

Abstract
Ion translocation by the sarcoplasmic reticulum Ca2+-ATPase depends on large movements of the A-domain, but the driving forces have yet to be defined. The A-domain is connected to the ion-binding membranous part of the protein through linker regions. We have determined the functional consequences of changing the length of the linker between the A-domain and transmembrane helix M3 (“A-M3 linker”) by insertion and deletion mutagenesis at two sites. It was feasible to insert as many as 41 residues (polyglycine and glycine-proline loops) in the flexible region of the linker without loss of the ability to react with Ca2+ and ATP and to form the phosphorylated Ca2E1P intermediate, but the rate of the energy-transducing conformational transition to E2P was reduced by >80%. Insertion of a smaller number of residues gave effects gradually increasing with the length of the insertion. Deletion of two residues at the same site, but not replacement with glycine, gave a similar reduction as the longest insertion. Insertion of one or three residues in another part of the A-M3 linker that forms an α-helix (“A3 helix”) in E2/E2P conformations had even more profound effects on the ability of the enzyme to form E2P. These results demonstrate the importance of the length of the A-M3 linker and of the position and integrity of the A3 helix for stabilization of E2P and suggest that, during the normal enzyme cycle, strain of the A-M3 linker could contribute to destabilize the Ca2E1P state and thereby to drive the transition to E2P.
The sarco(endo)plasmic reticulum Ca2+-ATPase (SERCA)2 is a membrane-bound ion pump that transports Ca2+ against a steep concentration gradient, utilizing the energy derived from ATP hydrolysis (1–3). It belongs to the family of P-type ATPases, in which the γ-phosphoryl group of ATP is transferred to a conserved aspartic acid residue during the reaction cycle. Both phospho and dephospho forms of the enzyme undergo transitions between so-called E1 and E2 conformations (Scheme 1). The E1 and E1P states display specificity for reaction with ATP and ADP, respectively (“kinase activity”), whereas E2P and E2 react with water and Pi instead of nucleotide (“phosphatase activity”). The E1 dephosphoenzyme of the Ca2+-ATPase binds two Ca2+ ions with high affinity from the cytoplasmic side, thereby triggering the phosphorylation from ATP. In E1P, the Ca2+ ions are occluded with no access to either side of the membrane, and Ca2+ is released to the luminal side after the conformational transition to E2P, likely in exchange for protons being countertransported. The structural organization and domain movements leading to Ca2+ translocation have recently been elucidated by crystallization of SERCA in various conformational states thought to represent intermediates in the pump cycle (4–7). SERCA is made up of 10 membrane-spanning mostly helical segments, M1–M10 (numbered from the N terminus), of which M4–M6 and M8 contribute liganding groups for Ca2+ binding, and a cytoplasmic headpiece separated into three distinct domains, named A (“actuator”), P (“phosphorylation”), and N (“nucleotide binding”). The A-domain appears to undergo considerable movement during the functional cycle. In the E1/E1P states, the highly conserved TGE183S loop of the A-domain is at great distance from the catalytic center containing nucleotide-binding residues and the phosphorylated Asp351 of the P-domain, but during the Ca2E1P → E2P transition, the A-domain rotates ∼90° around an axis perpendicular to the membrane, thereby moving the TGE183S loop into close contact with the catalytic site such that Glu183 can catalyze dephosphorylation of E2P (8, 9). During the dephosphorylation, Glu183 likely coordinates the water molecule attacking the aspartyl phosphoryl bond and withdraws a hydrogen. Hence, the movement of the A-domain during the Ca2E1P → E2P transition is the event that changes the catalytic specificity from kinase activity to phosphatase activity. During the dephosphorylation of E2P → E2, there is only a slight change of the position of the A-domain, and a large back-rotation is needed to reach the E1 form from E2; thus, the A-domain rotation defines the difference between the E1/E1P class of conformations and the E2/E2P class. Because the A-domain is physically connected to transmembrane helices M1–M3 through the linker segments A-M1, A-M2, and A-M3, the A-domain movement occurring during the Ca2E1P → E2P transition may be a key event in the opening of the Ca2+ sites toward the lumen, thus explaining the coupling of ATP hydrolysis to Ca2+ translocation. An important unanswered question is, however, how the movement of the A-domain is brought about. Which are the driving forces that destabilize Ca2E1P and/or stabilize E2P such that the energy-transducing Ca2E1P → E2P transition takes place? To answer this, it seems important to elucidate the exact roles of the linkers. Intriguing results have been obtained by Suzuki and co-workers, who demonstrated the importance of the A-M1 linker in connection with luminal release of Ca2+ from E2P (10). In this study, we have addressed the role of the A-M3 linker. An alignment of two crystal structures thought to resemble the Ca2E1P and E2·Pi forms (5), respectively, is shown in Fig. 1. The A-domain rotation is associated with formation of a helix (“A3 helix”) in the N-terminal part of the A-M3 linker, and this helix seems to interact with a helix bundle consisting of the P5–P7 helices of the P-domain, a feature exhibited by all published crystal structures of the E2 type (cf. supplemental Fig. S1 and Ref. 11). Moreover, when structures of similar crystallographic resolution are compared (as in Fig. 1), the non-helical part of the A-M3 linker in E2-type structures has a higher relative temperature factor (“B-factor”) than the corresponding segment in Ca2E1P (Fig. 1C, thick part colored orange-red for high temperature factor), thus suggesting a higher degree of freedom of movement relative to Ca2E1P. Hence, the A-M3 linker appears more strained in Ca2E1P compared with E2 forms, and the greater flexibility of the linker in E2 forms may promote the formation of the A3 helix.
