

Abstract
The surface expression and localization of AMPA receptors (AMPARs) at dendritic spines are tightly controlled to regulate synaptic transmission. Here we show that de novo exocytosis of the GluR2 AMPAR subunit occurs at the dendritic shaft and that new AMPARs diffuse into spines by lateral diffusion in the membrane. However, membrane topology restricts this lateral diffusion. We therefore investigated which mechanisms recruit AMPARs to spines from the shaft and demonstrated that inhibition of dynamin GTPase activity reduced lateral diffusion of membrane-anchored green fluorescent protein and super-ecliptic pHluorin (SEP)-GluR2 into spines. In addition, the activation of synaptic N-methyl-d-aspartate (NMDA) receptors enhanced lateral diffusion of SEP-GluR2 and increased the number of endogenous AMPARs in spines. The NMDA-invoked effects were prevented by dynamin inhibition, suggesting that activity-dependent dynamin-mediated endocytosis within spines generates a net inward membrane drift that overrides lateral diffusion barriers to enhance membrane protein delivery into spines. These results provide a novel mechanistic explanation of how AMPARs and other membrane proteins are recruited to spines by synaptic activity.
AMPA3 receptors (AMPARs) are of fundamental importance because they mediate the majority of fast excitatory synaptic transmission in the mammalian central nervous system (1). Most excitatory synapses are characterized morphologically by dendritic spines that contain an electron-dense postsynaptic density (PSD) at their head (2, 3). PSD is highly enriched in AMPARs and associated proteins equired for synaptic transmission and signal transduction (4-6). Activity-evoked changes in functional postsynaptic AMPARs mediate the two main forms of synaptic plasticity believed to underlie learning and memory in the hippocampus (7). Long term potentiation involves the activity-dependent recruitment of AMPARs to the postsynaptic membrane and a concurrent increase in AMPA-mediated transmission, whereas long term depression is a decrease in synaptic AMPAR function (8).
The number and subunit composition of synaptic AMPARs are stringently regulated, but despite intense investigation, the processes by which AMPARs are delivered to and retained at the PSD remain controversial. Using photoreactive antagonists and electrophysiology, it has been proposed that AMPARs are only inserted in the plasma membrane at the cell body and laterally diffuse long distances to synapses (9). In direct contrast, approaches using real-time imaging have suggested that AMPARs are inserted in the plasma membrane of the dendritic shaft close to, but not at, dendritic spines (10). It has also been suggested that AMPARs could be inserted directly into the plasma membrane of the PSD (11).
Independent of the route of delivery for new AMPARs to synapses, it is well established that lateral diffusion in the plasma membrane allows the exchange of receptors in and out of the PSD (12-14). Using palmitoylated membrane-anchored GFP (mGFP), which partitions to the inner leaflet of the plasma membrane, it has also been reported that diffusion is significantly retarded within spines compared with the shaft and that AMPAR activation increases the rates of mGFP diffusion in spines (15). In addition, we have shown previously that membrane protein movement into and out of spines is slow compared with lateral diffusion on non-spiny membrane (16), and modeling studies have predicted that spine length is a major determinant of the time a protein takes to reach the PSD (17). More recently, it has been proposed that endocytosis at specialized endocytic zones close to the PSD within spines is required to maintain the steady state complement of synaptic AMPARs (18).
Taken together these findings suggest that endocytosis and exocytosis as well as lateral diffusion and membrane topology may all play important roles in regulating membrane protein mobility in spines. The interrelationships between these processes, however, remain unclear. Here we used FRAP (fluorescence recovery after photobleaching) and multisite FLIP (fluorescence loss in photobleaching) to visualize super-ecliptic pHluorin-tagged GluR2 surface expression and AMPAR movement in real time. We examined how lateral diffusion is regulated in spines both by blocking dynamin GTPase activity and stimulating NMDARs. Combined with Monte Carlo simulations on lattices fitting theoretical spines, our data indicate that the membrane topology of spines alone is sufficient to constrain lateral diffusion. NMDAR activation facilitates AMPAR recruitment to spines by a process that involves the recruitment of plasma membrane, together with the constituent membrane proteins, from adjacent regions of the dendritic shaft being drawn into the spine to replace membrane that is internalized during endocytosis. In other words, our results suggest a mode of lateral diffusion that is neither free nor anomalous. Rather, we show the directional diffusion of membrane-embedded proteins toward the postsynapse driven by the endocytosis within the spine. These results provide a new mechanistic explanation of how synaptic activity can overcome topology-induced diffusion barriers to recruit new membrane proteins to the spine.
EXPERIMENTAL PROCEDURES
Computer Modeling—Monte Carlo simulations were performed with MATLAB software. Runs were built as perturbation/relaxation experiments. At the initial point (t = 0), an area of interest, the spine (or a flat circle) was empty of particles. During the course of the experimental simulation, particles undergoing the random walk and entering the region of interest were counted over time. Briefly, theoretical spines were built as the vertical concatenation of a truncated sphere (the spine head) placed on top of a cylinder (the spine neck). Each spine was located at the center of a flat square field 4 μm in length/side (see Fig. 2a). Spine features were assigned according to electron microscopy observations (2, 19), 20-250 nm for neck radius, 10-2000 nm for neck length, and 20-500 nm for head radius. The step size for spine shaping was 10 nm. For stubby spines, the neck length was 10-50 nm with equal neck and head radii. As a control, some runs were performed on a flat lattice with a central circle explored region.
FIGURE 2.

Spine membrane topology produces anomalous diffusion. a, three-dimensional representation of Monte Carlo simulations. Theoretical spines were produced by a vertical concatenation of a truncated sphere (head radius 450 nm) with a cylinder (neck radius 200 nm and neck length 750 nm) placed in the middle of a flat field (4 × 4 μm, half displayed in the figure). The step size for spine shaping was 10 nm. As a starting condition, particles (red dots) were randomly distributed in the flat field. The blue trajectory represents the successive positions of a particle during a 775-ms run, starting in the flat field and ending in the spine head. Over time, particles fill the theoretical spine; see right panel (t = 30 s). b, the black curve represents the number of particles in a spine (same features as in a) over time. The red curve shows the corresponding fit (time constant t is 5.78 s; efficient diffusion coefficient is 0.023 μm2/s; time exponent: a = 0.88). c, distribution of log(Deff/Dth) (where Deff = efficient diffusion coefficient and Dth = theoretical diffusion coefficient) for 256 experiments on flat and stubby spines and 512 experiments on mushroom-shaped spines. In mushroom-shaped spines, Deff is 10-1000 times smaller than Dth (medians: flat, -0.042; stubby, -0.271; mushroom-shaped, -1.41; stubby versus flat, p < 0.05; mushroom-shaped versus flat, p < 0.001). d and e, log(Deff/Dth) plotted versus spine features, respectively, neck radius/neck length and neck radius/head radius. Data were grouped (bin sizes, 0.2 (d) and 0.1 (e)) and mean ± S.E. plotted.
