

Abstract
A growing number of orphan G-protein-coupled receptors (GPCRs) have been reported to be activated by lipid ligands, such as lysophosphatidic acid, sphingosine 1-phosphate (S1P), and cannabinoids, for which there are already well established receptors. These new ligand claims are controversial due to either lack of independent confirmations or conflicting reports. We used the β-arrestin PathHunter™ assay system, a newly developed, generic GPCR assay format that measures β-arrestin binding to GPCRs, to evaluate lipid receptor and ligand pairing. This assay eliminates interference from endogenous receptors on the parental cells because it measures a signal that is specifically generated by the tagged receptor and is immediately downstream of receptor activation. We screened a large number of newly “deorphaned” receptors (GPR23, GPR92, GPR55, G2A, GPR18, GPR3, GPR6, GPR12, and GPR63) and control receptors against a collection of ∼400 lipid molecules to try to identify the receptor ligand in an unbiased fashion. GPR92 was confirmed to be a lysophosphatidic acid receptor with weaker responses to farnesyl pyrophosphate and geranylgeranyl diphosphate. The putative cannabinoid receptor GPR55 responded strongly to AM251, rimonabant, and lysophosphatidylinositol but only very weakly to endocannabinoids. G2A receptor was confirmed to be an oxidized free fatty acid receptor. In addition, we discovered that 3,3′-diindolylmethane, a dietary molecule from cruciferous vegetables, which has known anti-cancer properties, to be a CB2 receptor partial agonist, with binding affinity around 1 μm. The anti-inflammatory effect of 3,3′-diindolylmethane in RAW264.7 cells was shown to be partially mediated by CB2.
Over the past few years, a large number of orphan GPCRs3 have been shown to respond to lipid ligands for which there are already known receptors. For example, LPA was found to activate five more new orphan receptors as follows: GPR23 (1, 2), GPR92 (3, 4), GPR87 (5), P2Y5 (6), and P2Y10 (7), which are not very homologous to the three previously known high affinity LPA receptors in the endothelial differentiation gene (EDG) family. Five well established high affinity S1P receptors (S1P1-5 receptors) are also part of the EDG receptor family. Now five new receptors, GPR3, GPR6, GPR12, GPR63, and P2Y10, have been claimed to be novel S1P receptors (7-10). Similarly, GPR55 was discovered to be a new cannabinoid receptor adding to the two classical cannabinoid receptors CB1 and CB2 (11-14). Most of these claims are tentative at this point due to either lack of follow up studies or contradictory reports. For example, a recent study showed that farnesyl pyrophosphate (FPP) might be a more potent endogenous ligand than LPA for GPR92 (15). And GPR55 was also reported to be a lysophosphatidylinositol receptor (16).
Interestingly, lipid molecules that previously were not recognized as GPCR signaling mediators have also been shown to trigger signal transduction pathways via GPCRs. For example, G2A receptor was reported to be activated by oxidized free fatty acid 9S-hydroxyoctadecadienoic acid (9-HODE) (17), which was previously thought to activate nuclear hormone receptor peroxisome proliferator-activated receptor γ (18). This finding opens exciting new possibilities, but a lack of follow up reports leaves the original report unconfirmed. G2A was initially reported to be a lysophosphatidylcholine (LPC) receptor, but the report was later retracted (19). Whether it is a proton receptor like its sequence-related family members OGR1, TDAG8, and GPR4 has also been disputed (20, 21). G2A is highly expressed in lymphocytes and macrophages and has been postulated to be involved in cell proliferation control (22) or chemotaxis (23, 24), but some of these conclusions were reached using the retracted ligand LPC. It is essential to establish the true ligand of G2A to fully understand its function.
The pairing of GPCRs and ligands is highly error-prone because of the fact that GPCRs usually cannot be expressed and assayed as purified proteins (with the notable exception of rhodopsin) but instead require alternative complex cell-based assay systems (25). Even though most published papers do contain seemingly satisfactory internal controls, different reports often do not agree with each other, and paper retraction is not uncommon. The most widely used cell-based assays measure G protein-dependent secondary messenger formation, such as [cAMP] changes, Ca2+ flux, or reporter gene activation. In these systems, intact cells that express a milieu of endogenous receptors in addition to the overexpressed receptor of interest are used. The readout of GPCR activation is not receptor-specific (meaning it is a sum of all GPCRs in the assay tube), and may be several or many signaling steps downstream of receptor activation. Promiscuous G proteins such as Gα16 or G-protein chimeras such as Gqi5 are often used to artificially reroute the GPCR signaling to the Ca2+ pathway, which can also introduce artifacts. The signal difference between the receptor-overexpressed system and the wild type cell system is interpreted as the receptor-specific response. Because most host cell lines do express some combination of EDG receptors that respond to S1P and LPA, getting a silent parental cell response for such lipid molecules in traditional GPCR assays is very difficult. Thus, it is extremely important that multiple assay formats are used to validate claims of receptor-ligand pairing, especially for lipid ligands such as LPA and S1P.
Analogous to the orphan receptors, there are also some “orphan” compounds, compounds that have known in vivo effects but lack a defined molecular target. DIM is one such example. It is a dietary indole derived from digestion of indole-3-carbinol, found in Brassica vegetables such as broccoli and cauliflower. DIM is in clinical trials as a treatment for numerous forms of cancer because of its safety at high doses and its promising anti-tumor effect in vitro and in vivo (26, 27). It is also being investigated as a potential treatment for a variety of viral and antibiotic-resistant bacterial infections, because of its immunomodulatory effects (28, 29). The reported cellular effects of DIM are numerous (30), including cell cycle regulation, apoptosis induction, nuclear receptor-mediated gene transcriptional changes, induction of various drug-metabolizing cytochrome P450 enzymes, estrogen metabolism changes resulting in an increase in anticarcinogenic 2-hydroxylation of estrogen, inhibition of mitochondrial H+-ATP synthase resulting in induction of p21Cip1/Waf1 (31), stimulation of interferon-γ production and signaling (32), etc. The molecular target of the molecule is not well established, and it is unclear whether there might be many low affinity effectors or a single high affinity effector mediating the plethora of effects of DIM. DIM has been reported to directly bind (IC50 = 50 μm in one report (33) and Ki = 90 nm in another report (34)) and both agonize and antagonize aryl hydrocarbon receptor (35) and in turn modulate cytochrome P450 1A1 and estrogen metabolism and tumorigenesis (36). DIM also directly binds and antagonizes the androgen receptor with an IC50 in micromolar range (37). No other molecular target has been reported to our knowledge.
We recently completed an evaluation of a new generic GPCR assay system developed by DiscoveRx that measures β-arrestin translocation to the activated GPCR using β-galactosidase enzyme fragment complementation technology (38). This technology offers a tagged receptor assay whose readout, β-arrestin binding measured by reconstituted β-galactosidase activity, is immediately downstream of receptor activation. Thus, it is clearly advantageous over prior cell-based GPCR assays in proving specificity of ligand-dependent receptor activation, as it eliminates many of the issues mentioned above, such as endogenous receptor signal contamination and indirect readout. Furthermore, it is a generic GPCR assay that works for receptors that couple to all classes of G proteins, as it examines the desensitization and internalization pathway, not G protein-dependent signaling pathway. Compared with some other β-arrestin technologies that measure translocation by imaging, the readout is luminescence signal strength, which makes this an easy quantitative assay that does not require cell imaging, and amenable to high throughput screening.
To study orphan and novel receptor function, we first focused on a panel of recently deorphaned lipid GPCRs. We prepared a panel of 43 lipid molecules, including the various reported lipid ligands and a BioMol lipid collection of 345 compounds, and we tested them against a panel of 16 putative lipid GPCRs in the β-arrestin assay system in a transient transfection format. Our results shed light on the true ligands of these controversial receptors and also uncovered a novel receptor target for dietary molecule DIM.