SCHEME 1.
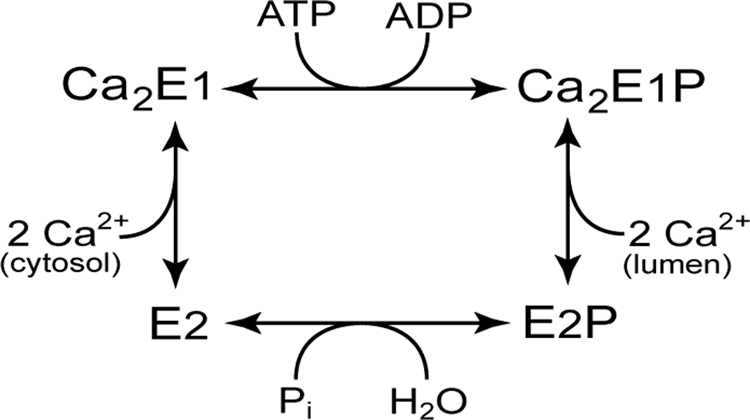
Ca2+-ATPase reaction cycle.
FIGURE 1.

A-M3 linker configuration in E1- and E2-type crystal structures. Crystal structures with Protein Data Bank codes 2zbd (Ca2E1P analog) and 1wpg (E2·Pi analog) are shown aligned. A, overview of structure 2zbd in bluish colors with green A-M3 linker and structure 1wpg in reddish colors with wheat A-M3 linker. B, magnification of the A-M3 linker (corresponding to the red box in A) with arrows indicating site 1, between Glu243 and Gln244, and site 2, between Gly233 and Lys234, in both conformations. The green A-M3 linker to the right is structure 2zbd. The wheat A-M3 linker to the left is structure 1wpg. Note the kinked A3 helix forming part of the latter structure. C, same A-M3 linker structures as in B but with the magnitude of the temperature factor (B-factor) indicated in colors (red > orange > yellow > green > blue) and by tube diameter. Because the two crystal structures selected here as E1- and E2-type representatives have similar crystallographic resolution (2.40 and 2.30 Å, respectively), the differences in temperature factor in specific regions provide direct information about chain flexibility.
Here, we have determined the functional consequences of changing the length (and thereby likely the strain) of the A-M3 linker. Polyglycine and glycine-proline loops of varying lengths were inserted at two different sites in the linker (Fig. 1), and deletions were also studied. Rather unexpectedly, we were able to insert as many as 41 residues in one of the sites without loss of expression or ability to react with Ca2+ and ATP, forming Ca2E1P, but the Ca2E1P → E2P transition was greatly affected.
EXPERIMENTAL PROCEDURES
Mutagenesis, Expression, and Assays for the Overall Reaction—Using the QuikChange site-directed mutagenesis kit, insertions, deletions, or point mutations were introduced into the cDNA encoding the rabbit fast twitch muscle Ca2+-ATPase (SERCA1a isoform) contained within the pMT2 vector (12). The mutations were validated by sequencing the cDNA throughout. The calcium phosphate precipitation method (13) was used for transfection with either wild-type or mutant cDNA into COS-1 cells (14). Differential centrifugation was used for isolation of microsomal vesicles containing either expressed wild-type or mutant Ca2+-ATPase (15). The concentration of expressed Ca2+-ATPase was determined by an enzyme-linked immunosorbent assay (16), and the amount of active enzyme (“active-site concentration”) was determined by measurement of the maximum capacity for phosphorylation with [γ-32P]ATP at 0 °C in the presence of 100 μm Ca2+ under conditions in which the dephosphorylation is slow relative to the phosphorylation. Transport of 45Ca2+ into the microsomal vesicles was measured by filtration (17), and the ATPase activity was determined by following the liberation of Pi (18, 19).