In a random walk a particle can haphazardly jump to one of four possible orthogonal positions at each time point. The length of the jump (L) and the time step (dt) define the theoretical diffusion coefficient (D) of the protein at the microscopic scale by the equation D = L2/4dt. The scale of the simulation was set such that the spacing between lattice sites was 50 nm, and the time step was 1.250 ms. Thus, the microscopic theoretical diffusion coefficient was 0.5 μm2/s (= (50 nm)2/4(1.25 ms)) as measured for mobile AMPARs (13). Nearest-neighbor positions on planar, cylindrical, and spherical lattices were computed as described in supplemental data Fig. 1. At the start point (t = 0), particles were distributed randomly on the flat region with a density of 20 particles/μm2 as described for extra-synaptic AMPARs (20). The number of particles expected in the spine at the end of the run was computed from the spine area with the above density and random movement of particles entering the square field during the run. As a boundary rule and to mimic an infinite pool of particles, when a computed position was out of field (IxI or IyI > 2 mm), a new trajectory started at a random position along the edge of the square field at the following time step. Particle counts in the area of interest were done every 100 ms. The time constant of relaxation was extracted from the curve using the following fit,
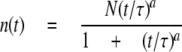 |
(Eq.1) |
where n(t) is the number of particle at time t, N is the number of particle in the area at the equilibrium, t is the half-time to equilibrium, and a is the time exponent, which gives a measure of the degree to which the motion is restricted. This fitting curve is adapted from theoretical FRAP experiments (21). The diffusion coefficient was then calculated from the time constant (t) and the spine area,
 |
(Eq.2) |
adapted from theoretical FRAP experiments under uniform disk illumination (22).
FIGURE 1.

SEP-GluR2 is inserted into the plasma membrane on the dendritic shaft. a, sample images of a FRAP-FLIP experiment in a neuron expressing SEP-GluR2. The large central boxed area was bleached once, whereas bleaching was maintained in the flanking regions. Scale bar = 5 μm. b, sample traces, with normalized mean intensity measured in regions of interest (white arrowheads in a). Two sample shaft regions, R1 and R2, with different recovery patterns, and spiny regions, S1 inside and S2 outside the region of interest, are shown. FRAP is indicated by the black arrow and the onset of FLIP by the green arrow. A recovery can be observed on the shaft but not in spines. Prior to FRAP, fluorescence variations were normalized to base-line intensity. The period after FRAP but before FLIP shows some recovery in the shaft due to rapid diffusion of mobile receptors. This diffusion of fluorescent receptors is prevented by the FLIP protocol, which bleaches all of the surface-expressed SEP-GluR2 diffusing in to the region of interest. c, mean ± S.E., sum of fluorescence in the entire FLIP region over 11 experiments at early and late FLIP time points, normalized to the early FLIP time point to avoid any variation due to differences in the area between separate sets of experiments (early FLIP, 1 ± 0.07; late FLIP, 1,21 ± 0.04; p < 0.05). d, mean (±S.E.) ΔFLIP in four different regions: i, where the fluorescence recovered on shaft (i.e. late FLIP was at least > early FLIP ± S.E.); ii, where no recovery was observed on the shaft; iii, in spines within the FLIP region; iv, in spines outside the FLIP regions (recovering shaft regions, 0.13 ± 0.01, n = 21, p < 0.005 versus nonrecovering shaft regions; nonrecovering shaft regions, 0.01 ± 0.03, n = 36; spines between FLIP regions, -0.02 ± 0.03, n = 28; spines outside FLIP regions, -0.52 ± 0.04, n = 17, p < 0.05 versus nonrecovering shaft regions). e, correlation plot between fluorescence recovery during FLIP (ΔFLIP) and internal pools of receptors (ΔNH4Cl) recorded on shaft regions of primary (black), secondary (red), and tertiary (green) dendrites. f, mean ± S.E., normalized fluorescence variations of SEP-GluR2 on dendritic shaft (black) and spines (red) during a 30-s application of pH 6.0 solution followed by washing and a 30-s application of 50 mm NH4Cl.
Cell Culture, Viral Transduction, and Transfection—Hippocampal cultures were prepared from embryonic (E18) rats, plated at high density on glass coverslips and grown in culture for 15-25 days in vitro. For viral expression of recombinant protein cultures at 21-25 days in vitro were transduced 24-36 h prior to experiments with attenuated Sindbis virus containing super-ecliptic pHluorin (SEP)-GluR2 or mGFP as described previously (16, 23). The membrane-anchored probe (mGFP) was produced by fusing the coding N-terminal region of GAP-43 (palmitoylation site) with the fluorescent probe eGFP as described previously (16, 24). Dynamin/mGFP co-transfection was achieved using Lipofectamine 2000 transfection according to manufacturer's protocols (invitrogen) in 15-19-day in vitro neurons.
Surface Immunostaining and Analysis—Following the appropriate pharmacological treatments, neurons were washed for 1 min in recording solution and incubated for 20 min at room temperature with 5 mg/ml anti-GluR2 (N-terminal, Zymed Laboratories) in recording solution. After washing to remove excess antibody, neurons were fixed with heated paraformaldehyde, 4% sucrose solution for 15 min at room temperature. Neurons were then incubated with 50 μm glycine and 0.3% bovine serum albumin for 10 min before incubation with a Cy3-conjugated secondary antibody. Confocal images of mGFP were used to produce masks after automated intensity thresholding (ImageJ). The masks were duplicated and the dendritic shaft was drawn manually to produce a first set of masks defining shaft regions. These shaft masks were then subtracted from the original masks to reveal spines in another set of masks. Masks were applied to corresponding GluR2 images, and following an automated intensity threshold, the number of puncta was measured. The membrane areas of the shaft regions and spines were assessed as described for the FRAP experiments (see below and supplemental data Fig. 2).
Live Cell Imaging, FRAP, and FLIP—Two main parameters can be determined from the fluorescence recovery curve in FRAP. First the difference between the pre-photobleaching level and the recovered steady state level after bleaching reflects the immobile fraction of protein. That is, the lower level of fluorescence after recovery from photobleaching is accounted for by bleached fluorescent protein that does not diffuse away from the area of photobleaching. These immobile proteins may be directly or indirectly tethered to cytoskeletal elements or restricted in confined membrane compartments. Secondly, the half-time to recovery indicates the mobility of the diffusible fraction of the protein under investigation.
Neurons were placed on the heated stage (37 °C) of a confocal Zeiss Axiovert microscope and bathed in extracellular recording solution containing (in mm): 140 NaCl, 5 KCl, 1.8 CaCl2, 0.8 MgCl2, 25 HEPES, and 0.9 g/liter glucose (pH 7.4). Pinhole settings and z focus were set to limit the contribution of spines projecting toward the z axis (Airy 1-2). The pH sensitivity was confirmed by perfusing cells with extracellular recording solution buffered at pH 6.0 or complemented with 50 mm NH4Cl.
FRAP-FLIP experiments were designed as a single photobleaching of a user-designed region of interest (central and flanking areas) followed, after a 1-min recovery, by maintaining (for 5 min) the bleaching of the flanking regions only. This protocol bleaches any SEP-GluR2 entering the region of interest via lateral membrane diffusion (25). The confocal power settings used ensured the effective photobleaching of all susceptible receptors (i.e. those in neutral pH and therefore emitting fluorescence) whether or not they were in the plane of acquisition. To measure fluorescence fluctuations on the shaft and spines, the length of the shaft under investigation and the spine heads were divided into 20-pixel regions, and the mean fluorescence was recorded over time. The mean of the first 10 time points (Fi) where considered as the base line and used to normalize the entire experiment (F/Fi). ΔFLIP is the difference between the mean of the last 10 measures of the FLIP sequence and the mean of the first 10 measures. ΔNH4Cl is the difference between the mean last 10 measures of NH4Cl application and the last 10 measures of the FLIP sequence. Differences in recovery were considered valid when the mean of the last 10 time points of FLIP was statistically significantly greater than the mean of the first 10 time points of FLIP. Fig. 1f represents the mean-fold fluctuation of fluorescence signal during pH solution switching.
