

Abstract
Cardiac myosin-binding protein C (cMyBP-C) is a regulatory protein expressed in cardiac sarcomeres that is known to interact with myosin, titin, and actin. cMyBP-C modulates actomyosin interactions in a phosphorylation-dependent way, but it is unclear whether interactions with myosin, titin, or actin are required for these effects. Here we show using cosedimentation binding assays, that the 4 N-terminal domains of murine cMyBP-C (i.e. C0-C1-m-C2) bind to F-actin with a dissociation constant (Kd) of ∼10 μm and a molar binding ratio (Bmax) near 1.0, indicating 1:1 (mol/mol) binding to actin. Electron microscopy and light scattering analyses show that these domains cross-link F-actin filaments, implying multiple sites of interaction with actin. Phosphorylation of the MyBP-C regulatory motif, or m-domain, reduced binding to actin (reduced Bmax) and eliminated actin cross-linking. These results suggest that the N terminus of cMyBP-C interacts with F-actin through multiple distinct binding sites and that binding at one or more sites is reduced by phosphorylation. Reversible interactions with actin could contribute to effects of cMyBP-C to increase cross-bridge cycling.
Cardiac myosin-binding protein C (cMyBP-C)2 is a thick filament accessory protein that performs both structural and regulatory functions within vertebrate sarcomeres. Both roles are likely to be essential in deciphering how a growing number of mutations found in the cMyBP-C gene, i.e. MYBPC3, lead to cardiomyopathies and heart failure in a substantial number of the world's population (1, 2).
Considerable progress has recently been made in determining the regulatory functions of cMyBP-C and it is now apparent that cMyBP-C normally limits cross-bridge cycling kinetics and is critical for cardiac function (3-5). Phosphorylation of cMyBP-C is essential for its regulatory effects because elimination of phosphorylation sites (serine to alanine substitutions) abolishes the ability of protein kinase A (PKA) to accelerate cross-bridge cycling kinetics and blunts cardiac responses to inotropic stimuli (6). The substitutions further impair cardiac function, reduce contractile reserve, and cause cardiac hypertrophy in transgenic mice (6, 7). By contrast, substitution of aspartic acids at these sites to mimic constitutive phosphorylation is benign or cardioprotective (8).
Although a role for cMyBP-C in modulating cross-bridge kinetics is supported by several transgenic and knock-out mouse models (6, 7, 9, 10), the precise mechanisms by which cMyBP-C exerts these effects are not completely understood. For instance, the unique regulatory motif or “m-domain” of cMyBP-C binds to the S2 subfragment of myosin in vitro (11) and binding is abolished by PKA-mediated phosphorylation of the m-domain (12). These observations have led to the idea that (un)binding of the m-domain from myosin S2 mediates PKA-induced increases in cross-bridge cycling kinetics. Consistent with this idea, Calaghan and colleagues (13) showed that S2 added to transiently permeabilized myocytes increased their contractility, presumably because added S2 displaced cMyBP-C from binding endogenous S2. However, other reports indicate that cMyBP-C can influence actomyosin interactions through mechanisms unrelated to S2 binding, because either purified cMyBP-C (14) or recombinant N-terminal domains of cMyBP-C (15) affected acto-S1 filament sliding velocities and ATPase rates in the absence of myosin S2. These results thus raise the possibility that interactions with ligands other than myosin S2, such as actin or myosin S1, contribute to effects of cMyBP-C on cross-bridge interaction kinetics.
The idea that cMyBP-C interacts with actin to influence cross-bridge cycling kinetics is supported by several studies that implicate the regulatory m-domain or sequences near it in actin binding (16-19). cMyBP-C is a member of the immunoglobulin (Ig) superfamily of proteins and consists of 11 repeating domains that bear homology to either Ig or fibronectin-like folds. Domains are numbered sequentially from the N terminus of cMyBP-C as C0 through C10. The m-domain, a unique sequence of ∼100 amino acids, is located between domains C1 and C2 and is phosphorylated on at least 3 serine residues by PKA (12). Although the precise structure of the m-domain is not known, small angle x-ray scattering data suggest that it is compact and folded in solution and is thus similar in size and dimensions to the surrounding Ig domains (20). Recombinant proteins encompassing the m-domain and/or a combination of adjacent domains including C0, C1, C2, and a proline-alanine-rich sequence that links C0 to C1 have been shown to bind actin (16, 18, 19).
The purpose of the present study was to characterize binding interactions of the N terminus of cMyBP-C with actin and to determine whether interactions with actin are influenced by phosphorylation of the m-domain. Results demonstrate that the N terminus of cMyBP-C binds to F-actin and to native thin filaments with affinities similar to that reported for cMyBP-C binding to myosin S2 (11). Furthermore, actin binding was reduced by m-domain phosphorylation, suggesting that reversible interactions of cMyBP-C with actin could contribute to modulation of cross-bridge kinetics.
EXPERIMENTAL PROCEDURES
Protein Expression and Purification—Recombinant murine cMyBP-C proteins containing various combinations of N-terminal domains with and without the m-domain were cloned and purified as described previously (18, 21). In some cases proteins were further purified by ion exchange chromatography (Bio-Rad). All proteins were centrifuged for 20 min at 120,000 rpm (511,000 × g) and 4 °C in a TLA 120.2 rotor in an Optima TLX ultracentrifuge (Beckman Coulter, Fullerton, CA) prior to use to remove any insoluble material. Final protein concentration was determined by UV spectrometry using extinction coefficients calculated from amino acid primary sequences and software from the Swiss Institute of Bioinformatics website (22). At least two separate preparations of recombinant cMyBP-C proteins were used for each experimental data set.
Rabbit skeletal myosin S1 was prepared by chymotryptic digestion of myosin in the presence of EDTA as described (23). Myosin S2Δ (comprised of the N-terminal 126 amino acids of myosin S2 (11)) was prepared by reverse transcriptase-PCR of RNA isolated from rat heart and subsequent cDNA cloning into the pQE-2 expression vector (Qiagen, Valencia, CA). Because the low percentage of aromatic amino acids in the S2Δ sequence precludes accurate measurement of concentration by UV spectrometry, the concentration of purified S2Δ was determined by amino acid analysis performed by the University of California Davis Molecular Structure Facility.