EXPERIMENTAL PROCEDURES
Reagents—Lipids were purchased from Avanti Polar Lipids, Sigma, Cayman Chemicals, Biomol, or Tocris Bioscience. BioMol compound libraries included bioactive lipids (201 compounds), endocannabinoids (60 compounds), and orphan ligands (84 compounds) libraries. Rimonabant (also known as SR141716) and SR141528 were synthesized at Novartis. Flash detection reagent for β-arrestin assay and coelenterazine for aequorin assay were purchased from DiscoveRx. FuGENE 6 and EDTA-free complete protease inhibitor mixture were from Roche Applied Science; [3H]CP55940 and [35S]GTPγS were from PerkinElmer Life Sciences. β-Arrestin assay technology was licensed from DiscoveRx. Aequorin technology was licensed from Euroscreen SA. CHO-hCB1 stable cell line was purchased from Euroscreen. CHO-hCB2 stable cell line was generated inhouse. hGPR55-HEK293 stable cell line was generated by Novartis GPCR-EP and kindly provided to us. cAMP HighRange HTRF kits were purchased from CisBio-US Inc.
β-Arrestin Assay—GPCRs of interest were cloned into the Prolink vector (DiscoveRx) for GPCR-prolink fusion protein production. Parental HEK293 cells that stably express β-arrestin2-β-gal-EA fusion protein (HEK293-BAEA) were detached and transiently transfected with the receptor of interest using FuGENE 6 transfection reagent in suspension mode. Transfected cells in assay medium (phenol red-free Dulbecco's modified Eagle's medium with 3% FBS) were plated into white solid 384-well plates at 15,000 cells/25 μl/well. After overnight incubation, 200 nl of test molecules were transferred into the cell plates by PinTool (GNF Systems) followed by 1-2 h of incubation at 37 °C, 5% CO2. Flash detection reagents were added at 12.5 μl/well. After 5 min to 1 h of incubation at room temperature, the cell plates were read on CLIPR (PerkinElmer Life Sciences) or Acquest (Molecular Devices) for luminescence signal.
Aequorin Assay—Parental CHO cells that stably express aequorin were transiently transfected with the receptor (in pcDNA3.1 vector) using the same protocol as with the β-arrestin assay. The transfected cells were seeded into 384-well black clear bottom plates (Greiner Bio-one) at 10,000 cells/25 μl/well in F-12 medium containing 3% FBS and incubated overnight. Coelenterazine (20 μm final) was added into the cell plates at 25 μl/well. The cell plates were returned to the incubator for 3 h. The compounds were diluted 1:20 into an intermediate plate with Tyrode buffer (130 mm NaCl, 2 mm CaCl2, 5 mm NaHCO3, 5 mm KCl, 1 mm MgCl2, 20 mm HEPES, pH 7.4). 12.5 μl/well of pre-diluted compounds were transferred into the cell assay plate and read on LumiLux (PerkinElmer Life Sciences) for flash luminescence.
An algorithm similar to area under curve was created inhouse to analyze the aequorin kinetic data, which was named Slope Threshold (Slope 100). The algorithm defines the beginning and the end of the luminescence intensity peak by comparing the difference in intensity of each time point with an earlier time point, and by determining if the difference exceeds a defined threshold, which was usually set at 100. If it does, then the intensity of the later time point is added into the Slope Threshold sum.
Radioligand Binding Assay—Membrane preparation using hCB1-CHO and hCB2-CHO cell lines was described previously (39). Saturation radioligand binding experiments were performed on each batch of the membrane preparation to determine the Kd (dissociation constant of the radioligand to the receptor) and the Bmax values (maximal receptor binding sites). The Kd value obtained from the saturation binding experiments that was used for the Ki calculation was 0.5 nm for human CB1 and human CB2. Competitive binding experiments were done with 0.5-0.8 nm of [3H]CP55940. Each reaction was carried out in round-bottom 96-well polystyrene assay plates, including test compound, membranes (3-10 μg/well), and [3H]CP55940 in assay buffer (50 mm Tris-HCl, 2.5 mm EDTA, 5 mm MgCl2, 0.05% bovine serum albumin, pH 7.4). The reaction was carried out at room temperature for 120 min before the membranes were harvested onto a Unifilter GF/B-96 filter plate using a Packard Filtermate Harvester. After nine washes with wash buffer (50 mm Tris-HCl, 2.5 mm EDTA, 5 mm MgCl2, 0.05% bovine serum albumin, pH 7.4), the filter was dried in a 37 °C oven for 30 min. MicroScint-20 was added and the plate sealed for scintillation counting on a TopCount.
GTPγS Binding Assay—The assay was carried out in 96-well filtration format as described (39).
cAMP Assay—CHO-CB1 or CHO-CB2 cells were detached and diluted in cell culture medium (F-12 with 10% FBS and 1% penicillin/streptomycin/glutamine) to a density of 40,000 cells/ml. 10,000 cells (25 μl) were seeded per well into the 384-well assay plate and incubated in 37 °C incubator overnight. 500 nl of compound was then transferred into each assay well, followed by 5 μl of forskolin addition for a final concentration of 60 μm. Cells were returned to a 37 °C incubator for 30 min. Finally, 15 μl each of HiRange d2-cAMP and anti-cAMP cryptate were dispensed per well. After >1 h room temperature incubation, data acquisition was done in the time-resolved fluorescence-resonance energy transfer (FRET) mode on Envision or ViewLux. The ratio between the acceptor fluorescence signal (A665 nm) and donor fluorescence signal (A620 nm) × 104, representing the FRET between the conjugated cAMP and the anti-cAMP antibody, was calculated and plotted on y axis. The higher the signal, the lower endogenous cAMP concentration is in the sample.
GPR55 Reporter Gene Assay—pGL3 basic vector (Promega) was modified to have three repeats of multiple response element consensus sequence, one cAMP-response element that is from a vasoactive intestinal peptide gene promoter, and serum-response element promoter before the luciferase reporter gene. HEK293-hGPR55 cells were transiently transfected with pGL3-CRE-MRE-SRE-luc reporter gene and plated at 15,000 cells/25 μl/well. After overnight incubation, test compounds were added to the cells with Pintool and incubated for 16-24 h. The reaction was then stopped by Bright-Glo addition (12 μl/well), and luminescence signal was read on CLIPR with 5 s of exposure.
IL-1β Taqman Reverse Transcription-PCR in RAW264.7 Cells—RAW264.7 cells were seeded into 6 well plates at 0.5 × 106 cells/well and incubated at 37 °C for 24 h. The cells were serum-starved in assay medium (Dulbecco's modified Eagle's medium + 1% FBS + penicillin/streptomycin) for 24 h, before receiving 18 h of compound treatment, followed by 12 h of LPS stimulation (100 ng/ml; Invivogen catalog number tlrl-eklps). Total RNA extraction, purification, and reverse transcription reaction were carried out using Qiagen RNeasy mini kit and ABI high capacity cDNA reverse transcription kit (catalog number 4368814). 200 ng/reaction of cDNA products from the reverse transcription reaction was used for Taqman PCR with TaqMan Universal PCR Master Mix (ABI catalog number 4304437). The primer and probe mix of mouse IL-1β (catalog number Mm01336189_m1) and mouse and tumor necrosis factor-α (Mm00443258_m1) were purchased from ABI. Mouse 36B4 primer 5′-AGA TGC AGC AGA TCC GCA-3′ (forward) and 5′-GTT CTT GCC CAT CAG CAC C-3′ (reverse) and probe 5′VIC-CGC TCC GAG GGA AGG CCG-TAMRA3′ were used as control.