In dynasore experiments, cells were incubated for 20 min prior to experiments done in recording solution complemented with 50 μm dynasore (from stock 50 mm in DMSO) or 1/1000 DMSO. For paired recordings of FRAP, spines and dendritic segments were imaged and bleached during the first 5 min of the experiment followed by a 3-min drug treatment and a 1-min wash and then were imaged/bleached a second time. Drug concentrations were: 50 μm NMDA, 90 mm KCl, with 100 μm glycine or 50 μm DAPV, and 1 μm TTX. For unpaired recordings, cells were incubated for 3 min with drug solutions, washed for 1 min, and imaged. Although NMDAR activation is likely to change the endocyotsis/exocyotsis rate of AMPARs, it is important to note that this was not a major issue in our analysis because the recovery kinetics was independent of initial baseline levels. Further, we did not detect any change in fluorescence in the control experiments performed on unbleached spines.
Fluorescence was collected by a 63× oil objective (N.A. = 1.4) using a 488 nm laser light for excitation. Full power laser excitation was used for photobleaching in FRAP, whereas only 10% power was used for FLIP and 1% power for imaging. Recovery curves were fitted as described previously (21). Diffusion coefficients were computed with
 |
(Eq.3) |
where t is the time constant extracted from fitted recovery curves. To accurately measure the membrane area of spines, we constructed fluorescence/membrane area calibration curves as described in supplemental data Fig. 3. In essence we considered the dendritic shaft as a cylinder and generated calibration curves relating the total fluorescence measured to the calculated membrane area.
FIGURE 3.

Dynamin GTPase activity within spines invokes an inward membrane protein flow. a, representative sample images of mGFP in dendritic spines during a FRAP experiment in which a single spine is bleached. The upper panels are sequential images taken at the times indicated from a control neuron, and the lower panels are from a dynasore-treated neuron. The white squares designate the bleached area. Scale bar = 1 μm. b, normalized FRAP traces of mGFP and corresponding fits from spines with comparable areas obtained in control (black) and dynasore (red)-treated cells. c, mean ± S.E. of mobile fraction of mGFP in spines (control, n = 22, 0.87 ± 0.06; dynasore, n = 24, 0.66 ± 0.03, p < 0.05) and on dendritic shaft (control, n = 7, 0.68 ± 0.05; dynasore, n = 8, 0.75 ± 0.07, not statistically significant). d and e, scatter plots of computed diffusion coefficients of mGFP in spines (medians in μm2/s, control, n = 22, 0.221; dynasore, n = 24, 0.078, p < 0.01) and on dendritic shaft (medians in μm2/s, control, n = 7, 2.39; dynasore, n = 8, 2.13, ns). f, distribution of computed diffusion coefficients of mGFP in spines compared with the ratio of neck and head radii. The logarithm of the diffusion coefficient is correlated with the ratio of neck and head radii in dynasore-treated cells but not in control condition (dynasore, r = 0.71; control, r = 0.03). g, medians of diffusion coefficients of mGFP for different spine classes.
For FLIP experiments, a user-defined FLIP region was selected in the vicinity of an isolated dendritic spine, and bleaching was maintained (after 10 basal iterations) in this region during the time course of the experiment. Fluorescence loss was recorded using ImageJ software by manually measuring distances from the FLIP region to the spine head and recording from and equivalent distance of the shaft. Fluorescence loss was fitted using a monoexponential decay with QtiPlot software. Files were converted with ImageJ (National Institutes of Health). Image and statistical analysis (Kruskal-Wallis one-way analysis of variance for multiple compared conditions; post hoc rank sum test) were performed using MATLAB software, and graphs were produced using QtiPlot software.
RESULTS
Exocytosis of GluR2 Occurs on the Dendritic Shaft—Super-ecliptic pHluorin is a pH-sensitive derivative of green fluorescent protein that is not excited at low pH, so it emits no fluorescent signal and is resistant to photobleaching under acidic conditions (26). In the late stages of the secretory pathway, extracellular domains of membrane proteins destined for the cell surface are maintained in low pH environment. Thus, SEP fused to the extracellular N terminus of the GluR2 AMPAR subunit is a useful tool for monitoring AMPAR subunit surface expression as the SEP is only exposed to a neutral pH on exocytosis (23, 25). When expressed using Sindbis virus in neurons, fluorophore-tagged GluR2 subunits (27) traffic to synapses and participate in synaptic transmission without changing the total number of synaptic receptors (28).
In virally transduced dispersed hippocampal neurons expressing SEP-GluR2, we photobleached surface receptors in a defined area of interest (FRAP); then, to avoid recovery due to lateral diffusion, the flanking regions were photobleached repetitively (FLIP, Fig. 1a).
Importantly, this protocol does not bleach intracellular, non-fluorescent SEP-GluR2 because the SEP is localized in acidic trafficking vesicles (16). This intracellular pool of SEP-GluR2, however, can be visualized by a brief exposure to NH4Cl, which reversibly collapses intracellular pH gradients (NH4Cl, Fig. 1b). Thus, in these experiments, any recovery in fluorescence in the membrane area of interest can be attributed to exocytosis within the region of interest. There are two possible sources of the non-photobleached receptors exocytosed to the membrane: they could be supplied by intracellular trafficking and de novo insertion or they could be recycled receptors that are in vesicles inside the shaft during FRAP.
Consistent with a recent report showing shaft insertion for GluR1 (10), which was published while this work was in progress, we observed a slow recovery of SEP-GluR2 fluorescence on shaft regions but never detected any fluorescence recovery in spines (see Fig. 1b, R1 and S1), indicating that new AMPARs had been inserted into the membrane on the dendritic shaft but had not exocytosed directly into the spines. 63% of the total number of shaft regions investigated did not show any recovery, as illustrated in the individual example R2 shown in Fig. 1, a and b (pooled data from multiple experiments are plotted in Fig. 1d). This suggests that AMPAR exocytosis occurs at specific locations or “hot spots” within the shaft plasma membrane and that spines are supplied with new GluR2-containing AMPARs by lateral diffusion from their sites of exocytosis on the shaft. To confirm that we had observed insertion rather than redistribution and clustering, we measured the total fluorescence in the central photobleached region at early and late time points of the FLIP protocol. Entirely consistent with the exocytosis of new receptors in the photobleached region, we observed a significant increase in the total fluorescence between these time points, indicating insertion of SEP-GluR2 during the time course of the FLIP experiment (Fig. 1c).
Spines close to the FLIP regions that had not been subjected to photobleaching lost fluorescence over time during FLIP (see S2 in Fig. 1, a and b), indicating that there was an exchange of the mobile fraction of their receptor content with AMPARs on the shaft. Consistent with our previous observations (16), spine S2 exchanged nearly all of its mobile receptors with photobleached ones within the 6-min time frame of this experiment. The cumulative data shown in Fig. 1d demonstrate that there was a significant and dramatic decrease in the fluorescence of adjacent spines.
Each FRAP-FLIP experiment was calibrated using a brief exposure to NH4Cl, and the amplitude of fluorescence recovery during the FLIP sequence (ΔFLIP) was correlated to the size of the intracellular, NH4Cl-sensitive (ΔNH4Cl) pool of receptors (Fig. 1, e and f). Acidic quenching effectively removed all SEP-GluR2 fluorescence from spines with no significant NH4Cl-sensitive fraction (Fig. 1, b and d; see also Ref. 16). These data show that the number of intracellular AMPARs in the spine is negligible compared with the surface population, ruling out the possibility of a reservoir of intracellular AMPARs stored in the spine to supply de novo surface synaptic receptors (also see supplemental Fig. 4). Although this certainly does not rule out the possibility of AMPAR recycling in spines (18), our results support a mechanism in which the recruitment of new GluR2-containing synaptic AMPARs occurs via insertion on the dendritic shaft followed by lateral diffusion to the spine head.