Bovine cardiac F-actin was prepared from ether powder as described (24). F-actin was maintained in a storage buffer (in mmol/liter: 50 KCl, 1 MgCl2, 2 Tris-HCl, pH 8.0, 0.2 CaCl2, 0.5 β-mercaptoethanol, 1 ATP, and 0.02% sodium azide) and kept at 4 °C until use. Native thin filaments (NTF) were purified from bovine heart left ventricle according to a modification of the protocol by Spiess et al. (25). Four volumes of buffer (in mmol/liter: 10 KPO4, pH 7.0, 100 KCl, 5 MgCl2, 1 EGTA, 1 NaN3, 1 DTT, and 1% Triton X-100) were added to ventricle minced in a Waring grinder and the tissue was homogenized using a Polytron PT3100. Protease inhibitor mixture (in mg/ml: 0.01 phenylmethylsulfonyl fluoride, 0.001 leupeptin, 0.001 pepstatin, 0.001 antipain) was added and the solution spun at 3,800 rpm for 13 min with a JS-4.2 rotor (Beckman). The pellet was homogenized, resuspended, and centrifuged again 4 more times with the last 3 in buffer lacking Triton X-100. Next, the pellet containing myofibrils was homogenized in 1.6 volumes of buffer plus 5 mm ATP to dissociate the thin and thick filaments. The solution was spun as above and the pellets (containing mostly thick filaments) discarded. The supernatant was then spun at 70,000 rpm for 15 min (Beckman Type 70 Ti rotor), the pellet discarded, then the supernatant spun again for 80 min. The pellets (containing mostly thin filaments) were resuspended in 25 ml of dialysis buffer (in mmol/liter: 20 Tris-HCl, 100 KCl, 5 MgCl2, 1 EGTA, 1 NaN3, 1 DTT) using a Dounce homogenizer and dialyzed overnight. The following day, the solution was brought to 200 mm KCl and 5 mm ATP and spun at 45,000 rpm for 15 min (Beckman Type 50.2 rotor) to remove any contaminating myosin. Pellets were discarded, and the supernatant was spun again for 150 min. The pellets were resuspended as above and dialyzed overnight. The purification cycle (centrifugation for 15 min, pellet discard, and centrifugation for 150 min) was repeated again the next day. Final pellets were overlaid with NTF buffer (in mmol/liter: 5 imidazole, pH 7.0, 50 KCl, 2 MgCl2, 0.2 ATP, 0.5 DTT) overnight to soften. The pellets were resuspended gently with a pipette and spun at 19,500 rpm for 30 min (Beckman JA-20 rotor) to remove debris and aggregated material. NTF concentration was measured by UV spectrometry using an extinction coefficient (280 nm) = 0.0529 μm-1 cm-1 and molecular weight = 62,500 per actin for the thin filament complex (7:1:1, actin:tropomyosin:troponin). Ca2+ regulation of purified NTF was confirmed prior to use in cosedimentation assays by measuring actin-activated S1-ATPase rates as described previously (15) except buffers contained 1 mm EGTA. Activity was 0.2 ± 0.1 s-1 in the absence of Ca2+ and 3.3 ± 0.9 s-1 (n = 6) in the presence of Ca2+ (pCa 3).
Phosphorylation of recombinant C1C2 was achieved by incubation with the catalytic subunit of PKA (Sigma P2645). C1C2 was dialyzed against a buffer containing (in mmol/liter: 20 HEPES, pH 7.4, 100 KCl, 10 MgCl2, 1 ATP, and 1 DTT). PKA was resuspended in 6 mg/ml DTT per the manufacturer's instructions and combined with an additional 1 mm ATP and C1C2 at 40 units of PKA/mg of C1C2. The reaction mixture was incubated at 4 °C for >5 h and then applied to a nickel-nitrilotriacetic acid column (Qiagen) to purify phosphorylated C1C2 and remove the PKA catalytic subunit. Phosphorylation status was assessed by Pro Q Diamond staining followed by Sypro Ruby staining (Invitrogen).
Cosedimentation Assays—Recombinant cMyBP-C proteins were dialyzed against a cosedimentation buffer (in mmol/liter: 20 imidazole, pH 7.4, 180 KCl, 1 MgCl2, 1 EGTA, 1 DTT). F-actin remained in its storage buffer prior to use. Recombinant cMyBP-C proteins (1-30 μm final concentrations) were combined with sufficient F-actin, ATP, and DTT to achieve final concentrations of 5 μm, 1 mm, and 1 mm, respectively, in a total final volume of 50 μl of cosedimentation buffer. For experiments with S2Δ, a 6-fold excess of S2Δ (30 μm) to actin was added to each tube. Reactions were allowed to equilibrate for 30 min at room temperature. Samples were then spun for 30 min at 100,000 rpm (390,000 × g) using a TLA 100 rotor at 4 °C (Beckman). The supernatants were removed from each tube and the pellets gently washed with 50 μl of cosedimentation buffer and dissolved in 100 μl of a 1:1 mixture of cosedimentation buffer to urea/thiourea sample buffer (26). Pellet fractions were loaded onto 10% polyacrylamide gels and run at constant 200 V for 50 min. Gels were stained for at least 1 h in 0.05% Coomassie R-250 staining solution, followed by destaining and drying overnight.
Quantification of Binding Data—Dried gels were scanned to a computer using a flatbed scanner and band intensities were measured using the gel analysis features of Image J (NIH, Bethesda, MD). The intensity ratio of recombinant protein to F-actin in each pellet was converted to a molar ratio (mole of cMyBP-C/mole of actin) using standard curves run on each gel that contained known amounts of cMyBP-C and actin in mol/mol ratios (Fig. 1). Standard curves were constructed by adding 40 pmol of actin to each of 7 tubes followed by addition of increasing amounts of recombinant protein (4-40 pmol) to achieve the desired mole ratio. A fixed volume from each standard tube (3.5 μl) was loaded onto the gel so that the amount of actin typically varied from 13 to 7.8 pmol across the wells of the standard curve. This range was selected to verify that staining intensity was linear over the amount of actin (10.6 pmol) loaded in each cosedimentation assay.
FIGURE 1.

Cosedimentation binding data for C0C2 and C1C2. A, representative high speed cosedimentation binding experiment. C0C2 (1-30 μm final concentrations) was mixed with 5 μm F-actin (final concentration) and spun for 30 min at 390,000 × g. The amount of C0C2 and actin in pellet fractions (left) was quantified by SDS-PAGE using a standard curve run on each gel with varying known molar ratios of C0C2 to actin (right). B, summary data (means ± S.D.) for C0C2 and C1C2 binding to actin. The Kd and Bmax values for C0C2 (circles) were 13.7 ± 5.5 μm and 0.92 ± 0.11 mol/mol (n = 7). Kd and Bmax values for C1C2 (squares) were 10.9 ± 2.2 μm and 1.03 ± 0.08 mol/mol (n = 6).