Phosphorylation with [γ-32P]ATP or 32Pi—Steady-state and kinetic measurements of phosphorylation from [γ-32P]ATP at 0 °C and measurements of equilibrium phosphorylation from 32Pi at 25 °C and dephosphorylation kinetics at 0 °C were carried out by hand-mixing as described previously (16, 17, 19, 20). Transient state kinetics at 25 °C were analyzed using the Bio-Logic QFM-5 quench-flow module (Bio-Logic SAS, Claix, France) (21). Details of reaction conditions are given in the figure legends. All phosphorylation and dephosphorylation assays were quenched with 0.5–2 volumes of 25% (w/v) trichloroacetic acid containing 100 mm H3PO4. The acid-precipitated enzyme was washed by centrifugation before SDS-PAGE on 7% gels at pH 6.0 (20, 22). Using the Packard Cyclone™ storage phosphor system, the 32P-labeled radioactive band corresponding to the Ca2+-ATPase was quantified. Background phosphorylation levels determined in the presence of EGTA and Ca2+ for assays carried out with [γ-32P]ATP and 32P, respectively, were subtracted from the data points.
Data Analysis—The experiments were generally conducted at least twice, and the complete set of data was analyzed by nonlinear regression. Ligand concentration dependences were fitted by the Hill equation (23). Monoexponential functions were fitted to the phosphorylation or dephosphorylation time courses using, in most cases, the SigmaPlot program (SPSS, Inc.). The time course of phosphorylation with [γ-32P]ATP starting from the Ca2E1 form was analyzed using the SimZyme program with a three-intermediate reaction scheme according to the previously described principles (21) as further detailed in the supplemental figure legends.
RESULTS
Mutants and the Overall Reaction—The A-M3 linker mutations studied here are indicated in Fig. 2. The importance of the length of the A-M3 linker was primarily examined by insertion and deletion mutagenesis at “site 1” between Glu243 and Gln244, where there is no secondary structure in any of the crystallized conformations (Fig. 1). The inserts were originally planned to vary in length from one to nine glycines and to include, in addition, 5G 1P 5G as the longest, 11-residue insert. However, by PCR error, an additional three mutants were obtained with 16 (5G 1P 4G 1P 5G), 26 (5G 1P (4G 1P)3 5G), and 41 (5G 1P (4G 1P)6 5G) residues inserted.
FIGURE 2.

Ca2+-ATPase activity. The rate of Ca2+-activated ATP hydrolysis was determined at 37 °C in 50 mm TES/Tris (pH 7.0), 100 mm KCl, 7 mm MgCl2, 1 mm EGTA, 0.9 mm CaCl2 (3 μm free Ca2+), 5 mm ATP, and 1 μm Ca2+ ionophore A23187. Following subtraction of the background activity determined in the absence of Ca2+, the catalytic turnover rate was calculated as the molar ratio of Pi liberated per active site/s. The active-site concentration was determined by phosphorylation with 5 μm [γ-32P]ATP for 10 s on ice in 40 mm MOPS/Tris (pH 7.0), 80 mm KCl, 5 mm MgCl2, and 0.1 mm CaCl2. The bars illustrate the catalytic turnover rates relative to the wild type (WT).
The deletion mutants studied lacked either Glu243 or Gln244 or both. As a control, testing the importance of the side chains of Glu243 and Gln244, a mutant with both of these residues substituted with glycine was also examined.
Studies were furthermore carried out with mutants carrying 1G and 3G inserts at “site 2” between Gly233 and Lys234 (cf. Fig. 1) or substitutions of Lys234. Mutants with substitutions of Gly233 were not studied here, as the effects of single substitutions of this residue have been described previously (20).
All 18 A-M3 linker mutants (even those with the very long inserts of 26 or 41 residues) could be expressed in COS-1 cells at a level sufficiently high for reliable measurements of the functional properties. The mutant with the longest insert generally showed an expression level of ∼50% that of the wild type. Mutant 233/234 3G3 gave the lowest expression level of all the mutants (∼30% of the wild type). Except for 233/234 1G and 233/234 3G, the mutant enzymes were able to catalyze ATP-driven 45Ca2+ uptake in the microsomal vesicles at a measurable rate. The overall reaction was characterized by determining the Ca2+-activated ATPase activity in the presence of saturating concentrations of Ca2+ and ATP as well as Ca2+ ionophore to make the vesicles leaky and thereby avoid back-inhibition by accumulated Ca2+. Fig. 2 depicts for all mutants the Ca2+-ATPase activity per unit of active enzyme expressed (catalytic turnover rate) relative to the value determined for the wild type. As the length of the insert at site 1 increased, the catalytic turnover rate decreased, roughly in proportion to the length of the insert. The mutant with the longest 41-residue insert displayed a catalytic turnover rate of only 0.3 relative to the wild type (see the values in supplemental Table S1). However, the mutant with only one glycine inserted at site 1 did not follow the general trend, as its activity was reduced significantly more than that of the mutant with a 3G insert. The deletion of either Glu243 or Gln244 resulted in a decrease in the relative Ca2+-ATPase activity to 0.7–0.8. Deletion of both residues reduced the relative activity to 0.4, whereas the mutant having these two residues replaced by glycines showed wild type-like Ca2+-ATPase activity. Substitution of Lys234 at site 2 with either glycine or alanine had only minor effects on the Ca2+-ATPase activity, whereas 1G and 3G inserts at site 2 had severe consequences (relative Ca2+-ATPase activity of 0.08–0.15, i.e. insignificant), consistent with the lack of measurable Ca2+ transport.