Spine Topology Limits Lateral Diffusion of Membrane Proteins—Dendritic spines restrict diffusion of ions, small soluble proteins (3), and membrane proteins (16, 17). However, it remains unclear whether membrane topology is sufficient to constrain protein diffusion in spines as modeled for nonplanar membranes (29) (also see supplemental data Fig. 1) and for the endoplasmic reticulum network (30). To investigate this, we preformed in silico experiments in which particles undergoing three-dimensional random walks were placed outside an area of interest (i.e. the spine) and allowed to diffuse freely over time (Fig. 2, a and b). This perturbation/relaxation model can be considered a computer-based FRAP experiment with an infinite pool of particles, where the spine neck radius and length and head radius can be adjusted. The diffusion coefficient extracted from fitted relaxation curves, called the efficient diffusion coefficient (Deff), was compared with the theoretical diffusion coefficient of particles (Dth; see “Experimental Procedures”). In the control non-spiny flat membrane areas, the computed efficient diffusion coefficient is close to the theoretical value (Fig. 2c, median of log(Deff/Dth) = -0.042). In mushroom-shaped spines the efficient diffusion coefficient is significantly smaller (10-1000-fold, 30 times smaller for medians) compared with non-spiny flat areas (Fig. 2c). These results are entirely consistent with our direct experimental observations of FRAP shown in Fig. 3 (see also Ref. 16). In these in silico experiments, no anchoring or subdomain confinements were imposed, suggesting that membrane topology alone is sufficient to produce anomalous diffusion. Entry into and exit from the spine head do not have a comparable probability to diffusion in non-spiny regions, and we propose that this imbalance produces anomalous diffusion. We attribute this to the neck region between the shaft and the spine head where diffusion is restricted. The narrower the neck the more it impedes protein entry into the spine head. Furthermore, once at the spine head the ratio between the neck radius and the head radius prevents diffusion out of the spine head. Thus, over short-time particles move freely, but over longer periods spine topology produces “non-free” diffusion. Consistent with this, analysis of the distribution of time exponents also indicates that anomalous diffusion occurs at spiny membranes (see supplemental data Fig. 2).
Comparing the correlation of log-(Deff/Dth) with the neck radius/neck length ratio and neck radius/head radius (Fig. 2, d and e), we found that the restriction of lateral diffusion could be attributed to the insulation of spine head by the neck as occurs for cytosolic molecules (31). Accordingly, the distribution of log-(Deff/Dth) computed for stubby spines, where the neck and head radii are equal, is shifted slightly through negative values (Fig. 2c, median, -0.271) (17).
On a cylindrical area such as a dendrite, diffusion is restricted along the circular dimension and free along the length, as expected for a semiclosed surface (i.e. there are boundaries in one dimension). Therefore, the displacement of a protein among the dendritic tree is a combination of free and confined motion, which is sufficient to produce globally restricted motion. Furthermore, on a spherically shaped membrane such as a spine head, proteins get trapped due to the topology. Thus, these computer-based modeling experiments establish that spine topology alone is sufficient to limit the exchange of membrane proteins between the shaft compartment and the spine head. Taken together, these data indicate that simple models of diffusion in nonplanar surfaces can generate confined motion consistent with membrane topology acting as a key regulator of protein diffusion.
Dynamin GTPase Activity in Spines Induces Membrane Protein Flow—Based on these data we hypothesized that exchange between the shaft and spine compartments must be facilitated to overcome the diffusion barrier imposed by the neck and to allow the dynamic regulation of synaptic protein content such as occurs in synaptic plasticity. Therefore, we investigated which mechanism could overcome this topology-induced restriction and produce a membrane drift to enhance membrane protein recruitment to the spine head.
Dynamins are large GTPases that induce membrane tubulation and pinching when activated by GTP (32). They are an integral part of the endocytic machinery in dendritic spines (33, 34) and are required to maintain synaptic AMPARs (18). We therefore used acute application of the recently described chemical dynamin inhibitor dynasore (35) (see supplemental data Fig. 5) to block dynamin GTPase activity and assessed lateral diffusion of mGFP, a protein that is targeted to the inner leaflet of the plasma membrane (16, 36) (Fig. 3, a and b). Importantly, dynasore treatment did not affect either the spine neck and head radii or the total spine length. Under control conditions there was only a very small immobile fraction of mGFP in spines, suggesting that spines are compartments that are distinct from the dendritic shaft, where the mobile fraction is 0.68 ± 0.05 (Fig. 3c, mean ± S.E., p < 0.05). We attribute the immobile or anomalous diffusing mGFP in the dendritic shaft to subdomain restriction caused by cytoskeletal elements as described for Kv2.1 potassium channels (37).
Dynasore significantly reduced the mobile fraction of mGFP in spines (control, n = 22, 0.87 ± 0.06; dynasore, n = 24, 0.66 ± 0.03, p < 0.05) consistent with mGFP immobilized in “frozen” endocytic pits. Dynasore treatment decreased the diffusion coefficient of mGFP in spines (medians in μm2/s, control, n = 22, 0.221; dynasore, n = 24, 0.078, p < 0.01) but had no effect on lateral diffusion of mGFP on the dendritic shaft (Fig. 3, d and e). In addition, dynasore treatment confirmed that the diffusion coefficient of mGFP in spines correlates to the ratio of the neck and head radii (Fig. 3f), as predicted by our modeling. Hence, dynasore affects only diffusion of mGFP in mushroom-shaped spines (Fig. 3g). These results suggest that dynamin GTPase activity facilitates lateral diffusion of membrane proteins in spines.
To test this concept further, we investigated the directional diffusion of mGFP in the neuronal plasma membrane. A small region of shaft was bleached repeatedly, and fluorescence loss was assessed in an adjacent isolated spine head and in an equidistant shaft region (Fig. 4, a and b). Under control conditions the kinetics of fluorescence loss was comparable in the spines and shaft regions. Dynasore dramatically slowed fluorescence loss in spines compared with shaft regions (see time constants, Fig. 4b). We assessed the direction of mGFP diffusion in non-spiny areas of dendritic shaft (flat, left versus right) and in spines versus shaft after vehicle or dynasore treatment. The mean time constant ratio (tspine/tshaft) was significantly increased in dynasore-treated neurons (flat, n = 5, 0.78 ± 0.16; spine/shaft control, n = 21, 1.21 ± 0.14; spine/shaft dynasore, n = 23, 3.24 ± 0.49; control versus dynasore, p < 0.05). In addition, although no correlation was observed between the time constant ratio and the neck to head diameter ratio under control conditions (Fig. 4, a and d), dynasore treatment revealed a clear relationship between diffusion and spine morphology compartmentalization (Fig. 4, a and e). This observation is entirely consistent with our FRAP and modeling data (Figs. 2e and 3f). Taken together our data support the concept that dynamin GTPase activity produces a membrane drift facilitating the transport of membrane material from shaft to spine in a manner that can overcome the restrictions imposed by spine topology.
FIGURE 4.

Dynamin-dependant membrane drift overcomes the diffusion barrier imposed by spine geometry. a, sample images of mGFP in dendritic spine and shaft during a control FLIP experiment. The central white region is continuously bleached, and fluorescence is recorded from the flanking spine (red box) and the equidistant shaft region (green box). b, normalized FLIP traces recorded in control and dynasore-treated cells from the FLIP region (black), the shaft (green), and the spine (red). Thin traces correspond to fits, and arrows indicate the time constant computed for the exponential decay fit (control, shaft, 6.02 s, spine, 4.46 s; dynasore, shaft, 4.11 s, spine, 9.98 s). c, mean ± S.E.; time constant ratio (spine/shaft, left/right for flat condition) measured during FLIP experiments (flat, n = 5, 0.78 ± 0.16; control, n = 21, 1.21 ± 0.14; dynasore, n = 23, 3.24 ± 0.49; control versus dynasore, p < 0.05). d and e, relationship between time constant ratios and spine geometry in control and dynasore-treated neurons. In dynasore-treated neurons the time constant ratio in FLIP decreases exponentially with the neck/head ratio (p < 0.05).