Cosedimentation binding data were plotted versus the total cMyBP-C concentration added and fit according to the following equation (27),
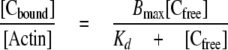 |
(Eq.1) |
where [Cbound] and [Cfree] are the bound and free concentrations of cMyBP-C proteins, respectively, [Actin] is the total actin concentration, Bmax is the maximal molar binding ratio (mole of cMyBP-C protein/mole of actin), and Kd is the dissociation constant (μm). Equation 1 can be rearranged by substituting Equation 2 in for [Cfree] and solving for [Cbound]/[Actin] to fit the data as a function of total cMyBP-C protein added ([Ctotal]), as shown in Equation 3.
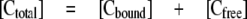 |
(Eq.2) |

Data were fit using the Microsoft Excel Solver package by varying the values of Bmax and Kd and minimizing the sum of squares between the actual and predicted binding ratios. Significance was calculated using analysis of variance and post hoc Bonferonni t tests. Results were considered significant at p < 0.01.
Analysis of Actin Cross-linking—Actin cross-linking was assessed by measurement of solution turbidity as described by Moos et al. (28). Samples containing mixtures of recombinant cMyBP-C proteins and F-actin were prepared as described above for cosedimentation assays and allowed to equilibrate for 30 min at room temperature. Light absorbance at λ = 350 nm was measured using a Beckman DU730 spectrophotometer. Samples containing F-actin alone served as blanks.
Electron Microscopy—5 μm F-actin and 30 μm recombinant proteins were mixed as described for cosedimentation assays with the exception that the centrifugation step prior to combining the proteins was omitted. Samples were incubated at room temperature for >30 min and 5-μl aliquots of each sample were applied to a freshly evaporated carbon film supported by holey formvar-covered grids. After adsorption to the carbon film (30 s), excess sample was rinsed off with 8 drops of 100 mm ammonium acetate. The ammonium acetate rinse was followed by application of 8 drops of 1% uranyl acetate for negative staining. Excess uranyl acetate was removed with filter paper after 30 s leaving a thin film of negative stain, which was then allowed to dry. Dried grids were examined and photographed at 80 kV in a JEOL-1200EXII electron microscope equipped with a 2k × 2k AMT HR60 High resolution Digital Camera (AMT, Danvers, MA).
RESULTS
The Regulatory Motif of cMyBP-C Binds to F-actin—We sought to quantify binding interactions between the cMyBP-C N terminus and actin by measuring binding affinity between the 4 N-terminal domains of cMyBP-C (i.e. C0C2 comprised of C0-C1-m-C2 and inclusive of the proline-alanine-rich linker sequence between C0 and C1) and F-actin using high speed cosedimentation assays. C0C2 is soluble at physiological ionic strength in the absence of actin and does not sediment during high speed centrifugation. However, when combined with F-actin, C0C2 readily pellets along with actin (Fig. 1), suggesting binding interactions between the two proteins. Binding was saturable at approximately a 1:1 molar ratio of C0C2 bound to actin (Bmax = 0.92 ± 0.11) and a Kd of 13.7 ± 5.5 μm (Fig. 1B and Table 1). C1C2 (i.e. C1-m-C2, which lacks the cardiac-specific C0 domain and the proline-alanine-rich region preceding C1) also bound to actin with comparable affinity (Kd = 10.9 ± 2.2 μm and Bmax = 1.03 ± 0.08), suggesting that the C0 domain and the Pro-Ala-rich region do not significantly influence interactions with actin under these assay conditions.
TABLE 1.
Summary data of dissociation constants (Kd) and molar binding ratios (Bmax) for binding of recombinant cMyBP-C proteins to F-actin
Data represent mean ± S.D.
| Protein | n | Kd | Bmax |
|---|---|---|---|
| μm | mol/mol actin | ||
| C0C2 | 7 | 13.7 ± 5.5 | 0.92 ± 0.11 |
| C1C2 (pH 7.4) | 6 | 10.9 ± 2.2 | 1.03 ± 0.08 |
| C1C2 (pH 8.0) | 6 | 8.3 ± 3.5 | 0.65 ± 0.10a |
| C1C2P | 7 | 13.3 ± 2.8 | 0.49 ± 0.16a |
| C1m | 4 | 8.7 ± 2.4 | 0.92 ± 0.14 |
| C0C1 | 7 | 40.4 ± 16.8a | 0.45 ± 0.10a |
| mC2 | 5 | 10.5 ± 4.7 | 0.59 ± 0.13a |
| C3mC4 | 5 | 11.2 ± 3.3 | 0.50 ± 0.09a |
Significant difference relative to C1C2 (p < 0.01).
To further map actin binding site(s) to specific domains of cMyBP-C, we first divided the 4 N-terminal domains of cMyBP-C into two non-overlapping recombinant fragments, C0C1 (inclusive of the Pro-Ala region) and mC2, and quantified binding of each subfragment to actin. As shown in Fig. 2, C0C1 binding to actin was significantly reduced compared with C0C2, with Kd = 40.4 ± 16.8 μm and Bmax = 0.45 ± 0.10. Although binding appeared saturable, the data could also be fit well to a straight line, suggesting that binding of C0C1 was weak and nonspecific or electrostatic in nature. By contrast, the binding affinity of mC2 (Fig. 2) was saturable with an affinity (Kd = 10.5 ± 4.7 μm) similar to that of C0C2, although the total amount of mC2 bound was reduced as shown by a decrease in Bmax (0.59 ± 0.13). To determine whether the m-domain was responsible for binding of mC2, we also investigated binding of the m-domain in combination with C1, i.e. C1m. As shown in Fig. 2, the Kd for C1m binding to actin was 8.7 ± 2.4 μm, not significantly different from that of C0C2. Bmax (0.92 ± 0.14) was also near 1.0 indicating a 1:1 binding stoichiometry comparable with C0C2. Thus, the m-domain in combination with C1 completely recapitulated the actin binding properties of the larger C0C2 fragment.
FIGURE 2.

Summary cosedimentation binding data for C1m, mC2, and C0C1. C1m (circles), Kd = 8.7 ± 2.4 μm, Bmax = 0.92 ± 0.14 (n = 4); mC2 (squares), Kd = 10.5 ± 4.7 μm, Bmax = 0.59 ± 0.13 (n = 5); C0C1 (triangles), Kd = 40.4 ± 16.8 μm, Bmax = 0.45 ± 0.10 (n = 7). Data are means ± S.D.
