with nothing or with proteinase K in the presence or absence of Triton X-100. The samples were resolved by 7.5% SDS-PAGE and immunoblotted for EGFR. Shown is a blot from a representative experiment (n = 6). B, quantification of EGFR degradation. EGFR levels (without proteinase K or Triton X-100) from A were measured using ImageJ (National Institutes of Health) and plotted as a percentage of total EGFR at 5 min. C, quantification of protease resistance. EGFR levels (with proteinase K treatment only) from A were measured using ImageJ and plotted as a percentage of total EGFR at 5 min. Data are plotted as the mean ± S.E. (n = 6). *, p < 0.05 (Student's t test).
In RAB7 knockdown cells, there is a time-dependent increase in the amount of EGFR resistant to proteinase K degradation (Fig. 11, A and C). As expected, in the siCON control cells, there is a peak of protease resistance after 30 min of EGF treatment (Fig. 11, A and C). These data from RAB7 knockdown cells are consistent with the EGFR entering the ILVs of LE/MVBs and accumulating there over time. Combined with previous data, this shows that RAB7 is not necessary for full maturation of a LE/MVB or cargo sequestration within this compartment.
DISCUSSION
To determine the role of RAB7 in the endocytic pathway, we used siRNA to deplete endogenous RAB7 and examine cargo trafficking and compartment biogenesis. Using this approach, we were able to assess RAB7 function while avoiding the potential complications of wild type or mutant RAB7 overexpression. We used the EGF·EGFR complex as a marker for endocytic trafficking. Under normal conditions, activated EGFR is internalized and delivered to early endosomes. It has been reported by others that receptor destined for the lysosome accumulates in RAB5-positive domains, which mature into LE/MVBs as cargo is sequestered in intraluminal vesicles (13, 38). This latter process is regulated by the coordinated actions of the ESCRT-I, -II, and -III complexes (39). Cargo reaches CD63-positive compartments, which other groups have shown fuse to lysosomes, resulting in proteolysis of the cargo (40).
In HeLa cells depleted of endogenous RAB7, the endocytic processes of cargo delivery to the ILVs of mature LE/MVB are unaffected. However at this stage, cargo delivery to lysosomes is slowed, resulting in the accumulation of enlarged LE/MVB tightly packed with ILVs. Our data allow us to draw the following conclusions. First, RAB7 is not required for trafficking through EEA1-positive compartments or into CD63-positive compartments. Second, MVB biogenesis and cargo sequestration are not adversely affected by the loss of RAB7. Finally, RAB7 is necessary for cargo delivery to lysosomes. In the absence of RAB7, the equilibrium of the late endocytic pathway is perturbed and cargo accumulates in the ILVs of MVB. We conclude that RAB7 is dispensable for delivery of cargo to the late endosome and biogenesis of MVBs but is required for efficient fusion of the LE/MVB to the lysosome.
Analysis of RAB7 has led to conflicting reports regarding its function in the endocytic pathway, with groups ascribing roles in early to late endosome trafficking and others suggesting that it regulates movement to the lysosome (7, 8, 11, 12, 16, 41). Live cell imaging experiments have sustained this controversy. Rink et al. (13) report that RAB5 positive endosomes grow in size concomitant with an increase in RAB5 signal intensity. At peak size, RAB7 replaces RAB5, and the compartment rapidly acquires degradative properties (13). Although the authors of this study do not demonstrate whether RAB7 is needed for trafficking through the early endosome, a related study by Vonderheit and Helenius (10) demonstrates that expression of a fluorescently tagged dominant negative RAB7 causes cargo (Semliki forest virus) to accumulate in the early endosome.
These published data provoke the question of why our data indicate that RAB7 is not required for movement of cargo from the early endosome to LE/MVB. First, we cannot discount the possibility that in normal cells RAB7 may regulate the early to late endosome transition, but in the absence of RAB7 another protein (presumably a RAB) can fulfill this obligation. Second, it is possible that despite the proposed association of RAB7 with the early endosome, it is not functional until later in the endocytic pathway. A third possibility is that the role of RAB7 may be cargo- or cell type-specific.