Ca2+ Interaction and Phosphorylation from [γ-32P]ATP—In the wild-type enzyme, only the form with two bound Ca2+ ions, Ca2E1, can be phosphorylated by ATP (Scheme 1). Like the wild type, all A-M3 linker mutants showed a Ca2+-dependent phosphorylation from [γ-32P]ATP, and the apparent Ca2+ affinity for activation was similar to that of the wild type or slightly higher (supplemental Fig. S2 and Table S1). Hence, there was a tendency of those mutants that displayed the largest reduction in Ca2+-ATPase activity to show a slight reduction in the K0.5 (ligand concentration giving a half-maximum effect) for Ca2+ activation. The rate of Ca2+ dissociation from Ca2E1 determined by taking advantage of the dependence of the ability to phosphorylate on the binding of the two Ca2+ ions (24) was for all mutants similar to that of the wild type (supplemental Fig. S3 and Table S1). Moreover, the time course of phosphorylation of the Ca2E1 form with [γ-32P]ATP was also wild type-like for most of the mutants, including those with the longest inserts at site 1. Only the two mutants with inserts at site 2 showed a slight reduction of the phosphorylation rate relative to the wild type (from 40 s-1 to 28 and 29 s-1) (supplemental Fig. S4 and Table S2).
Conformational Transition of the Phosphoenzyme—The Ca2E1P → E2P transition was examined by following the phosphoenzyme decay upon phosphorylation with [γ-32P]ATP under conditions in which Ca2E1P accumulates as the major steady-state intermediate in the wild-type enzyme (0 °C, presence of K+, neutral pH). An excess of EGTA was added to prevent formation of new phosphoenzyme by removing Ca2+, and the remaining phosphoenzyme was determined at various time intervals by acid quenching (Fig. 3, closed circles). Under these conditions, the Ca2E1P phosphoenzyme intermediate decays through the steps Ca2E1P → E2P → E2 + Pi. Depending on the rate constants, some of the ADP-insensitive E2P may accumulate during processing of the phosphoenzyme, and in a parallel set of experiments, the addition of ADP to remove Ca2E1P before acid quenching allowed quantification of the amount of E2P present at each time point (Fig. 3, open circles; see also supplemental Table S3 for the fraction of phosphoenzyme that initially was ADP-insensitive E2P). For most of the mutants, the major part of the phosphoenzyme was initially ADP-sensitive, as was the case for the wild type, but for the mutant with one glycine inserted at site 1, >50% of the phosphoenzyme was initially ADP-insensitive E2P. Following subtraction of the ADP-insensitive fraction (Fig. 3, open circles) from the total amount of phosphoenzyme (closed circles) to obtain the decay of Ca2E1P, the rate constant for the Ca2E1P → E2P transition could be determined by fitting a monoexponential decay function (supplemental Fig. S5). In Fig. 4, the rate constants are illustrated relative to that of the wild type, and the pattern observed here for the Ca2E1P → E2P transition is very similar to that seen for the ATPase activity in Fig. 2, with the rate constant decreasing with increasing length of the insert at site 1, down to 0.18 relative to the wild type for the longest inserts. Again, the insertion of one glycine at site 1 reduced the rate disproportionately (to 0.58, compare with 0.84 for the 3G insert), and there was also a quite significant reduction in the rate of Ca2E1P → E2P in the deletion mutants, with the E243del-Q244del double deletion mutant showing a relative rate constant as low as 0.18. Furthermore, the 1G and 3G inserts at site 2 led to a virtual block of Ca2E1P → E2P, thus explaining the very low ATPase activity of these mutants (Fig. 4; see also supplemental Table S2 for the values).
FIGURE 3.

Turnover and ADP sensitivity of phosphoenzyme. Phosphorylation was carried out with 5 μm [γ-32P]ATP for 10 s on ice in the presence of 40 mm MOPS/Tris (pH 7.0), 80 mm KCl, 5 mm MgCl2, 1 mm EGTA, 0.955 mm CaCl2 (10 μm free Ca2+), and 2 μm calcium ionophore A23187. Dephosphorylation was followed by acid quenching at the indicated time intervals after a chase with 6.7 mm EGTA (•). For comparison, the wild-type (WT) data from the upper left panel are indicated in all panels by the dashed line. The ADP-insensitive E2P fraction of the phosphoenzyme was determined by the addition of 0.9 mm ADP 2 s prior to acid quenching (○).
FIGURE 4.

Rate of Ca2E1P → E2P. The decay of the Ca2E1P phosphoenzyme calculated as the difference between the total amount of phosphoenzyme and the ADP-insensitive E2P fraction (difference between closed and open circles in Fig. 3) was fitted by a monoexponential decay function as illustrated in supplemental Fig. S5, and the extracted rate constants are shown relative to that of the wild type (WT).