Because C1m and mC2 both bound to actin with similar affinities, we next investigated whether the m-domain alone was sufficient to confer actin binding properties to a segment of cMyBP-C that does not bind actin. As shown in Fig. 3, the recombinant protein, C3C4, consisting of domains C3 and C4 of cMyBP-C, binds F-actin nonspecifically as determined by its linear actin binding relation. However, when the m-domain was inserted between domains C3 and C4 to create the C3mC4 protein, saturable binding to actin was observed. The Kd for binding was 11.2 ± 3.3 μm, not significantly different from C0C2 (Table 1). These data thus demonstrate that the regulatory m-domain of cMyBP-C binds specifically to actin and that binding is of sufficient affinity to contribute to the binding properties observed for C0C2. However, the total amount of C3mC4 bound to actin was significantly reduced compared with C0C2 (Bmax = 0.50 ± 0.09 versus 0.92 ± 0.11, p < 0.001).
FIGURE 3.

The MyBP-C m-domain binds specifically to actin. Cosedimentation binding data (means ± S.D.) for C3C4 (circles) comprised of domains C3 and C4 of cMyBP-C and for C3mC4 (squares) an engineered protein in which the m-domain sequence was inserted between domains 3 and 4 of cMyBP-C. C3C4 bound nonspecifically to actin as shown by the linear binding relation, whereas C3mC4 bound to actin in a saturable manner with Kd = 11.2 ± 3.3 μm, Bmax = 0.50 ± 0.09 (n = 5).
Multiple Actin Binding Sites of C0C2 Cross-link F-actin—A straight-forward explanation for the 1:1 molar binding stoichiometry observed for C0C2, C1C2, and C1m is that each molecule binds to a single binding site on an actin monomer. However, a different model is needed to account for the reduced binding ratios (∼1:2 actin) observed for C3mC4 or mC2. One possibility is that instead of binding to a single site on actin the C1 and m-domains bind to independent sites such that the total amount of protein bound to F-actin (1:1 for C1m, C1C2, and C0C2) reflects contributions from each of two independent sites (∼0.5 mol/mol actin for each site). To test this idea, we assessed the ability of various domains to cross-link actin filaments because cross-linking depends on the presence of multiple interaction sites with each site interacting with a different filament to form bundles or meshes (29). The extent of cross-linking can be quantified by measuring solution turbidity as assessed by light absorbance at 350 nm (28, 30). As shown in Fig. 4, combining C0C2 with F-actin under conditions identical to those used for cosedimentation assays significantly increased solution turbidity relative to C0C2 alone, consistent with the idea that addition of C0C2 to F-actin promotes formation of actin cross-links or bundles. C1C2 and C1m also increased turbidity, whereas C0C1, mC2, C3C4, and C3mC4 did not. Because proteins that did not increase turbidity all lacked one or both the C1 and m-domains, the results suggest that both C1 and the m-domains contain actin binding sites.
FIGURE 4.

Bundling of F-actin by C1 and the m-domain. F-actin was combined with recombinant cMyBP-C proteins under conditions identical to those used for cosedimentation binding assays (see “Experimental Procedures”) and solution turbidity was measured as light absorbance at λ = 350 nm. Proteins containing both C1 and the m-domain (i.e. C0C2, C1C2, and C1m) increased solution turbidity in a concentration-dependent manner, whereas C0C1, mC2, C3C4, and C3mC4 did not increase solution turbidity. Data are means ± S.D. A.U., arbitrary units.
To confirm that the observed increases in solution turbidity corresponded to actin bundling, electron microscopy was used to visualize F-actin filaments in the presence and absence of recombinant proteins. As shown in Fig. 5, F-actin in the absence of recombinant cMyBP-C proteins (Fig. 5A) appeared as single filaments deposited randomly on the grid surface. Punctate staining was also visible and was likely due to aggregation of actin monomers. By contrast, when combined with C0C2 or C1C2, F-actin formed thick tightly packed bundles (Fig. 5, B and C). Thick, regularly packed bundles were also readily apparent in the presence of C1m (Fig. 5D), although the extent of bundling as determined by the number and thickness of the bundles was somewhat reduced. The extent and quality of bundling was further diminished in the presence of C0C1 (Fig. 5E) or mC2 (Fig. 5F) because few bundles were observed and those that were seen were less regular and less tightly packed than those formed in the presence of C0C2, C1C2, or C1m. In particular, a regular side by side arrangement of filaments was not observed in the presence of C0C1. Instead, filaments appeared only loosely connected (Fig. 5H). This infrequent, loose association was most likely the result of nonspecific accumulation of filaments around protein aggregates rather than from true actin cross-linking. The bundles formed by mC2 (Fig. 5I) appeared somewhat better organized than those formed in the presence of C0C1, but their thickness and frequency were still significantly reduced compared with C0C2, C1C2, or C1m. Thus, tight, regularly packed bundles indicative of actin cross-linking were most commonly observed in the presence of C0C2, C1C2, and C1m, a result in good agreement with solution turbidity measurements. Although it is unlikely that the C1 and m-domain promote actin bundling in vivo because of reduced protein stoichiometry of cMyBP-C to actin in sarcomeres, the presence of regulatory proteins, and other constraints on sarcomere geometry, the results provide evidence for multiple independent actin binding sites on the C1 and m-domains of cMyBP-C.
FIGURE 5.

Electron micrographs of F-actin in the presence of recombinant cMyBP-C proteins. 5 μm F-actin was combined with 30 μm recombinant cMyBP-C proteins (1:6 ratio) and processed as described under “Experimental Proceduress” for visualization by electron microscopy. Scale bars represent 200 nm in A-F and 50 nm in G-I. A, F-actin alone; B, F-actin plus C0C2; C, F-actin plus C1C2; D, F-actin plus C1m; E, F-actin plus C0C1; F, F-actin plus mC2. Thick bundles of F-actin are apparent in the presence of C0C2 (B), C1C2 (C), and C1m (D), but are either not present in the absence of added proteins (A, F-actin alone) or reduced in the presence of C0C1 (E) or mC2 (F). Panels G-I show higher magnification images of F-actin bundles in the presence of C1m (G), C0C1 (H), and mC2 (I) to illustrate tighter more regular packing of F-actin in the presence of C1m relative to either C0C1 or mC2.
Phosphorylation of the MyBP-C M-domain Reduces Binding to F-actin—To investigate whether actin binding by cMyBP-C is affected by phosphorylation of the m-domain, we phosphorylated C1C2 using the catalytic subunit of PKA and assessed the ability of phosphorylated C1C2 (i.e. C1C2P) to bind actin or to cross-link filaments. Pro Q Diamond staining confirmed C1C2 phosphorylation following PKA treatment (Fig. 6A, inset). Compared with C1C2, C1C2P bound to actin with similar affinity (Kd 13.3 ± 2.8 μm). However, overall binding was significantly reduced as indicated by a decrease in Bmax (0.49 ± 0.16). Phosphorylation of C1C2 also abolished its ability to cross-link actin filaments because the low turbidity of F-actin solutions was unchanged upon addition of C1C2P (Fig. 6B). The latter reduction in the ability to induce actin cross-linking suggests that binding through at least one of the sites is eliminated by phosphorylation of the m-domain.