Data Analysis—EC50 or IC50 values were obtained by fitting the data with the sigmoidal dose-response curve-fitting tool of the GraphPad Prism software. Eight or twelve different concentrations were usually used and two or three data points per concentration. In radioligand binding assays, Ki was calculated using Equation 1 of Cheng and Prusoff,
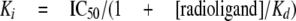 |
(Eq.1) |
RESULTS
In the evaluation of the β-arrestin PathHunter™ system, we confirmed the reported agonists and antagonists of a large number of known receptors, including Gs-, Gi-, and Gq-coupled receptors (Table 1). Furthermore, we were encouraged by the fact that transient transfection of receptors into β-arrestin-EA parental HEK293 cells gave robust signals, circumventing the need for labor-intensive stable cell line generation and selection. The EC50 values obtained in the transient β-arrestin assay for a subset of receptors were significantly higher than the EC50 values obtained from other assay formats (Table 1). This appears to be partly due to the low sensitivity resultant from transient transfection method. For example, β2-adrenergic receptor showed 4.6 nm EC50 in stable cell pool but 17-40 nm EC50 in transient assays. It is also possible that β-arrestin binding may require a higher degree of receptor activation than G-protein-dependent signaling or that the β-galactosidase detection system has lower sensitivity despite great amplification. Regarding antagonist or inverse agonist, no general trend of rightward shift of potency was observed, even in transient assay format (Table 1).
TABLE 1.
Activities of known receptor agonists and antagonists in β-arrestin assay versus other assay formats
The indicated agonist and antagonist compounds were tested on the corresponding receptor in various GPCR assay formats in dose-response curves. EC50, IC50, Ki, or Kb values, or the value ranges obtained in a number of experiments, were determined and tabulated to compare assay sensitivity.
| GPCR name | Agonist name | Transient β-arrestin assay, EC50 | Other assay format, EC50 (stable cell line unless noted) | Antagonist or inverse agonist name | Transient β-arrestin assay, IC50 | Other assay format, IC50 (stable cell line) |
|---|---|---|---|---|---|---|
| β2-Adrenergic receptor | Isoproterenol | 17-40 nm, 4.6 nm in stable pool | 1-9 nm in cAMP-HTRF assay | Propranolol | Kb = 0.79 nm | Kb = 1.5 nm |
| Orexin receptor 2 | Orexin A | 8.7-14.5 nm | 1.2-15.5 nm in FLIPR assay, 11.9 nm in IP1-HTRF assay, 14-31 nm in pERK Surefire assay | Compound 29 (Ref. 61) | 157 nm with 10 nm orexin A | 289 nm with 100 nm orexin A in FLIPR assay |
| CB2 | CP55940 | 1.6 nm | 0.28 nm in GTPγS assay, Ki = 0.22 nm in binding assay, 0.38 nm in cAMP-HTRF assay | SR144528 | 2.4 nm with no agonist | 0.8 nm with no agonist in GTPγS, Ki = 0.6 nm in binding |
| S1P1 | S1P | 26-43 nm | 0.9-2.6 nm in GTPγS assay | JTE-013 | 8.9 μm with 10 nm S1P | 1.9 μm with 10 nm S1P in GTPγS assay |
| H4 | Histamine | 46 nm | 55-77 nm in transient aequorin assay with Gα16 | |||
| GPR154 | Neuropeptide S | 10 nm | 19 nm in transient aequorin assay | |||
| LPA1 (EDG2) | LPA | 165 nm | 5.5 nm in FLIPR assay | Ki16425 | 120 nm with 10 μm LPA | 45-136 nm with 500 nm LPA in FLIPR assay |
A number of lipid molecules (see Table 2 for full names of all compounds) were purchased, dissolved into appropriate solvents, and arrayed into compound plates in serial dilutions. Additionally, the bioactive lipid library, endocannabinoid library, and orphan ligand library from BioMol were purchased and arrayed at a single concentration. The 16 selected human GPCRs (Table 2) were cloned into the Prolink expression vector, and β-arrestin assays were performed in transient transfection format to test the activity of compounds on each receptor.
TABLE 2.
List of receptors and lipid molecules used for screening
Each of the 16 human G-protein-coupled receptors was assayed against the 43 lipid molecules on the right. The lipid molecules were dissolved and stored according to the manufacturer's recommendations.
| Receptor | Lipid | Full name of lipid |
|---|---|---|
| CB2 | (±)11-HETE | 11-Hydroxy-5,8,12,14-eicosatetraenoic acid |
| G2A | (±)12-HETE | 12-Hydroxy-5Z,8Z,10E,14Z-eicosatetraenoic acid |
| GPR12 | (±)5-HETE | 5-Hydroxy-6E,8Z,11Z,14Z-eicosatetraenoic acid |
| GPR18 | (±)8-HETE | 8-Hydroxy-5Z,9E,11Z,14Z-eicosatetraenoic acid |
| GPR23 | (±)9-HETE | 9-Hydroxy-5,7,11,14-eicosatetraenoic acid |
| GPR3 | (±)9-HODE | 9-10Z,12E-Hydroxyoctadecadienoic acid |
| GPR34 | 12(S)-HETE | 12-Hydroxy-5Z,8Z,10E,14Z-eicosatetraenoic acid |
| GPR40 | 2-AG | 2-Arachidonoylglycerol |
| GPR43 | 5-Oxo-ETE | 5-Oxo-6,8,11,14-eicosatetraenoic acid |
| GPR55 | 9(S)-HODE | (9S)-10Z,12E-Hydroxyoctadecadienoic acid |
| GPR6 | 9(S)-HpODE | 9(S)-Hydroperoxy-10Z,12E-octadecadienoic acid |
| GPR63 | Anandamide | N-Arachidonylethanolamine |
| GPR84 | Arachidonic acid (20:4) | Arachidonic acid |
| GPR92 | C10 (10:0) | Decanoic acid |
| LPA2 | C11 (11:0) | Undecanoic acid |
| S1P1 | C12 | Sodium dodecanoate |
| C16 (16:0) | Palmitic acid | |
| C16 (16:3) | γ-Lindenic acid | |
| C2 (NaAc) | Sodium acetate | |
| C3 (3:0) | Propanoic acid | |
| C4 (4:0) | Butanoid acid | |
| C5 (5:0) | Valeric acid | |
| C6 (6:0) | Hexanoic acid | |
| DHA (22:6) | Docosahexaenoic acid | |
| DoPA (2 × 18:1) | 1,2-Dioleoyl-sn-glycero-3-phosphate | |
| Lanostadien | Lanostadien | |
| Lauric acid (12:0) | Lauric acid or dodecanoic acid | |
| Linoleic acid (18:2) | Linoleic acid | |
| LPA (16:0) | Lysophosphatidic acid | |
| LPA (18:1) | Lysophosphatidic acid | |
| LPC (16:0) | Lysophosphatidylcholine | |
| LPC (18:0) | Lysophosphatidylcholine | |
| LPC (chicken egg) (16:0, 18:0) | Lysophosphatidylcholine | |
| LPC (liver) | Lysophosphatidylcholine | |
| LPG (18:1) | Lysophosphatidylglycerol | |
| Lyso-PAF | Lyso-platelet-activating factor, alkyl lysophosphatidylcholine | |
| Lyso-PE (18:1) | Lysophosphatidylethanolamine | |
| Lyso-PS (18:1) | Lysophosphatidylserine | |
| NaGly | N-Arachidonylglycine | |
| PAF | Platelet-activating factor | |
| PC (18:1-16:1) | Phosphatidylcholine | |
| S1P | Sphingosine 1-phosphate | |
| SPC | Sphingosine phosphorylcholine |
GPR92 Is an LPA Receptor but Not GPR23—LPA2 (EDG4), GPR23, and GPR92 were included in the lipid molecule screen, with the well established LPA2 receptor serving as a positive control. LPA2 and GPR92 responded to LPA as expected (Fig. 1, A and B). LPA2 was activated more potently by LPA (18:1) with an EC50 of 93 nm, and less potently by LPA (16:0) with an EC50 of >10 μm. GPR92 was activated by LPA (18:1) and LPA (16:0) with EC50 of 339 nm and 5.8 μm, respectively. In contrast, GPR23 showed essentially no response to either form of LPA up to ∼100 μm (Fig. 1C). The small amount of signal was clearly insignificant as nonspecific responses to platelet-activating factor (PAF), LPC, and lyso-PAF reached much higher levels in the assay.
FIGURE 1.