E2P Phosphoenzyme Formed from Pi—The phosphorylation of E2 from 32Pi in the backward direction of the reaction cycle was examined under conditions (0.5 mm 32Pi (pH 6.0), 30% (v/v) dimethyl sulfoxide, no K+ present) that are highly favorable for E2P formation in the wild type (25). All mutants with insertions or deletions at site 1 were able to phosphorylate with 32Pi to levels >50% that of the wild type, whereas mutations at site 2 interfered strongly with the phosphorylation from 32Pi (supplemental Table S3, see column labeled “EP(Pi)/EP(ATP)”). Hence, only very small amounts of phosphoenzyme formed from 32Pi were observed for the mutants with 1G and 3G inserts at site 2. For K234G and K234A, the phosphorylation from 32Pi was <50% of the wild type, but clearly significant.
For the mutants that were able to become phosphorylated with Pi, we investigated the rate of E2P dephosphorylation (Fig. 5), and the relative rate constants corresponding to the decay of E2P are listed in supplemental Table S3 (see column labeled “Relative rc of E2P → E2”). Several of the site 1 insertion mutants, as well as the E243del-Q244del double deletion mutant, showed a reduced E2P dephosphorylation rate relative to the wild type. In most cases, this was only a slight effect, and it did not increase with increasing linker length. In fact, there was a tendency toward the opposite, with 243/244 1G and 243/244 5G showing the most pronounced reduction of the dephosphorylation rate, 243/244 1G to as little as 0.21 relative to the wild type, thus explaining the accumulation of E2P seen for this mutant in Fig. 3 and supplemental Table S3. The substitutions of Lys234 at site 2 did not reduce the E2P dephosphorylation rate at all.
FIGURE 5.

Dephosphorylation of E2P. Phosphorylation was carried out with 0.5 mm 32Pi for 10 min at 25 °C in the presence of 100 mm MES/Tris (pH 6.0), 10 mm MgCl2, 2 mm EGTA, and 30% (v/v) dimethyl sulfoxide (to increase Pi affinity). Following cooling on ice and 19-fold dilution into an ice-cold buffer containing 40 mm MOPS/Tris (pH 7.0), 2 mm EGTA, 5 mm MgCl2, 0.5 mm H3PO4, and 80 mm KCl, acid quenching was performed at the indicated time intervals (•). The lines illustrate the best fits of a monoexponential decay function to the data. The extracted rate constants are listed in supplemental Table S3. For comparison, the wild-type (WT) data from the upper left panel are indicated in all panels by the dashed line.
Ca2+ Binding Transition of the Dephosphoenzyme—In the wild type, the E2 → Ca2E1 transition is a relatively slow step compared with the phosphorylation of Ca2E1 by ATP, particularly at acidic pH values where two to three protons being countertransported have to dissociate in connection with the rearrangement that allows Ca2+ to bind (26). It is therefore possible to measure the rate of E2 → Ca2E1 by following the appearance of the ability to phosphorylate with [γ-32P]ATP upon the addition of Ca2+ to Ca2+-deprived enzyme in the E2 form (supplemental Fig. S6). The relative rate constants obtained with the A-M3 linker mutants are illustrated in Fig. 6 and listed in supplemental Table S2. For amino acid substitutions and small inserts (up to 11 in length) at site 1, deletions at site 1, and substitutions at site 2, we observed only small effects on the rate of the E2 → Ca2E1 transition. However, the longest inserts of 16, 26, and 41 residues at site 1 led to spectacular 5-, 8-, and 10-fold increases, respectively, in the rate constant for the E2 → Ca2E1 transition. The 3G insert at site 2 also had a major effect, increasing the rate by ∼15-fold.
FIGURE 6.

Rate of E2 → Ca2E1. The E2 → Ca2E1 transition was followed at 25 °C in 40 mm MES/Tris (pH 6.0), 80 mm KCl, 1 mm EGTA, and 1.1 mm CaCl2 by determining the amount of phosphorylatable enzyme appearing, as illustrated in supplemental Fig. S6. The bars illustrate the rate constants relative to the wild type (WT).
DISCUSSION
Mutants with Long Inserts at Site 1—Both the Ca2+-activated ATPase activity and the rate of the Ca2E1P → E2P transition were found to decrease in parallel with an increasing length of the insert at site 1, beginning at a length corresponding to approximately five glycines (Figs. 2 and 4). Under the conditions of our measurements of ATPase activity, the Ca2E1P → E2P transition is likely rate-limiting for the overall reaction, thus explaining the similar patterns exhibited by the overall reaction and the Ca2E1P → E2P partial reaction step in the dependence of the rate on the linker length. The slowing effect of long A-M3 linker inserts may be explained by a stabilization of Ca2E1P relative to E2P. Hence, in the wild type, strain of the A-M3 linker (cf. Fig. 1) could contribute to destabilize the Ca2E1P state and thereby to drive the transition to E2P (27), and by reducing the strain through an increase in the length of the peptide backbone chain, the long inserts would stabilize Ca2E1P. It is furthermore likely that the E2P form is destabilized by the long inserts, if they change the linker conformation and thereby interfere with the formation of the A3 helix, which has important stabilizing interactions with the P5–P7 helix bundle of the A-domain (see supplemental Fig. S1). An alternative interpretation could be that, because the A-M3 linker has to move in connection with the Ca2E1P → E2P transition (cf. Fig. 1 and supplemental Fig. S1), the long inserts would tend to slow the kinetics simply by virtue of the mass they represent. However, we furthermore observed an increased rate of the E2 → Ca2E1 transition for the longest inserts at site 1 (Fig. 6). Because the rotation of the A-domain during the E2 → Ca2E1 transition is facilitated rather than impeded by long inserts, the mass effect does not seem very important. The acceleration of the E2 → Ca2E1 transition is in accordance with the Ca2E1 form being stabilized relative to E2 by the longest inserts and seems to indicate that the dephospho forms of the enzyme generally behave in a way rather similar to the phospho forms.