Our data are consistent with RAB7 having an indirect rather than direct role in lysosome biogenesis (35). Since the loss of RAB7 causes a decrease in β-galactosidase activity and lysosome size as well as the redistribution of CD63 and LAMP-2, there is a clearly a role for RAB7 in maintaining the equilibrium of compartments in the late endocytic pathway. We favor the explanation that the altered distribution of endocytic marker proteins in RAB7 knock down cells is the consequence of lysosomes and LE/MVBs remaining segregated. In control cells, fusion occurs and forms LE/MVB-lysosome hybrid vesicles that have been reported by multiple groups as the site of cargo degradation (42, 43).
Additional findings that argue against a direct role for RAB7 in lysosome biogenesis are the modest reduction in lysosome size, number, and activity 72 h after the knockdown of RAB7. In contrast, in this same time frame, the loss of RAB7 almost completely blocks EGFR trafficking. These data provide the basis for arguing that regulating the movement of cargo is the primary function of RAB7, and its role in the biogenesis of the lysosome is secondary. However, our findings do not exclude the possibility that the continual presence of lysosome reflects the stability of this compartment.
It is important to note that in the absence of RAB7, MVB biogenesis is unaffected as ILVs form and sequester the EGFR. This is unexpected, given the finding that either overexpression or siRNA-mediated knockdown of the RAB7-interacting lysosomal protein (RILP) inhibits EGFR movement out of EEA1-positive compartments and prevents the formation of intraluminal vesicles of MVBs (44, 45). RILP was originally shown to coordinate with RAB7 to control lysosomal transport (46, 47). Importantly, however, RILP has also been shown to associate with proteins in the ESCRT-II complex that function in the formation of ILVs (44, 45, 48). That we do not see similar results with RAB7 knockdown suggests that RILP interacts with the ESCRT-II complex upstream of its interaction with RAB7.
We provide evidence for a role for RAB7 in the transfer of cargo from the LE/MVB to the lysosome. It has been shown by others that the maturation of an early endosome to a fusion-competent late endosome requires the exchange of RAB5 with RAB7 (10, 13). Our data show that RAB7 functions downstream of intraluminal vesiculation and ILV sequestration of cargo, suggesting that the RAB exchange occurs following LE/MVB maturation. This interpretation is consistent with the RAB5-RAB7 conversion event occurring at the peak in endosome diameter, followed directly by cargo degradation (13, 49). Further, we demonstrate RAB7 is required for efficient EGFR degradation and the ability of cargo to exit CD63-positive LE/MVBs and enter LAMP2-positive lysosomes. These findings support the idea that RAB7 is necessary for LE/MVB acquisition of fusion competence, probably through interactions with the homotypic fusion and vacuole protein sorting complex (42, 50–52). The functional requirement for RAB7 only at the terminal stage of the endocytic pathway ensures that LE/MVB fusion does not occur prior to the removal of recycling proteins or other regulated biological processes, such as signaling by receptor tyrosine kinases.
Our model for RAB7 function in the late endocytic pathway is analogous with the reported role of RAB7 in the parallel pathway of autophagy. Jager et al. (53) have shown that depletion of RAB7 does not affect the maturation of late autophagic vacuoles arising from the fusion between MVBs and autophagic vacuoles, but RAB7 was necessary for the final step of lysosome fusion (53).
In conclusion, we show that in the absence of RAB7, cargo enters a compartment that acquires the morphologic and protein composition characteristics of LE/MVBs but fails to fuse with lysosomes. Based on these data, we propose that the primary role of RAB7 is to regulate the movement of cargo from LE/MVB to lysosomes (Fig. 12). These findings are of obvious importance in understanding basic cell biology and provide important groundwork to identifying molecules that regulate EGFR signaling. Since cells with attenuated RAB7 sequester the EGFR in ILVs of the MVBs, this model can be used to assess whether receptor sequestration or degradation is the rate-limiting step in terminating EGFR signaling and if all signaling pathways are regulated in the same manner. An important question that remains is whether other RAB proteins are functionally involved in MVB maturation. Accordingly, it will also be interesting to determine if one RAB can compensate for the absence of a downstream RAB protein.