FIGURE 6.

Effects of phosphorylation of the m-domain and alkaline pH on C1C2 binding to actin. A, cosedimentation binding data (means ± S.D.) for phosphorylated C1C2 (i.e. C1C2P, filled circles). Pro Q Diamond staining (inset) confirmed phosphorylation of C1C2P following treatment with PKA. C1C2P bound to F-actin with Kd = 13.3 ± 2.8 μm, Bmax = 0.49 ± 0.16 (n = 7). Binding data for C1C2 (open circles) were redrawn from Fig. 1 for comparison. B, PKA treatment of C1C2 abolished the ability of C1C2P to increase the turbidity of F-actin solutions. C, cosedimentation binding data for C1C2 collected at pH 8.0 (closed circles), Kd = 8.3 ± 3.5 μm, Bmax = 0.65 ± 0.10 (n = 6). Binding data for C1C2 at pH 7.4 (open circles), were redrawn from Fig. 1 for comparison. D, turbidity of solutions containing F-actin and C1C2 was reduced at pH 8.0. A.U., arbitrary units.
Because the m-domain is highly basic (isoelectric point > 9.0) and phosphorylation is expected to reduce its positive charge, we also investigated whether decreasing the net charge of the m-domain by increasing pH could affect binding of C1C2 to actin. As shown in Fig. 6C, increasing solution pH to 8.0 had effects on binding similar to phosphorylation, i.e. total binding of C1C2 was reduced (Bmax 0.65 ± 0.10) and actin cross-linking was abolished (Fig. 6D). Together these results suggest that the actin binding properties of the N terminus of cMyBP-C can be modulated by electrostatic charge or by phosphorylation of the m-domain.
S2Δ Does Not Compete with cMyBP-C for Actin Binding—We next investigated whether myosin S2 competes with C1C2 to prevent its binding to actin because the regulatory m-domain is known to bind the first 126 N-terminal residues of myosin S2, referred to as S2Δ (11). Fig. 7A shows results of cosedimentation experiments performed with C1C2 in the presence of 30 μm S2Δ. The binding relationship between C1C2 and F-actin was not significantly affected by S2Δ because binding parameters obtained in the presence of S2Δ (Kd = 15.4 ± 3.0 μm and Bmax = 0.97 ± 0.10) were not significantly different for C1C2 in the absence of S2Δ (compare with Fig. 1, Kd = 10.9 ± 2.2 μm and Bmax = 1.03 ± 0.08). In separate experiments (Fig. 7B), increasing the concentration of S2Δ up to 50 μm while maintaining the concentration of C1C2 constant at 10 μm, also did not affect the amount of C1C2 or S2Δ found in each pellet. These results demonstrate that binding of the m-domain to F-actin is not diminished by competition of the m-domain with S2Δ.
FIGURE 7.

Cosedimentation binding of C1C2 to F-actin in the presence of S2Δ. A, cosedimentation binding experiments with 5 μm F-actin performed in the presence of 30 μm S2Δ. The presence of S2Δ did not significantly affect binding of C1C2 to F-actin (Kd = 15.4 ± 3.0 μm and Bmax = 0.97 ± 0.10, n = 4). B, cosedimentation binding assays performed with 5 μm F-actin and 10 μm C1C2 in the presence of 1-50 μm S2Δ. The amount of C1C2 (circles) and S2Δ (squares) found in the pellets did not change in the presence of increasing amounts of S2Δ. Data are means ± S.D.
The M-Domain Binds to Thin Filaments Independent of Ca2+—Results from this study thus far demonstrate that the 4 N-terminal domains of cMyBP-C bind to F-actin through interactions involving multiple domains. To determine whether these interactions persist in regulated thin filaments where the binding of cMyBP-C to actin may be affected by the presence of regulatory proteins (16), we performed cosedimentation binding assays using purified native thin filaments and C1C2. As shown in Fig. 8, binding of C1C2 to native thin filaments was saturable in the absence of Ca2+ (Kd = 12.5 ± 4.8 μm and Bmax = 0.90 ± 0.13) and binding was unchanged in the presence of Ca2+ (Kd = 8.4 ± 4.6 μm and Bmax = 0.90 ± 0.13). The Kd and Bmax values obtained for C1C2 binding to thin filaments in the presence or absence of Ca2+ were not significantly different from those obtained for binding to F-actin alone (Table 1). Together these data suggest that cMyBP-C binds with similar affinity to either F-actin or regulated thin filaments and that binding is not affected by Ca2+ activation of thin filaments.
FIGURE 8.

Cosedimentation binding of C1C2 to native thin filaments. NTF were prepared as described under “Experimental Procedures” from bovine heart and 5 μm purified NTF were used in cosedimentation binding assays. An SDS-PAGE of purified NTF is shown in the inset. Positions of actin, tropomyosin (Tm), troponin I (TnI), and troponin T (TnT) are indicated. Troponin C is not visible on the gel due to weak staining by Coomassie Blue. C1C2 bound to NTF in a saturable manner in the presence (open circles) or absence (closed circle) of Ca2+ (means ± S.D.). Kd and Bmax values were 8.4 ± 4.6 and 0.90 ± 0.13 μm (n = 6) for C1C2 in the presence of Ca2+ and 12.5 ± 4.8 and 0.90 ± 0.13 μm (n = 5) in the absence of Ca2+. Solutions lacking Ca2+ contained (in mmol/liter): 180 KCl, 20 imidazole, pH 7.4, 1 EGTA, 1 MgCl2, and 1 DTT. Solutions with Ca2+ contained additional CaCl2 to achieve 1 mm free Ca2+.
DISCUSSION
The major finding from this study is that the unique regulatory motif or m-domain of MyBP-C binds to F-actin and to native thin filaments in a specific, saturable manner with Kd = ∼10 μm. The interaction occurs at physiological ionic strength and is reduced by phosphorylation or by increasing pH, indicating that binding is influenced by electrostatic charge interactions. These data demonstrate that the N terminus of cMyBP-C can interact in a reversible manner with actin and thus raise the possibility that interactions of cMyBP-C with the thin filament contribute to intropic acceleration of cross-bridge cycling following phosphorylation of cMyBP-C.