The mutants with the longest inserts at site 1 showed a slight increase in the apparent Ca2+ affinity for activation of phosphorylation relative to the wild type. This probably does not represent any “true” increase in the intrinsic affinity of the E1 form for Ca2+, but is likely a consequence of both the stabilization of the Ca2E1 form relative to the low affinity E2 state and the reduced rate of Ca2E1P → E2P, causing the phosphoenzyme to accumulate at lower Ca2+ concentrations than normally required (24). The normal rate of Ca2+ dissociation from Ca2E1 (supplemental Fig. S3) supports the notion that the Ca2+-binding properties of the E1 form are wild type-like in all mutants studied here.
Anomalous Effect of the 1G Insert at Site 1—The mutant with one glycine inserted at site 1 deserves special consideration because it showed stronger effects on the ATPase activity and the Ca2E1P → E2P transition than the mutants with 3G and 5G inserts (Figs. 2 and 4) and in particular because it showed a significantly reduced rate of dephosphorylation of E2P to as little as 0.21 relative to the wild type (Fig. 5). By contrast, the mutants with the longest inserts at site 1 were very similar to the wild type with respect to the rate of dephosphorylation of E2P. A comparison of the available crystal structures thought to be analogs of the E2P ground state (7) and the E2·Pi product state (5) indicates some minor conformational difference between the A-M3 linkers of these E2 forms (supplemental Fig. S1). Mutation of Thr247, which is located at the C-terminal end of the A-M3 linker only three residues from site 1, affects E2P dephosphorylation (28) likely because the side chain of Thr247 forms a hydrogen bond with Glu340 of the P-domain in E2·Pi, but not in E2P, thus stabilizing the dephospho form relative to the phospho form. These subtle structural rearrangements accompanying the dephosphorylation obviously require flexibility of the A-M3 linker near site 1 and might therefore be more compromised by a single glycine insert compared with a longer insert that allows flexibility by a loop formation minimizing the push on the backbone in the vicinity.
Deletion Mutants—The deletions at site 1, E243del, Q244del, and E243del-Q244del, also had quite significant effects on the Ca2E1P → E2P transition and the ATPase activity, most pronounced for the double deletion E243del-Q244del, for which the rate of the Ca2E1P → E2P transition was reduced to 0.18 relative to the wild type, i.e. the same extent of reduction as seen for the longest insertions. Because the E243G-Q244G mutant was wild type-like, the effects of the deletions do not result from removal of the side chains, but must be a consequence of the shortening of the peptide backbone. Hence, the movement of the A-domain during the Ca2E1P → E2P transition not only requires strain in the A-M3 linker as a driving force that can be eliminated by lengthening of the linker, but in addition, there is a demand for the linker not to be too short. The explanation might be that the reduced flexibility of the shorter linker in the deletion mutants destabilizes the E2P form by interfering with the positioning of the A3 helix and thus with its important interactions with the P5–P7 helix bundle of the A-domain (cf. Fig. 1 and supplemental Fig. S1).
Mutants with 1G and 3G Inserts at Site 2—Of all the mutants studied here, those with 1G and 3G inserts at site 2 between Gly233 and Lys234 were the most strongly affected functionally, showing a complete block of the Ca2E1P → E2P transition, no significant ATPase activity or Ca2+ transport, and no phosphorylation of E2 backwards from Pi. Hence, these insertions seem to make the enzyme unable to attain the E2P state in the forward running as well as the backward running mode of the reaction cycle. The slight increase in apparent Ca2+ affinity of these mutants can be explained by the block of phosphoenzyme turnover as discussed above for the long inserts at site 1, and the E2 → Ca2E1 transition was furthermore greatly enhanced in the 3G insert mutant. The finding that relatively short insertions at site 2 are disruptive to the formation of E2P is in good accordance with a previous mutagenesis study in which replacement of the highly conserved Gly233 with larger residues was found to block the Ca2E1P → E2P transition and E2P formation from Pi (20). Looking at the various crystal structure analogs of E2 forms, it appears that the reason that interference at site 2 is so disturbing to E2P formation must be that this site is located in the middle of the A3 helix, right below (C-terminal to) a 90° kink of the helix (cf. Fig. 1 and supplemental Fig. S1). This kink appears to be necessary to accommodate the A-domain in its rotated position. The 1G and 3G inserts added next to Gly233 will probably destabilize the helix structure, and the previously described mutations of Gly233 to residues with side chains (20) will obviously prevent the helix from being kinked because the side chains of the two helix parts would clash in the mutants.