Activation of putative and established LPA receptors by LPA and other lipid molecules. Indicated human receptors (A, LPA2; B, GPR92; C, GPR23) were transiently transfected into the HEK293-BAEA parental cell line. 16-24 h later, test compounds (cpd) were added to the cells. 1 h later, the reaction was stopped by the addition of flash detection reagent, and β-galactosidase activity was measured. Relative luminescence unit (RLU) is plotted on the y axis, and the data are expressed as mean ± S.E.
We noticed that a small number of lipid molecules in the collection, namely LPC, PAF, lysophosphatidylethanolamine, lysophosphatidylglycerol, and lyso-PAF triggered significant signals at close to 100 μm on many receptors, even though they did not trigger responses from all receptors or the parental cells (data not shown). We do not know whether this represents some weak affinity of many receptors toward these molecules.
FPP and N-arachidonylglycine (NaGly) were recently reported to be endogenous ligands for GPR92 (15). In the report, LPA consistently showed weaker activity compared with FPP in several assays, including reporter gene assay, inositol phosphate production assay, and cAMP assay (15). We tested FPP, geranylgeranyl diphosphate, NaGly, and LPA using our transient β-arrestin and aequorin assay systems (Fig. 2). Our results showed that LPA is the most potent activator of the receptor, whereas FPP and geranylgeranyl diphosphate showed weaker but significant activities. NaGly showed only a minimal amount of activity, consistent with the published result (15).
FIGURE 2.

Response of human GPR92 to selected lipid molecules in the β-arrestin assay (A) and aequorin assay (B). HEK293-BAEA (A) or CHO-aequorin parental cells (B) were transiently transfected with GPR92. The next day, they were assayed for their responses to the indicated lipid molecules. GGPP, geranylgeranyl diphosphate.
3,3′-Diindolylmethane Is a CB2 Partial Agonist—CB2 receptor was included in the screen as a positive control for the cannabinoid receptor family. As expected, the two endocannabinoids in the collection, anandamide and 2-arachidonylglycerol (2-AG), showed up as hits for CB2. Arachidonic acid, a precursor molecule for 2-AG, also activated the receptor weakly, with the EC50 in the 10-100 μm range (data not shown). Interestingly, we identified DIM as a confirmed agonist hit for CB2 from the Biomol bioactive lipids collection.
To further confirm that DIM is indeed a CB2 agonist and to test its activity on the related CB1 receptor, we performed the β-arrestin assay, GTPγS binding assay, radioligand binding assay, and also cAMP assay on human CB1 and CB2 receptors (Fig. 3 and Fig. 4). The EC50/IC50/Ki values are summarized in Table 3. DIM agonist activity was confirmed in all four assays performed on CB2, with EC50 values ranging from 0.42 to 1.7 μm in different assays. The binding Ki is 1.1 ± 0.3 μm, as determined from four independent radioligand binding experiments. Similar to anandamide, DIM appears to be a partial agonist when compared with small molecule full agonist CP55940 in functional assays (Fig. 3, A, C and D). DIM also binds the CB1 receptor with a Ki of 4.3 ± 0.3 μm. However, this binding did not result in detectable functional activity in the cAMP assay (Fig. 4D). Significant but weak CB1 inverse agonist activity was detected in the GTPγS assay, with IC50 = 11.1 μm, and in the β-arrestin assay, with IC50 > 10 μm (Fig. 4, A and C). In addition, we tested DIM on putative cannabinoid receptor GPR55 and did not detect any activity (data not shown). Thus, we confirmed using multiple assay formats that DIM is a CB2 receptor partial agonist with ∼1 μm binding affinity and that it may also have some weak CB1 inverse agonist activity.
FIGURE 3.

DIM is a CB2 partial agonist. Response of the human CB2 receptor to the test molecules in the β-arrestin assay (A), radioligand competition binding assay (B), GTPγS binding assay (C), and cAMP assay (D). ○, CP55940; □, DIM; ▴, 2-AG; ▾, anandamide; and ♦, arachidonic acid. A, HEK293-BAEA parental cells were transiently transfected with CB2 and then assayed the next day. B and C, membrane preparations from a CB2-CHO stable cell line were used. D, CB2-CHO stable cell line was seeded and 16 h later stimulated with test compounds along with 60 μm of for skolin. After 30 min of stimulation, cells were lysed, and cAMP level was measured using cAMP-HTRF HighRange kit. The y axis plots the FRET ratio which is inversely proportional to the cellular cAMP concentration.
FIGURE 4.

DIM is a weak CB1 inverse agonist. Response of the human CB1 receptor to the test molecules in the β-arrestin assay (A), radioligand competition binding assay (B), GTPγS binding assay (C), and cAMP assay (D). ○, CP55940; □, DIM; ▴, 2-AG; ▾, anandamide; and ♦, arachidonic acid. A, HEK293-BAEA parental cells were transiently transfected with CB1 and then assayed the next day. B and C, membrane preparations from a CB1-CHO stable cell line were used. D, CB1-CHO stable cell line was seeded and 16 h later stimulated with test compounds along with 60 μm of forskolin. After 30 min of stimulation, cells were lysed, and cAMP level was measured using cAMP-HTRF HighRange kit. The y axis plots the FRET ratio that is inversely proportional to the cellular cAMP concentration.
TABLE 3.
The activity of DIM on CB1 and CB2 receptors in comparison with CP55940, anandamide, 2-AG, and arachidonic acid
The data are expressed as average ± S.D. of three or four experiments. Percentage of efficacy is calculated by normalizing the response range to that of a standard compound (CP55940 or 2-AG); the value shown is the average value of all the experiments.
| Receptor | Assay type | CP55940, EC/IC50 or Ki | DIM, EC/IC50 or Ki | Anandamide, EC/IC50 or Ki | 2-AG, EC/IC50 or Ki | Arachidonic acid, EC/IC50 or Ki |
|---|---|---|---|---|---|---|
| μm | μm | μm | μm | μm | ||
| CB2 | β-Arrestin assay (% of efficacy) | 0.0016 ± 0.0004 (100%) | 1.7 ± 1.3 (47%) | 0.74 ± 0.27 (51%) | 4.4 ± 3.2 (82%) | >10 |
| Ligand binding assay | 0.00022 ± 0.00008 | 1.1 ± 0.3 | 0.34 ± 0.13 | 0.59 ± 0.26 | 13 ± 3 | |
| GTPγS assay (% of efficacy) | 0.00028 ± 0.00005 (100%) | 0.98 ± 0.22 (37%) | 0.62 ± 0.32 (49%) | 0.32 ± 0.17 (95%) | 10 ± 4 (43%) | |
| cAMP-HTRF assay (% of efficacy) | 0.00038 ± 0.00008 (100%) | 0.42 ± 0.23 (43%) | ? (<20%) | 5.3 ± 2.3 (57%) | >10 | |
| CB1 | β-Arrestin assay (% of efficacy) | 0.00045 (100%) | >30, inhibition | 0.81 (67%) | 0.85 (>100%) | >10 |
| Ligand binding assay | 0.00024 ± 0.00008 | 4.3 ± 0.3 | 0.09 ± 0.01 | 0.25 ± 0.05 | 12 ± 5 | |
| GTPγS assay (% of efficacy) | ? | 11.1, inhibition | 0.44 (56%) | 0.32 (100%) | 4.2 (40%) | |
| cAMP-HTRF assay (% of efficacy) | 0.0005 | >30 | 3.5 (125%) | 3.4 (69%) | >10 |
CB2 Partially Mediates the Anti-inflammatory Effect of DIM in Murine Macrophages—DIM was reported to suppress inflammatory response to lipopolysaccharide (LPS) in murine monocyte/macrophage RAW264.7 cells (29). As CB2 is expressed in the cell line (data not shown), we wondered whether CB2 might be the target molecule that mediates the anti-inflammatory effect of DIM. In the absence of LPS, DIM caused a small decrease in IL-1β mRNA level in RAW264.7 cells (Fig. 5). When RAW264.7 cells were stimulated with 100 ng/ml of LPS, there was a dramatic increase in IL-1β and tumor necrosis factor-α mRNA levels, and DIM inhibited this increase (Fig. 5 and data not shown). The inhibition was of small magnitude but reproducible over many experiments. This DIM-dependent decrease in IL-1β mRNA level was significantly, but only partially, inhibited by a CB2-specific antagonist SR144528 (Fig. 5). Furthermore, CB1/CB2 dual agonist CP55940 also caused a significant but lower level of decrease in IL-1β mRNA level compared with DIM (Fig. 5). Thus, CB2 appears to be partially responsible for anti-inflammatory effect of DIM in this cellular system.