FIGURE 12.

Model for endocytic defects in the absence of RAB7. In wild-type HeLa cells, the EGFR is internalized via clathrin-coated pits from the plasma membrane, sorted through early endosomes to LE/MVBs, and delivered to lysosomes where it is degraded. LE/MVBs mature from early endosomes as cargo is sequestered within intraluminal vesicles. Based on our data in RAB7 knockdown cells, RAB7 is only needed to move cargo from the LE/MVB to lysosomes. Loss of RAB7 does not affect movement of the EGF·EGFR complex through the early endosome or the formation of ILVs in the MVB. Loss of RAB7 causes the accumulation of densely packed, enlarged MVBs, with a concomitant decrease in lysosome number.
Acknowledgments
We acknowledge the Imaging Facility at the Oklahoma Medical Research Foundation for support and use of equipment. We thank Jennifer McClintock, Dr. David Sherry, Dr. Leo Tsiokas, and Dr. Guangpu Li for assistance with the manuscript.
This work was supported, in whole or in part, by National Institutes of Health Grant P20 RR017703. The Fluoview1000 microscope was funded through National Institutes of Health, NEI, Grant EY09391 (to J. A. Summers Rada, Principal Investigator). This work was also supported by American Cancer Society Grant RSG-03-021-01 and Oklahoma Center for the Advancement of Science and Technology Grant HR03-014.
Footnotes
The abbreviations used are: LE, late endosome; MVB, multivesicular body; tTA, tetracycline transactivator; DMEM, Dulbecco's minimal essential medium; siRNA, small interfering RNA; Tfn, transferrin; PBS, phosphate-buffered saline; PNS, postnuclear supernatant; ESCRT, endosomal sorting complex required for transport; ILV, intraluminal vesicle; RILP, RAB7-interacting lysosomal protein; HA, hemagglutinin; EGF, epidermal growth factor; EGFR, EGF receptor.
References
- 1.Miaczynska, M., Pelkmans, L., and Zerial, M. (2004) Curr. Opin. Cell Biol. 16 400-406 [DOI] [PubMed] [Google Scholar]
- 2.Hurley, J. H., and Emr, S. D. (2006) Annu. Rev. Biophys. Biomol. Struct. 35 277-298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zerial, M., and McBride, H. (2001) Nat. Rev. Mol. Cell Biol. 2 107-117 [DOI] [PubMed] [Google Scholar]
- 4.Pfeffer, S., and Aivazian, D. (2004) Nat. Rev. Mol. Cell Biol. 5 886. [DOI] [PubMed] [Google Scholar]
- 5.Gurkan, C., Lapp, H., Alory, C., Su, A. I., Hogenesch, J. B., and Balch, W. E. (2005) Mol. Biol. Cell 16 3847-3864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chavrier, P., Parton, R. G., Hauri, H. P., Simons, K., and Zerial, M. (1990) Cell 62 317-329 [DOI] [PubMed] [Google Scholar]
- 7.Mukhopadhyay, A., Funato, K., and Stahl, P. D. (1997) J. Biol. Chem. 272 13055-13059 [DOI] [PubMed] [Google Scholar]
- 8.Press, B., Feng, Y., Hoflack, B., and Wandinger-Ness, A. (1998) J. Cell Biol. 140 1075-1089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schwartz, S. L., Cao, C., Pylypenko, O., Rak, A., and Wandinger-Ness, A. (2007) J. Cell Sci. 120 3905-3910 [DOI] [PubMed] [Google Scholar]