Comparison with Proteolytic Cleavage Studies—Proteolytic cleavage studies of the Na+,K+-ATPase originally demonstrated that tryptic and chymotryptic cleavage of the Na+,K+-ATPase at Arg262 (equivalent to Lys234 of the Ca2+-ATPase) and Leu266, respectively, i.e. the same region as site 2 of the Ca2+-ATPase defined here, can occur only in E1 states, whereas these sites are protected in E2 (29), in good agreement with the now known α-helical structure of this region in E2 (11). In the digested enzyme, the E1P form is stabilized relative to E2P, thus resembling the mutants studied here. In Ca2+-ATPase, specific proteolytic excision of MAATE243, i.e. the five residues just N-terminal to site 1, with proteinase K also led to inhibition of the Ca2E1P → E2P transition (30). In light of the present findings, this does not seem to be due to the loss of the side chains of the MAATE243 segment, but rather to the inability of the cleaved enzyme to create the strain in the A-M3 linker required for Ca2E1P → E2P transition.
Perspectives—We conclude from our results that the A-M3 linker length is quite important for the A-domain movements occurring during the major conformational changes of the Ca2+ transport cycle. The reduced rate of the Ca2E1P → E2P transition observed for the long inserts at site 1 supports the hypothesis that strain in the A-M3 linker is a driving force for the A-domain movement (27), and the effects of deletions and site 2 insertions likely reflect the importance of the position and integrity of the A3 helix for stabilization of E2P. A comparison of the E1-type crystal structures with Ca2+ bound in non-occluded and occluded states (the latter also with ATP/ADP and phosphate analogs bound) indicates that the stretching of the A-M3 linker causing the strain is a consequence of bending of the P-domain associated with Ca2+ occlusion and the resultant phosphorylation to form Ca2E1P, thus preparing the enzyme for the subsequent Ca2E1P → E2P transition (27).
We found it remarkable and surprising that an insert of as many as 41 residues in the A-M3 linker was compatible with high expression (i.e. close to normal protein folding) and function of the SERCA Ca2+ pump, albeit at a reduced rate. However, taking a broader view of P-type Ca2+ pumps, it is noteworthy that the plasma membrane Ca2+-ATPases from Nature's own hand have inserts in the A-M3 linker region close to site 1. These inserts, caused by gene differences as well as by alternative splicing of mRNA, vary in length (in fact up to 40–50 residues) and sequence according to isoform, but are generally highly charged and predicted to form an amphipathic helix. Interestingly, evidence has been reported that this region of the plasma membrane Ca2+-ATPase interacts with calmodulin, acidic membrane lipids that activate the pump rate, and possibly other regulatory proteins such as G-proteins (31, 32). Given the effects that we have observed on the rate-limiting conformational change, it is understandable and intriguing that, in some cases, Nature uses the A-M3 linker as a handle for regulatory interference with the pump cycle.
Supplementary Material
Acknowledgments
We thank Lene Jacobsen and Karin Kracht for expert technical assistance.
This work was supported in part by grants from the Danish Medical Research Council, the Novo Nordisk Foundation (Denmark), the Lundbeck Foundation (Denmark) (to A. N. H.), and the Danish National Research Foundation-PUMPKIN.
The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. S1–S6 and Tables S1–S3.
This article was selected as a Paper of the Week.
Footnotes
The abbreviations used are: SERCA, sarco(endo)plasmic reticulum Ca2+-ATPase; TES, N-tris(hydroxymethyl)methyl-2-aminoethanesulfonic acid; MOPS, 3-(N-morpholino)propanesulfonic acid; MES, 2-(N-morpholino)ethanesulfonic acid.
A slash between numbers followed by residues in single-letter code designates insertion between the two numbered residues of the indicated residues, e.g. 233/234 3G means that three glycines were inserted between residues 233 and 234. Deletions are indicated by single-letter code followed by “del,” e.g. E243del means deletion of Glu243.