FIGURE 5.

CB2 partially mediates the anti-inflammatory effect of DIM in RAW264.7 cells. RAW264.7 cells were serum-starved for 24 h, treated with test compound for 18 h, and followed by 12 h of LPS (100 ng/ml) stimulation when indicated. Total RNA was isolated and Taqman PCR performed for IL-1β and control gene 36B4 in each sample. IL-1β Ct values, after subtracting 36B4 Ct values, were base-line adjusted and graphed. Ct values are inversely proportional to the mRNA levels (Ct change of 1 equals 2-fold mRNA level change). CP55940 and SR144528 (SR) are CB1/CB2 dual agonist and CB2-specific antagonist, respectively. Each bar represents the mean ± S.E., n = 3. *, p < 0.05.
AM251, Rimonabant, and LPI Activate GPR55—In contrast to CB2, the GPR55 screen did not reveal the two endocannabinoid ligands, 2-AG and anandamide, as positive hits. However, AM251, a known CB1 antagonist/inverse agonist contained in the Biomol collection, was a confirmed agonist hit. We then tested a large number of cannabinoid receptor-related synthetic compounds or natural ligands on GPR55 in both the β-arrestin assay (Fig. 6A) and a reporter gene assay using pGL3-CRE-MRE-SRE-luc reporter gene (Fig. 6B). Rimonabant, also known as SR141716, a CB1 inverse agonist whose structure closely resembles AM251, also triggered significant agonist activity in both assays. Lysophosphatidylinositol (LPI), another reported ligand of GPR55 (16), was tested in the β-arrestin assay and was indeed active (Fig. 6A). It did not significantly active CB1 or CB2 (data not shown). Anandamide and Δ9-tetrahydrocannabinol (Δ9-THC) afforded a very small amount of activity (12% efficacy) in the β-arrestin assay (Fig. 6A), and 2-AG showed some activity in the reporter gene assay (Fig. 6B). Although the activities of anandamide and Δ9-tetrahydrocannabinol were very weak, we believe that they are real as the responses were enhanced to a more significant level when a low amount of AM251 (5 μm) was included in the assay (data not shown). As controls, the same compounds all triggered expected agonist or antagonist responses from the CB1 and CB2 receptors (Figs. 3 and 4 and data not shown). Noticeably, many compounds that were reported to have potent activity on GPR55, such as CP55940, HU210, abnormal cannabidiol (Abn-CBD, Fig. 6), and O-1602 (12, 13), did not show any detectable activity in our assays.
FIGURE 6.

Response of human GPR55 to test molecules in the β-arrestin assay (A) and reporter gene assay (B). A, HEK293-BAEA parental cells were transiently transfected with GPR55. After 16-24 h, the cells were assayed for their responses to the indicated lipid molecules. Two experiments were shown with different sets of compounds tested. B, GPR55-HEK293 stable cell line was transiently transfected with pGL3-SRE-MRE-CRE-luc. Test compounds were added 24 h later. Luciferase activity was assayed 16-24 h after compound stimulation using Bright-Glo and read on CLIPR for luminescence signal. Δ9-THC, Δ9-tetrahydrocannabinol; Abn-CBD, abnormal cannabidiol.
G2A Responds to Oxidized Free Fatty Acid 9-HODE and 11-HETE but Not LPC—Consistent with the Obinata et al. study (17), our screening revealed oxidized free fatty acids such as 9-HODE as agonist hits (Fig. 7A). G2A was also weakly activated by linoleic acid (nonoxidized precursor to 9-HODE), but with a higher EC50 value and a lower activation maximum. We found LPC, 13(S)-HODE, and lauric acid to be completely inactive on G2A (Fig. 7A). Because the EC50 values of these compounds appeared to be high in the β-arrestin assay, making accurate EC50 determinations difficult, we also tested these compounds in the aequorin assay. The aequorin assay is a more sensitive assay for G2A, giving lower EC50 values for all compounds tested. When we compared a variety of oxidized free fatty acids in the aequorin assay, we found that (±)9-HODE, 9(S)-HODE, 9-hydroperoxyoctadecadienoic acid (9-HpODE), and 11-HETE were the most potent agonists, with EC50 values ranging from 247 to 978 nm (Fig. 7B). Importantly, they show specific activity on G2A that is lacking in the parental cell line (data not shown). 8-HETE and 12-HETE showed very low or no activity. LPC and linoleic acid triggered massive responses from the parental CHO cell line in the aequorin assay (data not shown), making assessment of the G2A activity of these molecules in the aequorin assay impossible.
FIGURE 7.

Response of human G2A to lipid molecules in the β-arrestin assay (A) and aequorin assay (B). HEK293-BAEA or CHO-aequorin parental cells were transiently transfected with G2A. 16-24 h later, they were assayed for their responses to the indicated lipid molecules. 9-HpODE, 9-hydroperoxyoctadecadienoic acid.
GPR3, GPR6, GPR12, GPR63, and GPR18 Remain Orphans—We did not detect any hit for the putative S1P receptors GPR3, GPR6, GPR12, and GPR63. Although control S1P1 receptor responded to S1P very well with an EC50 of 34 nm, the above four receptors showed no activity whatsoever at up to 8 μm of S1P and 42 μm of sphingosine phosphorylcholine (data not shown). The assay also revealed anandamide and 2-AG as weak agonists on S1P1 receptor in the micromolar range (data not shown). This cross-reactivity of endocannabinoids on S1P1 is not surprising given the high receptor sequence homology. FTY720, a potent S1P1,3,4,5 receptor agonist, has been shown to have weak activity on the CB1 receptor (40).
We screened three reported free fatty acid (FFA) receptors GPR43 (41), GPR40 (42), and GPR84 (43) against the lipid collection. GPR43 responded to short chain FFA as expected. C2 and C3 carboxylic acids most potently activated the receptor and C4 fatty acid moderately activated the receptor, whereas C6 was almost inactive (data not shown). Mid-chain FFAs caused a decrease in signal at high concentrations (∼100 μm), likely due to nonspecific effects on cells at high concentrations, which may explain why we did not observe any positive response of C10-C12 on GPR40 and GPR84 (data not shown).
GPR18 and GPR34 were also included in the lipid screen; however, no positive hits were identified. The reported ligand for GPR18, NaGly (44), was not part of the original lipid collection and was later tested on GPR18 and found to be inactive (data not shown). We could not find any commercial source for the reported ligand for GPR34, lysophosphatidylserine 16:0 (45). Lysophosphatidylserine (18:1) did not trigger any signal increase at concentrations as high as 100 μm (data not shown).
DISCUSSION
We used the β-arrestin PathHunter™ assay as the main assay system to evaluate some of the recent deorphanization reports on lipid receptor and ligand pairs. This assay system is a relatively new technology that was not used in any of the previous reports on these orphan receptors. The distinct features of the system are as follows: 1) it is a universal, G protein-independent assay; and 2) it generates a receptor-specific signal proximal to receptor activation. Because of these two features, it is perfectly suited to tackle orphan receptors with little G protein signaling information. In Table 4, we summarize our findings, either in support of or arguing against published receptor and ligand pairs.
TABLE 4.