The results show that the m-domain of cMyBP-C alone is sufficient to bind actin because addition of the m-domain to two Ig domains (C3 and C4) that do not bind actin conferred saturable binding with an affinity similar to that of C1C2 (Fig. 3). Until now the only other binding partner identified for the m-domain was the S2 subfragment of myosin (11, 12). Binding of the m-domain to myosin S2 was inferred from binding of C1C2 (which contains the m-domain) to myosin minifilaments and to a smaller recombinant protein containing the first 126 N-terminal residues of myosin S2 (i.e. S2Δ) (11). The Kd for the interaction was ∼7 μm and binding was eliminated by phosphorylation of the m-domain (12). Interactions of the flanking domains, i.e. C1 and C2, with S2Δ were also demonstrated for each domain separately using NMR, but binding affinities were not determined in those studies and high protein concentrations (200-800 μm) were necessary to obtain NMR spectra (31, 32). In the present study the binding affinity of the m-domain (C3mC4, Fig. 3) for actin was similar to that reported for C1C2 binding to S2Δ. However, we found that S2Δ did not compete with actin for binding to C1C2 (Fig. 7, A and B), suggesting that the affinity of the m-domain for actin is greater than for S2. Because the present experiments were performed at higher ionic strength than those originally used to measure the affinity of C1C2 for S2 (11), the affinity of C1C2 for S2Δ may be reduced under high salt conditions. If so, then it is possible that the m-domain preferentially binds actin instead of S2 in vivo as well. Alternatively, binding sites for the two ligands may be separate and non-overlapping and binding of C1C2 to actin and/or S2 could occur independent of one another.
Additional actin binding site(s) outside of the m-domain were implied by the ability of C1C2 to cross-link actin because cross-linking depends on the presence of multiple binding sites. Because the minimal domains required for cross-linking were the C1 and m-domains, the results imply that a second binding site for actin exists within the C1 domain. C0 might also contribute to actin binding because the extent of cross-linking as assessed by solution turbidity was somewhat greater in the presence of C0C2 compared with either C1C2 or C1m (Fig. 4). However, the relative contribution of the C0 domain to actin binding is likely to be small because C1C2 (which lacks C0 and the proline-alanine-rich region preceding C1) bound to actin with an affinity comparable with C0C2 (Fig. 1). In addition, C0C1 bound poorly, if at all, to actin suggesting that C0 contributes little to actin binding under these conditions (Fig. 2). The finding that C0C1 bound only weakly was somewhat unexpected because cross-linking results as discussed above indicated a binding site within C1 that should still be able to cosediment with actin even if C0 does not. A possible explanation for the discrepancy is that C1 (and/or C0) binding to actin is strengthened by the presence of the m-domain. According to this scenario, C1 might bind more tightly to actin when stabilized at its C terminus by the m-domain as in C0C2 but not when the m-domain is absent as in C0C1. If so, then mutations linked to hypertrophic cardiomyopathy that are clustered near the C terminus of C1 (20, 32) could disrupt C1 interactions with the m-domain and reduce binding of C1 to actin.
Whereas the present results demonstrate that multiple binding sites for actin exist within the N terminus of cMyBP-C, the sites appear distinct and differentially affected by phosphorylation. This is because phosphorylation reduced, but did not abolish, binding of C1C2 to actin (Fig. 6), indicating that binding via at least one of the sites persists even following phosphorylation of the m-domain. Because cross-linking was eliminated either by phosphorylation of the m-domain or by increasing pH, the simplest explanation is that binding of the m-domain to actin is affected whereas binding via the C1 domain is not. This idea is depicted schematically in Fig. 9 in a model that accounts for actin cross-linking and the effects of pH and phosphorylation on actin binding. As shown in Fig. 9A, the N terminus of cMyBP-C binds to F-actin in the unphosphorylated state primarily through interactions that involve binding sites on the C1 and m-domains along with possible contributions from C0. However, upon phosphorylation of the m-domain or an increase in pH, reduced net positive charge of the m-domain reduces its affinity for actin and the m-domain becomes unbound (Fig. 9B). At the same time, binding of the C1 domain is largely unaffected by electrostatic interactions and it remains bound to actin, thereby accounting for continued C1C2-actin binding even following phosphorylation of the m-domain (Fig. 6). Note that binding of cMyBP-C to actin is depicted in Fig. 9 as less than the 1:1 stoichiometry measured here to reflect the occurrence of cMyBP-C in sarcomeres only at discreet locations along thick filament A-bands (33, 34).
FIGURE 9.

Model of cMyBP-C interactions with F-actin. cMyBP-C (green) is depicted as a series of 12 spherical domains representing domains C0 through C10 and the MyBP-C motif or m-domain (M) between domains C1 and C2. The Pro-Ala-rich region between the C0 and C1 domains is represented as a yellow rectangle. A, the N terminus of cMyBP-C interacts with the thin filament primarily through binding of the C1 domain and the m-domain as demonstrated by data in this study. The cardiac-specific C0 domain is also shown binding to actin based on data from this and other studies (16, 19). B, binding of the M-domain to the thin filament is reduced by phosphorylation of the m-domain or by increasing pH (reduced net positive charge of the m-domain).
Structural evidence in support of the m-domain in the unbound configuration (Fig. 9B) was recently obtained by Whitten et al. (16) who modeled the interaction of C0C2 with F-actin from neutron scattering data collected in solution at pH 8.0. In good agreement with binding data presented here, Whitten et al. (16) concluded that C0C2 bound to F-actin through interactions with C1 and C0, whereas the m-domain appeared unbound at the higher pH.
The model shown in Fig. 9 also complements recent functional observations that demonstrated that the C1 and m-domains constitute a functional subunit of cMyBP-C (21). Together the C1 and m-domains increased Ca2+ sensitivity of tension, rates of tension redevelopment at submaximal [Ca2+], and activated force at low levels of Ca2+ in permeabilized rat trabeculae. These functional observations suggested that the C1 and m-domains create a functional unit that can potently activate the thin filament. The present results extend these observations and suggest that activating effects of the C1 and m-domains are mediated by binding to actin.