- 10.Vonderheit, A., and Helenius, A. (2005) PLOS Biol. 3 1225-1238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ceresa, B. P., and Bahr, S. J. (2006) J. Biol. Chem. 281 1099-1106 [DOI] [PubMed] [Google Scholar]
- 12.Meresse, S., Gorvel, J. P., and Chavrier, P. (1995) J. Cell Sci. 108 3349-3358 [DOI] [PubMed] [Google Scholar]
- 13.Rink, J., Ghigo, E., Kalaidzidis, Y., and Zerial, M. (2005) Cell 122 735-749 [DOI] [PubMed] [Google Scholar]
- 14.Poteryaev, D., Fares, H., Bowerman, B., and Spang, A. (2007) EMBO J. 26 301-312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Feng, Y., Press, B., and Wandinger-Ness, A. (1995) J. Cell Biol. 131 1435-1452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vitelli, R., Santillo, M., Lattero, D., Chiariello, M., Bifulco, M., Bruni, C. B., and Bucci, C. (1997) J. Biol. Chem. 272 4391-4397 [DOI] [PubMed] [Google Scholar]
- 17.Fukuda, M., Kanno, E., Ishibashi, K., and Itoh, T. (2008) Mol. Cell. Proteomics 7 1031-1042 [DOI] [PubMed] [Google Scholar]
- 18.Damke, H., Baba, T., van der Bliek, A. M., and Schmid, S. L. (1995) J. Cell Biol. 131 69-80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shah, A. H., Cianciola, N. L., Mills, J. L., Sonnichsen, F. D., and Carlin, C. (2007) J. Cell Biol. 179 965-980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sorkin, A. D., Teslenko, L. V., and Nikolsky, N. N. (1988) Exp. Cell Res. 175 192-205 [DOI] [PubMed] [Google Scholar]
- 21.King, A. C. (1984) Biochem. Biophys. Res. Commun. 124 585-591 [DOI] [PubMed] [Google Scholar]
- 22.Lopez-Alcala, C., Alvarez-Moya, B., Villalonga, P., Calvo, M., Bachs, O., and Agell, N. (2008) J. Biol. Chem. 283 10621-10631 [DOI] [PubMed] [Google Scholar]
- 23.Kornilova, E., Sorkina, T., Beguinot, L., and Sorkin, A. (1996) J. Biol. Chem. 271 30340-30346 [DOI] [PubMed] [Google Scholar]
- 24.Robbins, A. R. (1979) Proc. Natl. Acad. Sci. U. S. A. 76 1911-1915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Driskell, O. J., Mironov, A., Allan, V. J., and Woodman, P. G. (2007) Nat. Cell Biol. 9 113-120 [DOI] [PubMed] [Google Scholar]
- 26.Fullman, R. L. (1953) J. Metals 5 447-452 [Google Scholar]
- 27.Williams, M. A. (1985) Quantitative Methods in Biology: Practical Methods in Electron Microscopy, pp. 52-71, Elsevier North-Holland Biomedical Press, Amsterdam, The Netherlands
- 28.Malerod, L., Stuffers, S., Brech, A., and Stenmark, H. (2007) Traffic 8 1617-1629 [DOI] [PubMed] [Google Scholar]
- 29.Shim, J.-H., Xiao, C., Hayden, M. S., Lee, K.-Y., Trombetta, E. S., Pypaert, M., Nara, A., Yoshimori, T., Wilm, B., Erdjument-Bromage, H., Tempst, P., Hogan, B. L. M., Mellman, I., and Ghosh, S. (2006) J. Cell Biol. 172 1045-1056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Berkers, J. A., van Bergen en Henegouwen, P. M., and Boonstra, J. (1991) J. Biol. Chem. 266 922-927 [PubMed] [Google Scholar]
- 31.Huang, F., Khvorova, A., Marshall, W., and Sorkin, A. (2004) J. Biol. Chem. 279 16657-16661 [DOI] [PubMed] [Google Scholar]
- 32.Vieira, A. V., Lamaze, C., and Schmid, S. L. (1996) Science 274 2086-2089 [DOI] [PubMed] [Google Scholar]