Summary of lipid receptor-ligand test results
| Receptor | Reported ligands | Confirmed | Refs. |
|---|---|---|---|
| CB1 | Various cannabinoids | CP55940, anandamide, 2-AG | |
| CB2 | Various cannabinoids | CP55940, anandamide, 2-AG, DIM | |
| GPR55 | Various cannabinoids, AM251, rimonabant, LPI, CP55940, HU210, Abn-CBD, O-1602 | AM251, rimonabant, LPI, endocannabinoids very weak activity | 11-14 |
| S1P1 | S1P | Yes | |
| LPA2 | LPA | Yes | |
| GPR92 | LPA, FPP | LPA, FPP, geranylgeranyl diphosphate | 3, 4, 15 |
| G2A | 9-HODE, 11-HETE | Yes | 17 |
| GPR43 | Short chain FFA | Yes | 41 |
| GPR3 | S1P | No | 8 |
| GPR6 | S1P | No | 8, 9 |
| GPR12 | S1P, sphingosine phosphorylcholine | No | 8, 62 |
| GPR18 | NaGly | No | 44 |
| GPR23 | LPA | No | 1, 2 |
| GPR63 | S1P | No | 10 |
| GPR34 | Lysophosphatidylserine (16:0) | Unable to test | 45 |
| GPR40 | Mid and long chain FFA | Unable to test | 42 |
| GPR84 | Mid chain FFA | Unable to test | 43 |
Although a positive result in the β-arrestin system, combined with previous data from different assay formats, leaves little doubt on the authenticity of the receptor-ligand pair, a negative result in the β-arrestin system does not definitely prove that the receptor is not activated by the ligand. There are several reasons why an experiment could theoretically fail to pick up a true ligand. They include the following. 1) The receptor is not expressed or trafficked properly to the cell surface in our transient HEK293 transfection system; we did not observe this problem for any well established receptors so far, but it remains a theoretical possibility. 2) The receptor expression is down-regulated due to the presence of ligand in the serum-containing growth medium; we routinely used charcoal-filtered serum as an alternative serum source to check for this possibility. 3) The receptor happens to belong to rare cases where the activated receptor does not bind to β-arrestin2 and get internalized. 4) The ligand tested was not soluble or appropriately prepared to activate the receptor. Because a 5-30-fold right shift of the agonist titration curve has been observed (Table 1), it is possible that we may not have observed a weak ligand activation because of limitation on solubility of certain lipid ligands.
A very interesting observation from the screen was the identification of DIM, an anti-cancer compound, as a novel CB2 receptor agonist. This molecule is known to directly modulate intracellular aryl hydrocarbon receptor (33-35) and androgen receptor (37), but to our knowledge, no literature has linked it with a cell-surface GPCR before. Our results suggest that the activation of CB2 by DIM is likely the molecular mechanism behind some of the reported anti-inflammatory effects of DIM. CB2 receptor is highly expressed in the immune cells in the periphery, such as macrophages and T cells. Interestingly, a number of human leukemia and lymphoma cell lines were also reported to express CB2 (46). These cell lines, which include Jurkat, Molt-4, and Sup-T1, are susceptible to apoptosis induced by a variety of cannabinoids. In fact, cannabinoids have been shown to inhibit growth in several tumor xenograft models, to curb growth or induce apoptosis in a number of transformed cell lines, and inhibit tumor angiogenesis and metastasis (47, 48). Thus, whether CB2 could also mediate anti-tumor effect of DIM warrants further investigation. CB2 has been implicated in a large number of physiological functions. Emerging areas of investigation on CB2 include pain (49), neuroinflammation (50), hepatic fibrosis (51), gastrointestinal motility and inflammation (52), atherosclerosis (53), immune function (54), demyelinating disease (55), ischemia (56), bone metabolism (57), and reproduction (58). Our study not only raises the possibility that CB2 could be mediating some of the beneficial effects of DIM in vivo but also suggests that perhaps DIM, a marketed nutritional supplement, might be useful for treating many more diseases than what is currently appreciated.
Among all the reported novel LPA and S1P receptors, we could only confirm GPR92 to be a true LPA receptor. This is an interesting example where divergent receptors bind the same ligand; GPR92 has a low sequence homology with the classical LPA1-3 receptors (21-22%). The affinity of GPR92 for LPA has not been directly compared with classical LPA receptors before. Kotarsky et al. (3) reported a 6.4 nm binding affinity and 9.3 μm EC50 of LPA on GPR92 in the cell-based reporter gene assay. Our study directly compared GPR92 and EDG4 (LPA2) in the same assay format and found GPR92 to be almost as potent. GPR92 is highly expressed in gastrointestinal CD8+ cytotoxic lymphocytes (3), small and medium diameter neurons in dorsal root ganglion, and also embryonic stem cells (4). In recombinant systems, it influences cellular cytoskeletal arrangement, such as stress fiber formation and neurite retraction (4). Its exact function in vivo awaits further studies.
There have been several papers and patents reporting that GPR55 is the third cannabinoid receptor (11-14). Although all reports agreed that GPR55 is an unusual G13-coupled receptor, the exact activity and EC50 values of various compounds differ in different reports, perhaps reflecting the differences in assay formats and stable cell lines, as well as the lack of a good standard assay for G13-coupled receptors. Consistent with our finding, a patent from GlaxoSmithKline reported AM251 to be a hit in a yeast-based GPR55 screen (WO01/86305). It is interesting to note that AM251 and rimonabant are both inverse agonist compounds that have no agonist activity on CB1; however, they are purely agonist compounds on GPR55. In the study of Ryberg et al. (12), a large number of CB1- and CB2-related synthetic or naturally occurring molecules were shown to be potent low nanomolar activators of GPR55 in the GTPγS assay. AM251 was also active with an EC50 of 39 nm in their study. However, many of these molecules were found to be inactive in a separate study using intracellular Ca2+ as the readout (14). In our β-arrestin assay system and reporter gene assay system, AM251, rimonabant, and LPI are strong agonists, whereas endocannabinoids showed only very weak activity. CP55940, HU210, O-1602, and abnormal cannabidiol showed no activity, possibly reflecting differences between intact cell-based assays and membrane-based GTPγS assay. The function of GPR55 is under active investigation. A recent report (59) suggested that it plays a role in mechanical hyperalgesia associated with inflammatory and neuropathic pain.
G2A, OGR1, TDAG8, and GPR4 are close sequence homologues constituting a receptor family. Whereas the latter three receptors are now established as pH-sensing receptors, the natural ligand for G2A remains uncertain. Our result is consistent with the Obinata et al. study (17) in showing that G2A has no activity whatsoever toward LPC, and that oxidized FFAs such as 9-HODE are its true ligands. This finding expands the known naturally occurring signaling lipids to include oxidized FFAs. Plasma FFAs and low density lipoprotein are continuously exposed to oxidative stress to generate hydroxyl species. Cell-associated phospholipids can also be subjected to oxidative pressure by cellular stress. Interestingly, G2A receptor expression was previously shown to be up-regulated by stress-inducing and cell-damaging agents (60). Thus, G2A might serve as a stress sensor in the immune system to trigger appropriate responses in a stressful environment. 9-HODE was previously reported to directly bind peroxisome proliferator-activated receptor γ with a Kd of 10-20 μm (18). The EC50 of 9-HODE on G2A was in hundreds of nanomolar range in the aequorin assay and low micromolar range in the β-arrestin assay. Thus, it is possible that some of the effects of oxidized low density lipoprotein or oxidized FFAs that were previously thought to be mediated via nuclear hormone receptors are actually mediated by GPCR G2A.
In conclusion, we used a novel GPCR assay system to test the authenticity of lipid receptor-ligand pairs that generated a number of interesting findings, including support for and against various published ligands. This information may serve as a critical first step in elucidating the functions of these orphan receptors, as well as helping to understand the mechanism of action of emerging medicinal compounds such as DIM.
Acknowledgments
We are grateful to Xiang-Ju Gu for providing the BioMol libraries; Novartis GPCR-EP for providing GPR55-HEK293 stable cell line; Cecilia Jiang for providing the pGL3-CRE-MRE-SRE-luc construct; Shin-Shay Tian and Jing Li for providing certain lipids and GPR40/43 cDNA clones; GNF gene core and Novartis GPCR-EP for providing GPCR cDNA clones; and GNF Advanced Automation Technology group for technical assistance. We specifically thank Tim R. Smith for assistance with the aequorin assays, and Mitch Hull for writing software for the aequorin data analysis.