We sought to reproduce our dynasore results using the more conventional dominant negative strategy to block dynamin activity (38, 39). We expressed mCherry-tagged wild type dynamin 2 (Dyn2WT) or the GTPase dominant negative mutant dynamin 2 (Dyn2K44A) together with mGFP and assessed lateral mobility in spines. Both Dyn2WT and Dyn2K44A displayed similar distributions in dendrites and spines in 19-day in vitro cultured hippocampal neurons, as illustrated for Dyn2K44A in Fig. 5a. As observed with dynasore, disruption of dynamin GTPase activity by Dyn2K44A caused a significant decrease in the mGFP diffusion coefficient in dendritic spines, whereas Dyn2WT had no significant effect (Fig. 5b; medians in μm2/s, Dyn2WT, n = 18, 0.475; Dyn2K44A, n = 21, 0.070, p < 0.01 versus Dyn2WT). Surprisingly, both Dyn2WT and Dyn2K44A decreased the immobile fraction of mGFP in dendritic spines but not in the shaft (Fig. 5c; control, n = 20, 0.79 ± 0.03; Dyn2WT, n = 24, 0.64 ± 0.06; Dyn2K44A, n = 21, 0.65 ± 0.03, p < 0.05 versus control). The reasons for this are unclear, but it is possible that the effects could be due to prolonged expression. We next used the same FLIP protocol as described for dynasore to test the effects of Dyn2WT and Dyn2K44A on the lateral diffusion of mGFP. Dyn2WT had no effect on the kinetics of fluorescence loss, whereas Dyn2K44A had effects similar to those of dynasore in that it reduced fluorescence loss from spines (Fig. 5, d and e; control, 1.49 ± 0.16; Dyn2WT, 1.42 ± 0.28; Dyn2K44A, 2.98 ± 0.30; Dyn2WT versus Dyn2K44A, p < 0.05). These results are again consistent with the spine being a morphologically distinct membrane compartment and the idea that dynamin activity within the spine drives membrane drift and the recruitment of membrane proteins.
FIGURE 5.

Dominant negative dynamin inhibits membrane drift into spines. a, sample of images of the dendritic shaft of a neuron expressing mCherry-tagged dynamin 2K44A and mGFP presented as an inverted color map for monochromatic images. Scale bar = 1 μm. b, scatter plots of computed diffusion coefficients of mGFP in spines and dendritic shafts. The median values in μm2/s are: on spines, control (mGFP alone), n = 20, 0.302; Dyn2WT, n = 18, 0.475; Dyn2K44A, n = 21, 0.070, p < 0.01 versus Dyn2WT); on dendritic shaft, control (mGFP alone), n = 5, 1.68; Dyn2WT, n = 5, 1.47; Dyn2K44A, n = 5, 1.17, ns). c, mean ± S.E. of mobile fraction of mGFP in spines (control (mGFP alone), n = 20, 0.79 ± 0.03; Dyn2WT, n = 24, 0.64 ± 0.06; Dyn2K44A, n = 21, 0.65 ± 0.03, p < 0.05 versus control) and on the dendritic shaft (control (mGFP alone), n = 5, 0.65 ± 0.06; Dyn2WT, n = 5, 0.62 ± 0.05; Dyn2K44A, n = 5, 0.65 ± 0.04). d, normalized mGFP FLIP traces recorded in Dyn2WT- and Dyn2K44A-transfected neurons from the FLIP region (black), the shaft (gray), and the spine (red). Thin traces are corresponding fits, and arrows indicate the time constant computed for the exponential decay fit (Dyn2WT, shaft, 6.45 s, spine, 4.45 s; Dyn2K44A, shaft, 4.29 s, spine, 12.41 s). e, mean ± S.E. of time constant ratio (spine/shaft) measured during FLIP experiments (control (mGFP alone), 1.49 ± 0.16; Dyn2WT, 1.42 ± 0.28; Dyn2K44A, 2.98 ± 0.30; Dyn2WT versus Dyn2K44A, p < 0.05).
Synaptic NMDA Receptor Activation Promotes Membrane Protein Recruitment to Spines via Membrane Drift—We next examined whether membrane protein recruitment is sensitive to NMDAR activation, as these receptors are critical for both long term depression and potentiation (40). We first used mGFP as a membrane-bound protein marker in FRAP experiments where the recovery kinetics were recorded before and after drug treatments in the same spine (paired recordings). One caveat for this experimental paradigm is that the mobile fraction could not be assessed, because the spine was photobleached twice successively so that the immobile fraction, which was photobleached in the first round, was lost from the second recovery curve (Fig. 6a, steady state level). NMDA application (50 μm, 3 min) caused a significant decrease in the half-time to recovery (Fig. 6a) leading to an increase of the diffusion coefficient (medians in μm2/s, n = 23, pre, 0.18; post, 0.33, p < 0.01 versus pre, Fig. 6b). In contrast, preincubation with dynasore occluded the NMDA-induced facilitation of diffusion (medians in μm2/s, dynasore, n = 22, pre, 0.07; post, 0.04, Fig. 6, a and b). This suggests that dynamin activity mediates the facilitated diffusion evoked by NMDA. KCl (90 mm, 3 min) with glycine (100 μm) also facilitated mGFP lateral diffusion in spines measured as the ratio of diffusion coefficient post-application to pre-application. Again, dynasore abolished this facilitation (Fig. 6c; medians, KCl, n = 22, 2.95; KCl + dynasore, n = 20, 0.77, p < 0.01 versus KCl). Both NMDA- and KCl-induced facilitation of lateral diffusion was prevented by co-application of 50 μm DAPV (Fig. 6c; medians, NMDA + DAPV, n = 18 1.00; KCl + DAPV, n = 19, 1.02) confirming the involvement of NMDARs. NMDA application in calcium-free buffer did not increase mGFP diffusion in dendritic spines and had no effect on the mobile fraction (data not shown). Neither TTX (1 μm, 3 min) nor DAPV alone altered the diffusion coefficient of mGFP in spines (Fig. 6c; medians, TTX, n = 20, 1.04; DAPV, n = 17, 0.86). Note, the control condition here is a paired recording with no drug application (Fig. 6c; median, control, n = 17, 1.038).
FIGURE 6.

Synaptic activity-dependant regulation of lateral diffusion of membrane proteins in spines. a, sample paired recording traces of normalized fluorescence from mGFP in spines during FRAP experiments before (black) and after (gray) NMDA stimulation. The lower panel shows traces with dynasore (red) pretreatment. b, scatter plots of computed diffusion coefficients for mGFP in spines pre- and post-NMDA application in control (black) and dynasore (red)-treated neurons (medians in μm2/s, control, n = 23, pre, 0.18, post, 0.33, p < 0.01; dynasore, n = 22, pre, 0.07, post, 0.04, ns). c, scatter plots of diffusion coefficients pre- and post-drug treatment computed for mGFP in spines (medians, NMDA, n = 22, 2.21, p < 0.01 versus control; NMDA + dynasore, n = 22, 0.63, NMDA + DAPV, n = 18, 1.01; KCl, n = 22, 2.95, p < 0.01 versus control; KCl + dynasore, n = 20, 2.95, p < 0.01 versus control; KCl + DAPV, n = 19, 1.02; TTX, n = 20, 1.04; DAPV, n = 17, 0.86; control, n = 17, 1.03). d, unpaired recording sample traces of normalized fluorescence from SEP-GluR2 in spines during FRAP. The top left panel shows the control. e, mean ± S.E. of the mobile fraction of SEP-GluR2. Control, n = 30, 0.61 ± 0.03; dynasore, n = 21, 0.49 ± 0.02, p < 0.05 versus control; NMDA, n = 19, 0.46 ± 0.04, p < 0.05 versus control; KCl, n = 19, 0.47 ± 0.04, p < 0.05 versus control; KCl + dynasore, n = 20, 0.45 ± 0.03, p < 0.05 versus control; KCl + DAPV, n = 22, 0.60 ± 0.03, ns). f, scatter plots of computed diffusion coefficients of SEP-GluR2 in spines (median in μm2/s, control, n = 30, 0.074; dynasore, n = 21, 0.055, p < 0.05 versus control; NMDA, n = 19, 0.070, ns; KCl, n = 19, 0.144, p < 0.01 versus control; KCl + dynasore, n = 20, 0.049, p < 0.01 versus KCl, p < 0.05 versus control; KCl + DAPV, n = 22, 0.064, ns.