Binding of the C1 and m-domains to actin could influence the activation state of the thin filament and/or cross-bridge cycling through several mechanisms. One possibility is that binding of C1 and the m-domain to actin directly activates the thin filament by affecting the position of tropomyosin as proposed by Whitten et al. (16), who predicted that the position of C0C2 on F-actin would clash sterically with tropomyosin in the Ca2+ off state. Binding of C1 and the m-domain could thus compete with and displace tropomyosin, thereby facilitating cross-bridge binding and cycling. A related prediction of this scenario is that binding of cMyBP-C to thin filaments may be increased in the presence of Ca2+ when the position of tropomyosin on the thin filament is shifted (35-37). However, we found that C1C2 bound to thin filaments regardless of the presence or absence of Ca2+ (Fig. 8), suggesting that steric clash with tropomyosin does not significantly limit C1C2 binding to actin. Consistent with these observations, Yamamoto (38) also found that binding of skeletal MyBP-C to thin filaments was robust regardless of whether or not Ca2+ was present, although somewhat more binding was apparent in the presence of Ca2+. Because skeletal thin filaments are more completely activated by Ca2+ alone, whereas cardiac thin filaments also require strongly bound cross-bridges (39, 40), differences between our results and those of Yamamoto (38) may reflect differences in the relative sensitivity of skeletal and cardiac thin filaments to activation by Ca2+.
Although displacement of tropomyosin by the C1 and m-domains could provide an explanation for the activating effects of the N terminus of cMyBP-C when added exogenously to motility assays or permeabilized myocytes (18, 21, 41), an apparent paradox is how phosphorylation of the m-domain, which decreases binding to actin as shown here (Fig. 6) can account for increased cross-bridge cycling in vivo (6). That is, how can movement of the m-domain to the unbound state (Fig. 9B) account for acceleration of cross-bridge cycling following phosphorylation by PKA (6)? Although experiments presented here do not directly address this question, several explanations are possible. The first is that binding of the m-domain to actin in the unphosphorylated state could block or alter myosin-S1 cross-bridge binding and thereby limit cross-bridge attachment to the thin filament. Phosphorylation of the m-domain and unbinding from actin could thus promote myosin binding and thereby accelerate cross-bridge cycling. Alternatively, unbinding of the m-domain from actin could free the m-domain for interactions with other ligands, such as myosin S1 or S2, myosin light chains (32, 42), or thin filament regulatory proteins. Such interactions could affect cross-bridge cycling kinetics by influencing the position or docking of myosin S1 heads onto the thin filament or by activating the thin filament in a manner analogous to the way in which TnI unbinding actin promotes thin filament activation (43, 44).
A significant result from this study is that binding of the C1 domain to actin persists even following phosphorylation and unbinding of the m-domain from actin. This result suggests that cMyBP-C can tether the thick and thin filaments together regardless of the phosphorylation state of the m-domain. If so, then cMyBP-C could structurally couple thick and thin filaments together. Such coupling could potentially influence cross-bridge cycling kinetics by creating a drag that slows sarcomere shortening as considered by Hofmann and colleagues (45) or by modulating sarcomere lattice spacing. In support of the latter idea, phosphorylation of myofilament proteins by PKA increased sarcomere lattice spacing in cMyBP-C knock-out mice, but not in wild-type mice (46), suggesting that cMyBP-C normally restricts lattice expansion.
In summary, we have shown that the C1 and m-domains of cMyBP-C bind to actin. The m-domain of cMyBP-C binds reversibly to actin in a phosphorylation-dependent manner, whereas C1 binding to actin is unaffected by phosphorylation. Reversible binding and unbinding of the m-domain to actin could thereby influence cross-bridge cycling in response to inotropic agonists, whereas phosphorylation independent binding of C1 to actin could maintain close coupling of thick and thin filaments regardless of phosphorylation state. Such coupling could ensure optimal lattice spacing as sarcomere shortening ensues and thereby help to maintain efficient cross-bridge interactions throughout the cardiac cycle.
Acknowledgments
We thank Dr. Michael Regnier and Galina Flint for the gift of myosin and An-yue Tu, Elaine Hoye, and David Dai for assistance with protein cloning and expression. We also thank Dr. Howard White for advice on purification of native thin filaments and Dr. Maria Razumova for critical reading of the manuscript.
This work was supported, in whole or in part, by National Institutes of Health Grants SC1HL096017 (to R. W. K.) and HL080367 (to S. P. H.). This work was also supported by a National Science Foundation Graduate Research Fellowship (to J. F. S.).
Footnotes
The abbreviations used are: cMyBP-C, cardiac myosin-binding protein C; NTF, native thin filaments; PKA, protein kinase A; DTT, dithiothreitol.
References
- 1.Richard, P., Charron, P., Carrier, L., Ledeuil, C., Cheav, T., Pichereau, C., Benaiche, A., Isnard, R., Dubourg, O., Burban, M., Gueffet, J. P., Millaire, A., Desnos, M., Schwartz, K., Hainque, B., and Komajda, M. (2003) Circulation 107 2227-2232 [DOI] [PubMed] [Google Scholar]
- 2.Dhandapany, P. S., Sadayappan, S., Xue, Y., Powell, G. T., Rani, D. S., Nallari, P., Rai, T. S., Khullar, M., Soares, P., Bahl, A., Tharkan, J. M., Vaideeswar, P., Rathinavel, A., Narasimhan, C., Ayapati, D. R., Ayub, Q., Mehdi, S. Q., Oppenheimer, S., Richards, M. B., Price, A. L., Patterson, N., Reich, D., Singh, L., Tyler-Smith, C., and Thangaraj, K. (2009) Nat. Genet. 41 187-191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Harris, S. P., Bartley, C. R., Hacker, T. A., McDonald, K. S., Douglas, P. S., Greaser, M. L., Powers, P. A., and Moss, R. L. (2002) Circ. Res. 90 594-601 [DOI] [PubMed] [Google Scholar]