- 33.Dinneen, J. L., and Ceresa, B. P. (2004) Exp. Cell Res. 294 509-522 [DOI] [PubMed] [Google Scholar]
- 34.White, I., Bailey, L., Aghakhani, M., Moss, S., and Futter, C. (2006) EMBO J. 25 1-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bucci, C., Thomsen, P., Nicoziani, P., McCarthy, J., and van Deurs, B. (2000) Mol. Biol. Cell 11 467-480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mullock, B. M., Bright, N. A., Fearon, C. W., Gray, S. R., and Luzio, J. (1998) J. Cell Biol. 140 591-601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bache, K. G., Stuffers, S., Malerod, L., Slagsvold, T., Raiborg, C., Lechardeur, D., Walchli, S., Lukacs, G. L., Brech, A., and Stenmark, H. (2006) Mol. Biol. Cell 17 2513-2523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Russell, M. R. G., Nickerson, D. P., and Odorizzi, G. (2006) Curr. Opin. Cell Biol. 18 422-428 [DOI] [PubMed] [Google Scholar]
- 39.Babst, M. (2005) Traffic 6 2-9 [DOI] [PubMed] [Google Scholar]
- 40.Bright, N. A., Gratian, M. J., and Luzio, J. P. (2005) Curr. Biol. 15 360-365 [DOI] [PubMed] [Google Scholar]
- 41.Mukhopadhyay, A., Barbieri, A. M., Funato, K., Roberts, R., and Stahl, P. D. (1997) J. Cell Biol. 136 1227-1237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Luzio, J. P., Pryor, P. R., and Bright, N. A. (2007) Nat. Rev. Mol. Cell Biol. 8 622-632 [DOI] [PubMed] [Google Scholar]
- 43.van Meel, E., and Klumperman, J. (2008) Histochem. Cell Biol. 129 253-266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Progida, C., Spinosa, M. R., De Luca, A., and Bucci, C. (2006) Biochem. Biophys. Res. Commun. 347 1074-1079 [DOI] [PubMed] [Google Scholar]
- 45.Wang, T., and Hong, W. (2006) Biochem. Biophys. Res. Commun. 350 413-423 [DOI] [PubMed] [Google Scholar]
- 46.Jordens, I., Fernandez-Borja, M., Marsman, M., Dusseljee, S., Janssen, L., Calafat, J., Janssen, H., Wubbolts, R., and Neefjes, J. (2001) Curr. Biol. 11 1680-1685 [DOI] [PubMed] [Google Scholar]
- 47.Cantalupo, G., Alifano, P., Roberti, V., Bruni, C. B., and Bucci, C. (2001) EMBO J. 20 683-693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Progida, C., Malerod, L., Stuffers, S., Brech, A., Bucci, C., and Stenmark, H. (2007) J. Cell Sci. 120 3729-3737 [DOI] [PubMed] [Google Scholar]
- 49.Del Conte-Zerial, P., Brusch, L., Rink, J. C., Collinet, C., Kalaidzidis, Y., Zerial, M., and Deutsch, A. (2008) Mol. Syst. Biol. 4 1-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Peplowska, K., Markgraf, D. F., Ostrowicz, C. W., Bange, G., and Ungermann, C. (2007) Dev. Cell 12 739-750 [DOI] [PubMed] [Google Scholar]
- 51.Seals, D. F., Eitzen, G., Margolis, N., Wickner, W. T., and Price, A. (2000) Proc. Natl. Acad. Sci. U. S. A. 97 9402-9407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ungermann, C., Price, A., and Wickner, W. (2000) Proc. Natl. Acad. Sci. U. S. A. 97 8889-8891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Jager, S., Bucci, C., Tanida, I., Ueno, T., Kominami, E., Saftig, P., and Eskelinen, E.-L. (2004) J. Cell Sci. 117 4837-4848 [DOI] [PubMed] [Google Scholar]