These results suggest that both whole cell and synaptic activation of NMDARs produce a dynamin-dependant recruitment of membrane protein to spines. We therefore examined whether AMPARs are subject to dynamin-dependant membrane drift by imaging SEP-GluR2 in unpaired (to compare mobile fractions) recordings (Fig. 6d). Dynasore reduced the mobile fraction of GluR2-containing AMPARs in spines (Fig. 6e; means ± S.E., control, n = 30, 0.61 ± 0.03; dynasore, n = 21, 0.49 ± 0.02, p < 0.05 versus control) and slowed fluorescence recovery (Fig. 6f; medians in μm2/s, control, n = 30, 0.074; dynasore, n = 21, 0.055, p < 0.05 versus control) suggesting that, like mGFP, SEP-GluR2 is recruited in spines by dynamin-dependant membrane drift. General NMDAR activation (NMDA; 50 μm, 3 min) did not alter SEP-GluR2 diffusion (Fig. 6f; median in μm2/s, NMDA, n = 19, 0.070) but decreased the mobile fraction (Fig. 6e; mean ± S.E., NMDA, n = 19, 0.46 ± 0.04, p < 0.05 versus control), consistent with clustering and stabilization of AMPARs (41). The synaptic NMDAR activation protocol (KCl + glycine), on the other hand, significantly facilitated lateral diffusion of AMPARs in spines (Fig. 6f; median in μm2/s, KCl, n = 19, 0.144, p < 0.01 versus control) as reported from single particle tracking experiments (42). This facilitation of diffusion is concomitant with a decrease in the mobile fraction (Fig. 6e; mean ± S.E., KCl, n = 19, 0.47 ± 0.04, p < 0.05 versus control). Both dynasore and DAPV prevented this facilitation of AMPAR lateral diffusion (medians in μm2/s, KCl + dynasore, n = 20, 0.049, p < 0.01 versus control; KCl + DAPV, n = 22, 0.064, p < 0.01 versus KCl). Dynasore, but not DAPV, decreased the mobile fraction of SEP-GluR2 (Fig. 6e; mean ± S.E., KCl + dynasore, n = 20, 0.45 ± 0.03, p < 0.05 versus control; KCl + DAPV, n = 22, 0.66 ± 0.03), suggesting that some receptors are trapped in pits and that synaptic NMDARs are responsible for AMPAR stabilization in spines. Taken together, our data indicate that global NMDAR activation stabilizes AMPARs and prevents lateral recruitment, whereas activation of synaptic NMDARs facilitate dynamin-dependant recruitment and clustering of AMPARs in spines.
To confirm our observations with SEP-GluR2 in FRAP experiments, we immunolabeled native surface GluR2-containing AMPARs following pharmacological treatment and measured the staining density on spine and shaft regions (Fig. 7, a and b). As expected, dynasore treatment reduced GluR2 spine enrichment, whereas NMDA application had no effect (control, 1.42 ± 0.09; dynasore, 1.16 ± 0.08, p < 0.05 versus control; NMDA, 1.38 ± 0.08, not statistically significant versus control, Fig. 7b). Preferential activation of synaptic NMDA receptors using KCl and glycine (adapted from Ref. 43) enhanced GluR2 staining on the surface of dendritic spines, an effect that was blocked by both DAPV and dynasore (KCl, 1.90 ± 0.15, p < 0.01 versus control; KCl + DAPV, 1.33 ± 0.06, p < 0.05 versus KCl; KCl + dynasore, 1.19 ± 0.08, p < 0.05 versus KCl, Fig. 7b). Following a 10-min wash, no further effect of KCl with glycine was observed (data not shown). Consistent with our live imaging observations, these immunocytochemical results suggest that dynamin activity is required for the recruitment of AMPARs to dendritic spines following synaptic activation of NMDARs.
FIGURE 7.

Synaptically regulated spine enrichment in native GluR2-containing AMPARs. a, sample image of a dendritic shaft with spines of a neuron expressing mGFP (1). Scale bar = 1 μm. 2; binary mask image was obtained from panel 1. Panel 3, surface staining for GluR2 AMPARs subunit in panel 1. Panels 4-6, outline images of GluR2 surface staining after Boolean operation with mask from panel 2, showing manually eroded spines and remaining shaft. Outlined images are shown of GluR2 surface staining on the shaft and spines (white arrows) of neurons after dynasore or KCl treatment relative to the control displayed in a4. b, mean ± S.E. of the ratio of puncta density (density on spines/density on shaft, puncta/μm2) for GluR2 surface staining in treated neurons (n = 10 for each condition). Control, 1.42 ± 0.09; dynasore, 1.16 ± 0.08, p < 0.05 versus control; NMDA, 1.38 ± 0.08, ns versus control; NMDA + DAPV, 1.45 ± 0.09; NMDA + dynasore, 1.15 ± 0.10, p < 0.05 versus control; KCl, 1.90 ± 0.15, p < 0.01 versus control; KCl + DAPV, 1.33 ± 0.06, p < 0.05 versus KCl; KCl + dynasore, 1.19 ± 0.08, p < 0.05 versus KCl). c, schematic illustrating how AMPARs are inserted on the dendritic shaft and then recruited into dendritic spines by a dynamin-dependant membrane and protein flow. This lateral recruitment overcomes the restricted diffusion imposed by spine geometry and is regulated by synaptic NMDA receptors.
DISCUSSION
The mechanisms by which synaptic membrane proteins are trafficked to spines are not well understood. For example, it has been proposed that new AMPARs are inserted into the plasma membrane at the cell soma and migrate by lateral diffusion to postsynaptic sites (9). In direct contrast another study has reported that AMPARs are inserted directly into the postsynaptic density (11). Other groups have suggested that the sites of membrane insertion differ depending on the subunit composition of the AMPAR with GluR1 inserted into dendritic shaft membrane and GluR2 inserted directly in the spine (44). Here we demonstrate that SEP-GluR2 is inserted into the dendritic shaft membrane and enters spines via lateral diffusion.
Neurons contain in the order of 10,000 synapses so spine membrane represents a significant proportion of the total plasma membrane area of a neuron. We did not detect any significant population of intracellular SEP-GluR2 in the spine, thus ruling out the possibility of a reservoir of naive AMPARs awaiting surface expression. Rather, our data suggest that new GluR2-containing synaptic AMPARs are recruited to the synapse via lateral diffusion to the spine head from sites of exocytosis in adjacent regions of dendritic shaft membrane. A similar model has been proposed very recently using rapid live imaging techniques and the SEP-GluR1 subunit (10).