- 4.Korte, F. S., McDonald, K. S., Harris, S. P., and Moss, R. L. (2003) Circ. Res. 93 752-758 [DOI] [PubMed] [Google Scholar]
- 5.Stelzer, J. E., Fitzsimons, D. P., and Moss, R. L. (2006) Biophys. J. 90 4119-4127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tong, C. W., Stelzer, J. E., Greaser, M. L., Powers, P. A., and Moss, R. L. (2008) Circ. Res. 103 974-982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nagayama, T., Takimoto, E., Sadayappan, S., Mudd, J. O., Seidman, J. G., Robbins, J., and Kass, D. A. (2007) Circulation 116 2399-2408 [DOI] [PubMed] [Google Scholar]
- 8.Sadayappan, S., Osinska, H., Klevitsky, R., Lorenz, J. N., Sargent, M., Molkentin, J. D., Seidman, C. E., Seidman, J. G., and Robbins, J. (2006) Proc. Natl. Acad. Sci. U. S. A. 103 16918-16923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Palmer, B. M., Georgakopoulos, D., Janssen, P. M., Wang, Y., Alpert, N. R., Belardi, D. F., Harris, S. P., Moss, R. L., Burgon, P. G., Seidman, C. E., Seidman, J. G., Maughan, D. W., and Kass, D. A. (2004) Circ. Res. 94 1249-1255 [DOI] [PubMed] [Google Scholar]
- 10.Stelzer, J. E., Patel, J. R., Walker, J. W., and Moss, R. L. (2007) Circ. Res. 101 503-511 [DOI] [PubMed] [Google Scholar]
- 11.Gruen, M., and Gautel, M. (1999) J. Mol. Biol. 286 933-949 [DOI] [PubMed] [Google Scholar]
- 12.Gruen, M., Prinz, H., and Gautel, M. (1999) FEBS Lett. 453 254-259 [DOI] [PubMed] [Google Scholar]
- 13.Calaghan, S. C., Trinick, J., Knight, P. J., and White, E. (2000) J. Physiol. 528 151-156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Saber, W., Begin, K. J., Warshaw, D. M., and VanBuren, P. (2008) J. Mol. Cell Cardiol. 44 1053-1061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shaffer, J. F., Razumova, M. V., Tu, A. Y., Regnier, M., and Harris, S. P. (2007) FEBS Lett. 581 1501-1504 [DOI] [PubMed] [Google Scholar]
- 16.Whitten, A. E., Jeffries, C. M., Harris, S. P., and Trewhella, J. (2008) Proc. Natl. Acad. Sci. U. S. A. 105 18360-18365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Squire, J. M., Luther, P. K., and Knupp, C. (2003) J. Mol. Biol. 331 713-724 [DOI] [PubMed] [Google Scholar]
- 18.Razumova, M. V., Shaffer, J. F., Tu, A. Y., Flint, G. V., Regnier, M., and Harris, S. P. (2006) J. Biol. Chem. 281 35846-35854 [DOI] [PubMed] [Google Scholar]
- 19.Kulikovskaya, I., McClellan, G., Flavigny, J., Carrier, L., and Winegrad, S. (2003) J. Gen. Physiol. 122 761-774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jeffries, C. M., Whitten, A. E., Harris, S. P., and Trewhella, J. (2008) J. Mol. Biol. 377 1186-1199 [DOI] [PubMed] [Google Scholar]
- 21.Razumova, M. V., Bezold, K. L., Tu, A. Y., Regnier, M., and Harris, S. P. (2008) J. Gen. Physiol. 132 575-585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gasteiger, E., Gattiker, A., Hoogland, C., Ivanyi, I., Appel, R. D., and Bairoch, A. (2003) Nucleic Acids Res. 31 3784-3788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Margossian, S. S., and Lowey, S. (1982) Methods Enzymol. 85 55-71 [DOI] [PubMed] [Google Scholar]
- 24.Pardee, J. D., and Spudich, J. A. (1982) Methods Enzymol. 85 164-181 [DOI] [PubMed] [Google Scholar]
- 25.Spiess, M., Steinmetz, M. O., Mandinova, A., Wolpensinger, B., Aebi, U., and Atar, D. (1999) J. Struct. Biol. 126 98-104 [DOI] [PubMed] [Google Scholar]
- 26.Fritz, J. D., Swartz, D. R., and Greaser, M. L. (1989) Anal. Biochem. 180 205-210 [DOI] [PubMed] [Google Scholar]
- 27.Wilkinson, K. D. (2004) Methods Mol. Biol. 261 15-32 [DOI] [PubMed] [Google Scholar]
- 28.Moos, C., Mason, C. M., Besterman, J. M., Feng, I. M., and Dubin, J. H. (1978) J. Mol. Biol. 124 571-586 [DOI] [PubMed] [Google Scholar]
- 29.Korn, E. D. (1982) Physiol. Rev. 62 672-737 [DOI] [PubMed] [Google Scholar]
- 30.Ishikawa, M., Murofushi, H., and Sakai, H. (1983) J. Biochem. (Tokyo) 94 1209-1217 [DOI] [PubMed] [Google Scholar]
- 31.Ababou, A., Gautel, M., and Pfuhl, M. (2007) J. Biol. Chem. 282 9204-9215 [DOI] [PubMed] [Google Scholar]
- 32.Ababou, A., Rostkova, E., Mistry, S., Le Masurier, C., Gautel, M., and Pfuhl, M. (2008) J. Mol. Biol. 384 615-630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Luther, P. K., Bennett, P. M., Knupp, C., Craig, R., Padron, R., Harris, S. P., Patel, J., and Moss, R. L. (2008) J. Mol. Biol. 384 60-72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bennett, P., Craig, R., Starr, R., and Offer, G. (1986) J. Muscle Res. Cell Motil. 7 550-567 [DOI] [PubMed] [Google Scholar]
- 35.McKillop, D. F., and Geeves, M. A. (1993) Biophys. J. 65 693-701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lehman, W., Craig, R., and Vibert, P. (1994) Nature 368 65-67 [DOI] [PubMed] [Google Scholar]
- 37.Pirani, A., Vinogradova, M. V., Curmi, P. M., King, W. A., Fletterick, R. J., Craig, R., Tobacman, L. S., Xu, C., Hatch, V., and Lehman, W. (2006) J. Mol. Biol. 357 707-717 [DOI] [PubMed] [Google Scholar]
- 38.Yamamoto, K. (1986) FEBS Lett. 208 123-127 [DOI] [PubMed] [Google Scholar]
- 39.Gordon, A. M., Homsher, E., and Regnier, M. (2000) Physiol. Rev. 80 853-924 [DOI] [PubMed] [Google Scholar]
- 40.Moss, R. L., Razumova, M., and Fitzsimons, D. P. (2004) Circ. Res. 94 1290-1300 [DOI] [PubMed] [Google Scholar]
- 41.Kunst, G., Kress, K. R., Gruen, M., Uttenweiler, D., Gautel, M., and Fink, R. H. (2000) Circ. Res. 86 51-58 [DOI] [PubMed] [Google Scholar]
- 42.Margossian, S. S. (1985) J. Biol. Chem. 260 13747-13754 [PubMed] [Google Scholar]
- 43.Galinska-Rakoczy, A., Engel, P., Xu, C., Jung, H., Craig, R., Tobacman, L. S., and Lehman, W. (2008) J. Mol. Biol. 379 929-935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Brown, L. J., Sale, K. L., Hills, R., Rouviere, C., Song, L., Zhang, X., and Fajer, P. G. (2002) Proc. Natl. Acad. Sci. U. S. A. 99 12765-12770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hofmann, P. A., Greaser, M. L., and Moss, R. L. (1991) J. Physiol. 439 701-715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Colson, B. A., Bekyarova, T., Locher, M. R., Fitzsimons, D. P., Irving, T. C., and Moss, R. L. (2008) Circ. Res. 103 244-251 [DOI] [PMC free article] [PubMed] [Google Scholar]