Footnotes
The abbreviations used are: GPCR, G-protein-coupled receptor; 2-AG, 2-arachidonoylglycerol; CHO, Chinese hamster ovary; DIM, 3,3′-diindolylmethane; EDG, endothelial differentiation gene; FFA, free fatty acids; FPP, farnesyl pyrophosphate; FRET, fluorescence-resonance energy transfer; GTPγS, guanosine 5′-O-3 thiotriphosphate; HEK293, human embryonic kidney 293; HEK293-BAEA, HEK293 cells stably express β-arrestin2-EA fusion protein; 9-HODE, 9-hydroxyoctadecadienoic acid; 11-HETE, 11-hydroxy-5,8,12,14-eicosatetraenoic acid; HTRF, homogeneous time-resolved fluorescence; LPA, lysophosphatidic acid; LPC, lysophosphatidylcholine; LPI, lysophosphatidylinositol; LPS, lipopolysaccharide; NaGly, N-arachidonylglycine; PAF, platelet-activating factor; S1P, sphingosine 1-phosphate; FBS, fetal bovine serum; IL, interleukin; FLIPR, fluorometric imaging plate reader.
References
- 1.Noguchi, K., Ishii, S., and Shimizu, T. (2003) J. Biol. Chem. 278 25600-25606 [DOI] [PubMed] [Google Scholar]
- 2.Lee, C. W., Rivera, R., Dubin, A. E., and Chun, J. (2007) J. Biol. Chem. 282 4310-4317 [DOI] [PubMed] [Google Scholar]
- 3.Kotarsky, K., Boketoft, A., Bristulf, J., Nilsson, N. E., Norberg, A., Hansson, S., Owman, C., Sillard, R., Leeb-Lundberg, L. M., and Olde, B. (2006) J. Pharmacol. Exp. Ther. 318 619-628 [DOI] [PubMed] [Google Scholar]
- 4.Lee, C. W., Rivera, R., Gardell, S., Dubin, A. E., and Chun, J. (2006) J. Biol. Chem. 281 23589-23597 [DOI] [PubMed] [Google Scholar]
- 5.Tabata, K., Baba, K., Shiraishi, A., Ito, M., and Fujita, N. (2007) Biochem. Biophys. Res. Commun. 363 861-866 [DOI] [PubMed] [Google Scholar]
- 6.Pasternack, S. M., von Kügelgen, I., Aboud, K. A., Lee, Y. A., Rüschendorf, F., Voss, K., Hillmer, A. M., Molderings, G. J., Franz, T., Ramirez, A., Nürnberg, P., Nöthen, M. M., and Betz, R. C. (2008) Nat. Genet. 40 329-334 [DOI] [PubMed] [Google Scholar]
- 7.Murakami, M., Shiraishi, A., Tabata, K., and Fujita, N. (2008) Biochem. Biophys. Res. Commun. 371 707-712 [DOI] [PubMed] [Google Scholar]
- 8.Uhlenbrock, K., Gassenhuber, H., and Kostenis, E. (2002) Cell. Signal. 14 941-953 [DOI] [PubMed] [Google Scholar]
- 9.Ignatov, A., Lintzel, J., Kreienkamp, H. J., and Schaller, H. C. (2003) Biochem. Biophys. Res. Commun. 311 329-336 [DOI] [PubMed] [Google Scholar]
- 10.Niedernberg, A., Tunaru, S., Blaukat, A., Ardati, A., and Kostenis, E. (2003) Cell. Signal. 15 435-446 [DOI] [PubMed] [Google Scholar]
- 11.Baker, D., Pryce, G., Davies, W. L., and Hiley, C. R. (2006) Trends Pharmacol. Sci. 27 1-4 [DOI] [PubMed] [Google Scholar]
- 12.Ryberg, E., Larsson, N., Sjögren, S., Hjorth, S., Hermansson, N. O., Leonova, J., Elebring, T., Nilsson, K., Drmota, T., and Greasley, P. J. (2007) Br. J. Pharmacol. 152 1092-1101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Johns, D. G., Behm, D. J., Walker, D. J., Ao, Z., Shapland, E. M., Daniels, D. A., Riddick, M., Dowell, S., Staton, P. C., Green, P., Shabon, U., Bao, W., Aiyar, N., Yue, T. L., Brown, A. J., Morrison, A. D., and Douglas, S. A. (2007) Br. J. Pharmacol. 152 825-831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lauckner, J. E., Jensen, J. B., Chen, H. Y., Lu, H. C., Hille, B., and Mackie, K. (2008) Proc. Natl. Acad. Sci. U. S. A. 105 2699-2704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Oh, D. Y., Yoon, J. M., Moon, M. J., Hwang, J. I., Choe, H., Lee, J. Y., Kim, J. I., Kim, S., Rhim, H., O'Dell, D. K., Walker, J. M., Na, H. S., Lee, M. G., Kwon, H. B., Kim, K., and Seong, J. Y. (2008) J. Biol. Chem. 283 21054-21064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Oka, S., Nakajima, K., Yamashita, A., Kishimoto, S., and Sugiura, T. (2007) Biochem. Biophys. Res. Commun. 362 928-934 [DOI] [PubMed] [Google Scholar]
- 17.Obinata, H., Hattori, T., Nakane, S., Tatei, K., and Izumi, T. (2005) J. Biol. Chem. 280 40676-40683 [DOI] [PubMed] [Google Scholar]
- 18.Nagy, L., Tontonoz, P., Alvarez, J. G., Chen, H., and Evans, R. M. (1998) Cell 17 229-240 [DOI] [PubMed] [Google Scholar]
- 19.Kabarowski, J. H., Zhu, K., Le, L. Q., Witte, O. N., and Xu, Y. (2001) Science 293 702-705; Correction (2005) Science 307, 206 [DOI] [PubMed] [Google Scholar]
- 20.Murakami, N., Yokomizo, T., Okuno, T., and Shimizu, T. (2004) J. Biol. Chem. 279 42484-42491 [DOI] [PubMed] [Google Scholar]
- 21.Radu, C. G., Nijagal, A., McLaughlin, J., Wang, L., and Witte, O. N. (2005) Proc. Natl. Acad. Sci. U. S. A. 102 1632-1637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lin, P., and Ye, R. D. (2003) J. Biol. Chem. 278 14379-14386 [DOI] [PubMed] [Google Scholar]
- 23.Radu, C. G., Yang, L. V., Riedinger, M., Au, M., and Witte, O. N. (2004) Proc. Natl. Acad. Sci. U. S. A. 101 245-250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Peter, C., Waibel, M., Radu, C. G., Yang, L. V., Witte, O. N., Schulze-Osthoff, K., Wesselborg, S., and Lauber, K. (2008) J. Biol. Chem. 283 5296-5305 [DOI] [PubMed] [Google Scholar]
- 25.Siehler, S. (2008) Biotechnol. J. 3 471-483 [DOI] [PubMed] [Google Scholar]
- 26.Ge, X., Yannai, S., Rennert, G., Gruener, N., and Fares, F. A. (1996) Biochem. Biophys. Res. Commun. 228 153-158 [DOI] [PubMed] [Google Scholar]
- 27.Aggarwal, B. B., and Ichikawa, H. (2005) Cell Cycle 4 1201-1215 [DOI] [PubMed] [Google Scholar]
- 28.Xue, L., Pestka, J. J., Li, M., Firestone, G. L., and Bjeldanes, L. F. (2008) J. Nutr. Biochem. 19 336-344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cho, H. J., Seon, M. R., Lee, Y. M., Kim, J., Kim, J. K., Kim, S. G., and Park, J. H. (2008) J. Nutr. 138 17-23 [DOI] [PubMed] [Google Scholar]
- 30.Weng, J. R., Tsai, C. H., Kulp, S. K., Wang, D., Lin, C. H., Yang, H. C., Ma, Y., Sargeant, A., Chiu, C. F., Tsai, M. H., and Chen, C. S. (2007) Cancer Res. 67 7815-7824 [DOI] [PubMed] [Google Scholar]