It is important to emphasize that our results do not exclude the possibility of SEP-GluR2 recycling within the spine (18). FRAP of SEP-GluR2 does not allow analysis of these events because any recycled receptors will be photobleached while being surface-expressed. The fact that we did not see any recovery in spines from receptors exocytosed in the nearby shaft membrane during the time course of our experiments was unexpected. We attribute this to the newly inserted receptors diffusing into the flanking FLIP regions and being bleached and also to the shaft having a much larger membrane area than the spines, thus acting as a buffer causing a lengthy time lag beyond that which we could keep the cells viable on the microscope stage before sufficient numbers of fluorescent receptors were present to be incorporated and visualized in spines. Interestingly, we observed no correlation between the ranking of the dendritic section or distance from the soma in recovery after photobleaching of the dendritic shaft membrane. Overall, the data in Fig. 1 show that during FRAP the shaft plasma membrane is gradually replenished by SEP-GluR2 AMPAR, whereas the spine plasma membrane does not, consistent with AMPAR exocytosis only to shaft plasma membrane.
Using mathematical modeling approaches, we have demonstrated that spine topology alone is sufficient to restrict the entry of new receptors into the postsynaptic density via lateral diffusion. This finding is consistent with previous modeling for nonplanar membranes (29, 30) and experimental observations showing the restricted diffusion of cytosolic ions and small soluble proteins (3) as well as membrane proteins (16) into spines. Intriguingly, we show that the neck-imposed diffusion barrier can be overcome by a mechanism that involves dynamin GTPase activity, which we hypothesized produces and inward membrane drift and consequently facilitates diffusion of membrane proteins into the spine. This begs the question of how dynamin GTPase activity can produce membrane drift into spines.
Dynamins are involved in a variety of membrane reorganization phenomenon such as podosome invasion, dorsal waves, lamellipodia accumulation, and membrane budding (32). To date, in spines, dynamins have been implicated only in endocytosis (18, 34, 45), so we reasoned that the most likely explanation for our observations is that endocytosis produces the membrane drift. A recent study indicates that dynamin 3 plays in an important role in maintaining postsynaptic AMPARs at the PSD but shows that this function does not involve GTPase activity (18). Therefore, based on this observation, together with our data using the inactive dynamin 2 mutant K44A, we propose that the membrane drift results we show here are likely mediated by dynamin 2. If this is the case, dynamin 3 is important for maintaining the existing complement of synaptic AMPARs, presumably by recycling, whereas dynamin 2 plays a role in the recruitment of new AMPARs to the synapse.
Following endocytosis vesicles migrate to the dendritic shaft to fuse with endosomes (46, 47). Significantly, prolonged exposure to dynasore did not alter spine size (normalized size, mean ± S.E., 20 min, control, 1.12 ± 0.07; dynasore, 1.06 ± 0.09; 50 min, control, 0.98 ± 0.11; dynasore, 1.01 ± 0.12), suggesting that there was no net increase in membrane recruitment at steady state. The simplest explanation for our results is that under basal conditions the drift of membrane entering the spine, carrying plasma membrane and proteins, exactly matches the spine membrane that is internalized and trafficked to endosomes located in the shaft. This implies that under nonstimulated conditions there is a mechanism to maintain the status quo. Under conditions that lead to enhanced synaptic efficacy, the incoming membrane, together with recycling, acts to increase the membrane area, resulting in the structural spine growth associated with long term potentiation (48, 49).
Although we cannot exclude changes to the nascent excitability of the cells having confounding influences, a transient high potassium and glycine protocol, which favors activation of synaptic NMDARs, facilitated SEP-GluR2 diffusion into spines, whereas global activation of both synaptic and nonsynaptic NMDARs did not invoke inward SEP-GluR2 diffusion into spines. These results infer that global NMDAR activation, probably via a mechanism involving calcium entry through the NMDAR (41), may stabilize or “freeze” membrane AMPARs and inhibit lateral diffusion, whereas synaptic NMDAR activation leads to endocytosis within the spine that facilitates recruitment and clustering of AMPARs in spines. The observation that NMDA application causes the recruitment of mGFP but not of SEP-GluR2 in dendritic spines (Fig. 6, c and f) is, at first sight, counter-intuitive. The most likely explanation is that NMDA stimulates the membrane drift to drive material in spines but, simultaneously under these experimental conditions, prevents AMPAR recruitment by retaining these receptors on the shaft by a calcium entry-dependent freezing of mobile AMPARs as reported previously (41). Such a mechanism might provide a selective filter to allow the specific mobilization of plasma membrane proteins during synaptic plasticity. For example, the balance between mobilization and retention may differ under different stimulation conditions. Taken together our results suggest that naive AMPARs are inserted into the dendritic shaft plasma membrane and diffuse laterally to the PSD in a process facilitated by bulk membrane drift caused by the internalization of membrane material within the spine. We propose that this mechanism is regulated by synaptic NMDARs to control the AMPAR complement at synapses (Fig. 7c).
The number of AMPARs entering the spine by lateral recruitment would depend on the number of AMPARs exocytosed in the vicinity of the spine. Therefore, the number of receptors available to be positioned at the PSD will depend crucially on regulated AMPAR insertion (and internalization) outside of the spine and the ability of these receptors to move freely. Hence, mechanisms of receptor anchoring or release from subcompartments such as the PSD by Stargazin (50) or synaptic activity (51) are also of critical importance in the correct regulation of synaptic AMPARs.
In conclusion, our data support the hypothesis that, as for intracellular ions (3), spine topology is sufficient to form a distinct compartment that limits free lateral diffusion of membrane proteins. Our data further suggest that dynamin GTPase activity within the spine can overcome this restriction via a mechanism involving bulk inward membrane drift mediated via membrane endocytosis within the spine and consequently facilitated diffusion of membrane proteins. Thus, these results provide a novel mechanistic explanation of how the number of AMPARs at the PSD can be regulated by synaptic activity.
Supplementary Material
Acknowledgments
We are grateful to Stéphane Martin and Jonathan Hanley for their comments on the manuscript and to Atsushi Nishimune and Yasuko Nakamura for the mGFP and SEP-GluR2 constructs. We thank Emma Jenkins and Philip Rubin for mCherry tagging. We also thank Marc McNiven and Agnès Hémar for sharing their dynamin 2 constructs.
This work was supported by the Medical Research Council (MRC), the Wellcome Trust, and the European Union (GRIPPANT and ENI-NET grants).
The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. 1-5 and a Bibliography.
Footnotes
The abbreviations used are: AMPA, α-amino-3-hydroxyl-5-methyl-4-isoxazole-propionate; NMDA, N-methyl-d-aspartate; NMDAR, NMDA receptor; PSD, postsynaptic density; mGFP, membrane-anchored green fluorescent protein; FRAP, fluorescence recovery after photobleaching; FLIP, fluorescence loss in photobleaching; ΔFLIP, difference between the means of early and late FLIP sequences; TTX, tetrodotoxin; DAPV, d-2-amino-5-phosphonovalerate; Dyn2, dynamin 2; SEP, super-ecliptic pHluorin.