References
- 1.Hasselbach, W., and Makinose, M. (1963) Biochem. Z. 339 94-111 [PubMed] [Google Scholar]
- 2.de Meis, L., and Vianna, A. L. (1979) Annu. Rev. Biochem. 48 275-292 [DOI] [PubMed] [Google Scholar]
- 3.Toyoshima, C., and Inesi, G. (2004) Annu. Rev. Biochem. 73 269-292 [DOI] [PubMed] [Google Scholar]
- 4.Toyoshima, C., Nakasako, M., Nomura, H., and Ogawa, H. (2000) Nature 405 647-655 [DOI] [PubMed] [Google Scholar]
- 5.Toyoshima, C., Nomura, H., and Tsuda, T. (2004) Nature 432 361-368 [DOI] [PubMed] [Google Scholar]
- 6.Olesen, C., Sørensen, T.-L. M., Nielsen, R. C., Møller, J. V., and Nissen, P. (2004) Science 306 2251-2255 [DOI] [PubMed] [Google Scholar]
- 7.Olesen, C., Picard, M., Winther, A. M., Gyrup, C., Morth, J. P., Oxvig, C., Møller, J. V., and Nissen, P. (2007) Nature 450 1036-1042 [DOI] [PubMed] [Google Scholar]
- 8.Clausen, J. D., Vilsen, B., McIntosh, D. B., Einholm, A. P., and Andersen, J. P. (2004) Proc. Natl. Acad. Sci. U. S. A. 101 2776-2781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Anthonisen, A. N., Clausen, J. D., and Andersen, J. P. (2006) J. Biol. Chem. 281 31572-31582 [DOI] [PubMed] [Google Scholar]
- 10.Daiho, T., Yamasaki, K., Danko, S., and Suzuki, H. (2007) J. Biol. Chem. 282 34429-34447 [DOI] [PubMed] [Google Scholar]
- 11.Schack, V. R., Morth J. P., Toustrup-Jensen, M. S., Anthonisen, A. N., Nissen, P., Andersen, J. P., and Vilsen, B (2008) J. Biol. Chem. 283 27982-27990 [DOI] [PubMed] [Google Scholar]
- 12.Kaufman, R. J., Davies, M. V., Pathak, V. K., and Hershey, J. W. (1989) Mol. Cell. Biol. 9 946-958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen, C., and Okayama, H. (1987) Mol. Cell. Biol. 7 2745-2752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gluzman, Y. (1981) Cell 23 175-182 [DOI] [PubMed] [Google Scholar]
- 15.Maruyama, K., and MacLennan, D. H. (1988) Proc. Natl. Acad. Sci. U. S. A. 85 3314-3318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vilsen, B., Andersen, J. P., and MacLennan, D. H. (1991) J. Biol. Chem. 266 16157-16164 [PubMed] [Google Scholar]
- 17.Vilsen, B., Andersen, J. P., Clarke, D. M., and MacLennan, D. H. (1989) J. Biol. Chem. 264 21024-21030 [PubMed] [Google Scholar]
- 18.Baginski, E. S., Foa, P. P., and Zak, B. (1967) Clin. Chem. 13 326-332 [PubMed] [Google Scholar]
- 19.Sørensen, T., Vilsen, B., and Andersen, J. P. (1997) J. Biol. Chem. 272 30244-30253 [DOI] [PubMed] [Google Scholar]
- 20.Andersen, J. P., Vilsen, B., Leberer, E., and MacLennan, D. H. (1989) J. Biol. Chem. 264 21018-21023 [PubMed] [Google Scholar]
- 21.Sørensen, T.-L. M., Dupont, Y., Vilsen, B., and Andersen, J. P. (2000) J. Biol. Chem. 275 5400-5408 [DOI] [PubMed] [Google Scholar]
- 22.Weber, K., and Osborn, M. (1969) J. Biol. Chem. 244 4406-4412 [PubMed] [Google Scholar]
- 23.Clausen, J. D., McIntosh, D. B., Woolley, D. G., Anthonisen, A. N., Vilsen, B., and Andersen, J. P. (2006) J. Biol. Chem. 281 9471-9481 [DOI] [PubMed] [Google Scholar]
- 24.Andersen, J. P., Sørensen, T.-L. M., Povlsen, K., and Vilsen, B. (2001) J. Biol. Chem. 276 23312-23321 [DOI] [PubMed] [Google Scholar]
- 25.de Meis, L., Martins, O. B., and Alves, E. W. (1980) Biochemistry 19 4252-4261 [DOI] [PubMed] [Google Scholar]
- 26.Forge, V., Mintz, E., and Guillain, F. (1993) J. Biol. Chem. 268 10953-10960 [PubMed] [Google Scholar]
- 27.Toyoshima, C. (2009) Biochim. Biophys. Acta, in press
- 28.Clausen, J. D., and Andersen, J. P. (2004) J. Biol. Chem. 279 54426-54437 [DOI] [PubMed] [Google Scholar]
- 29.Jorgensen, P. L., and Andersen, J. P. (1988) J. Membr. Biol. 103 95-120 [DOI] [PubMed] [Google Scholar]
- 30.Møller, J. V., Lenoir, G., Marchand, C., Montigny, C., le Maire, M., Toyoshima, C., Juul, B. S., and Champeil, P. (2002) J. Biol. Chem. 277 38647-38659 [DOI] [PubMed] [Google Scholar]
- 31.Falchetto, R., Vorherr, T., and Carafoli, E. (1992) Protein Sci. 1 1613-1621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Strehler, E., and Zacharias, D. A. (2001) Physiol. Rev. 81 21-50 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.