- 31.Gong, Y., Sohn, H., Xue, L., Firestone, G. L., and Bjeldanes, L. F. (2006) Cancer Res. 66 4880-4887 [DOI] [PubMed] [Google Scholar]
- 32.Xue, L., Firestone, G. L., and Bjeldanes, L. F. (2005) Oncogene 24 2343-2353 [DOI] [PubMed] [Google Scholar]
- 33.Jellinck, P. H., Forkert, P. G., Riddick, D. S., Okey, A. B., Michnovicz, J. J., and Bradlow, H. L. (1993) Biochem. Pharmacol. 45 1129-1136 [DOI] [PubMed] [Google Scholar]
- 34.Bjeldanes, L. F., Kim, J. Y., Grose, K. R., Bartholomew, J. C., and Bradfield, C. A. (1991) Proc. Natl. Acad. Sci. U. S. A. 88 9543-9547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chen, I., Safe, S., and Bjeldanes, L. F. (1996) Biochem. Pharmacol. 51 1069-1076 [DOI] [PubMed] [Google Scholar]
- 36.Chen, I., McDougal, A., Wang, F., and Safe, S. (1998) Carcinogenesis 19 1631-1639 [DOI] [PubMed] [Google Scholar]
- 37.Le, H. T., Schaldach, C. M., Firestone, G. L., and Bjeldanes, L. F. (2003) J. Biol. Chem. 278 21136-21145 [DOI] [PubMed] [Google Scholar]
- 38.Yan, Y. X., Boldt-Houle, D. M., Tillotson, B. P., Gee, M. A., D'Eon, B. J., Chang, X. J., Olesen, C. E., and Palmer M. A. (2002) J. Biomol. Screen. 7 451-459 [DOI] [PubMed] [Google Scholar]
- 39.Dyson, A., Peacock, M., Chen, A., Courade, J. P., Yaqoob, M., Groarke, A., Brain, C., Loong, Y., and Fox, A. (2005) Pain 116 129-137 [DOI] [PubMed] [Google Scholar]
- 40.Paugh, S. W., Cassidy, M. P., He, H., Milstien, S., Sim-Selley, L. J., Spiegel, S., and Selley, D. E. (2006) Mol. Pharmacol. 70 41-50 [DOI] [PubMed] [Google Scholar]
- 41.Brown, A. J., Goldsworthy, S. M., Barnes, A. A., Eilert, M. M., Tcheang, L., Daniels, D., Muir, A. I., Wigglesworth, M. J., Kinghorn, I., Fraser, N. J., Pike, N. B., Strum. J. C., Steplewski, K. M., Murdock, P. R., Holder, J. C., Marshall, F. H., Szekeres, P. G., Wilson, S., Ignar, D. M., Foord, S. M., Wise, A., and Dowell, S. J. (2003) J. Biol. Chem. 278 11312-11319 [DOI] [PubMed] [Google Scholar]
- 42.Briscoe, C. P., Tadayyon, M., Andrews, J. L., Benson, W. G., Chambers, J. K., Eilert, M. M., Ellis, C., Elshourbagy, N. A., Goetz, A. S., Minnick, D. T., Murdock, P. R., Sauls, H. R., Jr., Shabon, U., Spinage, L. D., Strum, J. C., Szekeres, P. G., Tan, K. B., Way, J. M., Ignar, D. M., Wilson, S., and Muir, A. I. (2003) J. Biol. Chem. 278 11303-11311 [DOI] [PubMed] [Google Scholar]
- 43.Wang, J., Wu, X., Simonavicius, N., Tian, H., and Ling, L. (2006) J. Biol. Chem. 281 34457-34464 [DOI] [PubMed] [Google Scholar]
- 44.Kohno, M., Hasegawa, H., Inoue, A., Muraoka, M., Miyazaki, T., Oka, K., and Yasukawa, M. (2006) Biochem. Biophys. Res. Commun. 347 827-832 [DOI] [PubMed] [Google Scholar]
- 45.Sugo, T., Tachimoto, H., Chikatsu, T., Murakami, Y., Kikukawa, Y., Sato, S., Kikuchi, K., Nagi, T., Harada, M., Ogi, K., Ebisawa, M., and Mori, M. (2006) Biochem. Biophys. Res. Commun. 341 1078-1087 [DOI] [PubMed] [Google Scholar]
- 46.McKallip, R. J., Lombard, C., Fisher, M., Martin, B. R., Ryu, S., Grant, S., Nagarkatti, P. S., and Nagarkatti, M. (2002) Blood 100 627-634 [DOI] [PubMed] [Google Scholar]
- 47.Guzmán, M. (2003) Nat. Rev. Cancer 3 745-755 [DOI] [PubMed] [Google Scholar]
- 48.Flygare, J., and Sander, B. (2007) Semin. Cancer Biol. 18 176-189 [DOI] [PubMed] [Google Scholar]
- 49.Yamamoto, W., Mikami, T., and Iwamura, H. (2008) Eur. J. Pharmacol. 583 56-61 [DOI] [PubMed] [Google Scholar]
- 50.Benito, C., Tolón, R. M., Pazos, M. R., Núñez, E., Castillo, A. I., and Romero J. (2008) Br. J. Pharmacol. 153 277-285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lotersztajn, S., Teixeira-Clerc, F., Julien, B., Deveaux, V., Ichigotani, Y., Manin, S., Tran-Van-Nhieu, J., Karsak, M., Zimmer, A., and Mallat, A. (2008) Br. J. Pharmacol. 153 286-289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wright, K. L., Duncan, M., and Sharkey, K. A. (2008) Br. J. Pharmacol. 153 263-270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Steffens, S., Veillard, N. R., Arnaud, C., Pelli, G., Burger, F., Staub, C., Karsak, M., Zimmer, A., Frossard, J. L., and Mach, F. (2005) Nature 434 782-786 [DOI] [PubMed] [Google Scholar]
- 54.Klein, T. W., Newton, C., Larsen, K., Lu, L., Perkins, I., Nong, L., and Friedman, H. (2003) J. Leukocyte Biol. 74 486-496 [DOI] [PubMed] [Google Scholar]
- 55.Arévalo-Martín, A., García-Ovejero, D., Gómez, O., Rubio-Araiz, A., Navarro-Galve, B., Guaza, C., Molina-Holgado, E., and Molina-Holgado, F. (2008) Br. J. Pharmacol. 153 216-225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Pacher, P., and Haskó, G. (2008) Br. J. Pharmacol. 153 252-262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bab, I., Ofek, O., Tam, J., Rehnelt, J., and Zimmer, A. (2008) J. Neuroendocrinol. 20 Suppl. 1, 69-74 [DOI] [PubMed] [Google Scholar]
- 58.Maccarrone, M. (2008) Br. J. Pharmacol. 153 189-198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Staton, P. C., Hatcher, J. P., Walker, D. J., Morrison, A. D., Shapland, E. M., Hughes, J. P., Chong, E., Mander, P. K., Green, P. J., Billinton, A., Fulleylove, M., Lancaster, H. C., Smith, J. C., Bailey, L. T., Wise, A., Brown, A. J., Richardson, J. C., and Chessell, I. P. (2008) Pain 139 225-236 [DOI] [PubMed] [Google Scholar]
- 60.Weng, Z., Fluckiger, A. C., Nisitani, S., Wahl, M. I., Le, L. Q., Hunter, C. A., Fernal, A. A., Le Beau, M. M., and Witte, O. N. (1998) Proc. Natl. Acad. Sci. U. S. A. 95 12334-12339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hirose, M., Egashira, S., Goto, Y., Hashihayata, T., Ohtake, N., Iwaasa, H., Hata, M., Fukami, T., Kanatani, A., and Yamada, K. (2003) Bioorg. Med. Chem. Lett. 13 4497-4499 [DOI] [PubMed] [Google Scholar]
- 62.Ignatov, A., Lintzel, J., Hermans-Borgmeyer, I., Kreienkamp, H. J., Joost, P., Thomsen, S., Methner, A., and Schaller, H. C. (2003) J. Neurosci. 23 907-914 [DOI] [PMC free article] [PubMed] [Google Scholar]