References
- 1.Shepherd, J. D., and Huganir, R. L. (2007) Annu. Rev. Cell Dev. Biol. 23 613-643 [DOI] [PubMed] [Google Scholar]
- 2.Harris, K. M., and Kater, S. B. (1994) Annu. Rev. Neurosci. 17 341-371 [DOI] [PubMed] [Google Scholar]
- 3.Nimchinsky, E. A., Sabatini, B. L., and Svoboda, K. (2002) Annu. Rev. Physiol. 64 313-353 [DOI] [PubMed] [Google Scholar]
- 4.Henley, J. M. (2003) Neurosci. Res. 45 243-254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Henley, J. (2001) Biochem. Soc. Trans. 29 485-488 [DOI] [PubMed] [Google Scholar]
- 6.Kim, E., and Sheng, M. (2004) Nat. Rev. Neurosci. 5 771-781 [DOI] [PubMed] [Google Scholar]
- 7.Neves, G., Cooke, S. F., and Bliss, T. V. (2008) Nat. Rev. Neurosci. 9 65-75 [DOI] [PubMed] [Google Scholar]
- 8.Malenka, R. C. (2003) Ann. N. Y. Acad. Sci. 1003 1-11 [DOI] [PubMed] [Google Scholar]
- 9.Adesnik, H., Nicoll, R. A., and England, P. M. (2005) Neuron 48 977-985 [DOI] [PubMed] [Google Scholar]
- 10.Yudowski, G. A., Puthenveedu, M. A., Leonoudakis, D., Panicker, S., Thorn, K. S., Beattie, E. C., and von Zastrow, M. (2007) J. Neurosci. 27 11112-11121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gerges, N. Z., Backos, D. S., Rupasinghe, C. N., Spaller, M. R., and Esteban, J. A. (2006) EMBO J. 25 1623-1634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Groc, L., Gustafsson, B., and Hanse, E. (2006) Trends Neurosci. 29 132-139 [DOI] [PubMed] [Google Scholar]
- 13.Tardin, C., Cognet, L., Bats, C., Lounis, B., and Choquet, D. (2003) EMBO J. 22 4656-4665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Matsuzaki, M., Ellis-Davies, G. C., Nemoto, T., Miyashita, Y., Iino, M., and Kasai, H. (2001) Nat. Neurosci. 4 1086-1092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Richards, D. A., De Paola, V., Caroni, P., Gahwiler, B. H., and McKinney, R. A. (2004) J. Physiol. 558 503-512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ashby, M. C., Maier, S. R., Nishimune, A., and Henley, J. M. (2006) J. Neurosci. 26 7046-7055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Holcman, D., and Triller, A. (2006) Biophys. J. 91 2405-2415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lu, J., Helton, T. D., Blanpied, T. A., Racz, B., Newpher, T. M., Weinberg, R. J., and Ehlers, M. D. (2007) Neuron 55 874-889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Harris, K. M., Jensen, F. E., and Tsao, B. (1992) J. Neurosci. 12 2685-2705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tanaka, J., Matsuzaki, M., Tarusawa, E., Momiyama, A., Molnar, E., Kasai, H., and Shigemoto, R. (2005) J. Neurosci. 25 799-807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Feder, T. J., Brust-Mascher, I., Slattery, J. P., Baird, B., and Webb, W. W. (1996) Biophys. J. 70 2767-2773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Axelrod, D., Koppel, D. E., Schlessinger, J., Elson, E., and Webb, W. W. (1976) Biophys. J. 16 1055-1069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ashby, M. C., De La Rue, S. A., Ralph, G. S., Uney, J., Collingridge, G. L., and Henley, J. M. (2004) J. Neurosci. 24 5172-5176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Leaney, J. L., Benians, A., Graves, F. M., and Tinker, A. (2002) J. Biol. Chem. 277 28803-28809 [DOI] [PubMed] [Google Scholar]
- 25.Ashby, M. C., Ibaraki, K., and Henley, J. M. (2004) T. Neurosci. 27 257-261 [DOI] [PubMed] [Google Scholar]
- 26.Sankaranarayanan, S., De Angelis, D., Rothman, J. E., and Ryan, T. A. (2000) Biophys. J. 79 2199-2208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Perestenko, P. V., and Henley, J. M. (2003) J. Biol. Chem. 278 43525-43532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shi, S., Hayashi, Y., Esteban, J. A., and Malinow, R. (2001) Cell 105 331-343 [DOI] [PubMed] [Google Scholar]
- 29.Aizenbud, B. M., and Gershon, N. D. (1982) Biophys. J. 38 287-293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sbalzarini, I. F., Hayer, A., Helenius, A., and Koumoutsakos, P. (2006) Biophys. J. 90 878-885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bloodgood, B. L., and Sabatini, B. L. (2005) Science 310 866-869 [DOI] [PubMed] [Google Scholar]
- 32.Kruchten, A. E., and McNiven, M. A. (2006) J. Cell Sci. 119 1683-1690 [DOI] [PubMed] [Google Scholar]
- 33.Gray, N. W., Fourgeaud, L., Huang, B., Chen, J., Cao, H., Oswald, B. J., Hemar, A., and McNiven, M. A. (2003) Curr. Biol. 13 510-515 [DOI] [PubMed] [Google Scholar]
- 34.Racz, B., Blanpied, T. A., Ehlers, M. D., and Weinberg, R. J. (2004) Nat. Neurosci. 7 917-918 [DOI] [PubMed] [Google Scholar]
- 35.Macia, E., Ehrlich, M., Massol, R., Boucrot, E., Brunner, C., and Kirchhausen, T. (2006) Dev. Cell 10 839-850 [DOI] [PubMed] [Google Scholar]
- 36.Moriyoshi, K., Richards, L. J., Akazawa, C., O'Leary, D. D., and Nakanishi, S. (1996) Neuron 16 255-260 [DOI] [PubMed] [Google Scholar]
- 37.Tamkun, M. M., O'Connell, K. M., and Rolig, A. S. (2007) J. Cell Sci. 120 2413-2423 [DOI] [PubMed] [Google Scholar]
- 38.Damke, H., Baba, T., Warnock, D. E., and Schmid, S. L. (1994) J. Cell Biol. 127 915-934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.McNiven, M. A., Kim, L., Krueger, E. W., Orth, J. D., Cao, H., and Wong, T. W. (2000) J. Cell Biol. 151 187-198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dudek, S. M., and Bear, M. F. (1993) J. Neurosci. 13 2910-2918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Borgdorff, A. J., and Choquet, D. (2002) Nature 417 649-653 [DOI] [PubMed] [Google Scholar]
- 42.Groc, L., Heine, M., Cognet, L., Brickley, K., Stephenson, F. A., Lounis, B., and Choquet, D. (2004) Nat. Neurosci. 7 695-696 [DOI] [PubMed] [Google Scholar]
- 43.Pickard, L., Noel, J., Duckworth, J. K., Fitzjohn, S. M., Henley, J. M., Collingridge, G. L., and Molnar, E. (2001) Neuropharmacology 41 700-713 [DOI] [PubMed] [Google Scholar]
- 44.Passafaro, M., Piech, V., and Sheng, M. (2001) Nat. Neurosci. 4 917-926 [DOI] [PubMed] [Google Scholar]
- 45.Blanpied, T. A., Scott, D. B., and Ehlers, M. D. (2002) Neuron 36 435-449 [DOI] [PubMed] [Google Scholar]
- 46.Cooney, J. R., Hurlburt, J. L., Selig, D. K., Harris, K. M., and Fiala, J. C. (2002) J. Neurosci. 22 2215-2224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Park, M., Salgado, J. M., Ostroff, L., Helton, T. D., Robinson, C. G., Harris, K. M., and Ehlers, M. D. (2006) Neuron 52 817-830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Harris, K. M., Fiala, J. C., and Ostroff, L. (2003) Philos. Trans. R. Soc. Lond. B Biol. Sci. 358 745-748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Matsuzaki, M., Honkura, N., Ellis-Davies, G. C., and Kasai, H. (2004) Nature 429 761-766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bats, C., Groc, L., and Choquet, D. (2007) Neuron 53 719-734 [DOI] [PubMed] [Google Scholar]
- 51.Ehlers, M. D. (2007) Neuron 55 686-689 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.