

Abstract
The B7 costimulatory molecules govern many aspects of T cell immune responses by interacting with CD28 for costimulation, but also with CTLA-4 for immune suppression. Although blockade of CTLA-4 with Ab in humans undergoing cancer immune therapy has led to some cases of inflammatory bowel disease, spontaneous animal models of colitis that depend upon modulation of B7 interactions have not been previously described. In this study, we demonstrate that mice expressing a soluble B7-2 Ig Fc chimeric protein spontaneously develop colitis that is dependent on CD28-mediated costimulation of CD4+ T cells. We show that the chimeric protein has mixed agonistic/antagonist properties, and that it acts in part by blocking the cell intrinsic effects on T cell activation of engagement of CTLA-4. Disease occurred in transgenic mice that lack expression of the endogenous B7 molecules (B7 double knock-out mice), because of the relatively weak costimulatory delivered by the chimeric protein. Surprisingly, colitis was more severe in this context, which was associated with the decreased number of Foxp3+ regulatory T cells in transgenic B7 double knock-out mice. This model provides an important tool for examining how B7 molecules and their effects on CTLA-4 modulate T cell function and the development of inflammatory diseases.
It is well known that T cells play a pivotal role in the pathogenesis of autoimmune diseases, including inflammatory bowel disease (IBD)3 (1). Because optimal T cell activation requires CD28-mediated costimulatory signals (2– 4), in addition to the primary signal through the TCR, the CD28 ligands B7-1 (CD80) and B7-2 (CD86) have been reported to be a critical factor for the induction of T cell mediated autoimmunity (5–7).
Although a complete lack of B7 costimulation abolishes T cell-mediated colitis (8, 9), it has not been fully elucidated how varying the level of B7 costimulation affects the pathogenesis of colitis. Transfer of mouse CD4+CD45RBhigh T cells to immune deficient RAG−/− recipients provides a useful model for the T cell-mediated initiation of IBD (1, 10). We recently reported that a partial reduction in costimulation in RAG−/− recipients that lacked either B7-1 or B7-2 caused accelerated colitis through the enhanced activation and differentiation of effector T cells (8). Although the transferred T cells proliferated comparably to the wild type when costimulated by a single B7 molecule, either B7-1 or B7-2, the wild type amount of IL-2 secretion, and subsequent up-regulation of CTLA-4 expression by the activated T cells, required both B7 molecules. Like CD28, CTLA-4 binds to B7-1 and B7-2, but it mediates inhibitory signals, as demonstrated by the lethal inflammatory disease that occurs in CTLA-4-deficient mice (11–13). Furthermore, in the recent clinical trials for patients with metastatic melanoma, CTLA-4 blockade caused grade III/IV severe autoimmunity in several sites, including colitis (14), which strongly suggests a critical role of CTLA-4 in maintaining peripheral tolerance and in preventing IBD. We therefore concluded that the absence of a single B7 molecule leads to accelerated colitis induction in part because of decreased CTLA-4 induction by T cells.
In addition to the regulation that is mediated by the induction of CTLA-4 expression, B7 molecules further contribute to immune regulation by participating in the homeostatic maintenance of regulatory T cells (Treg) that express CD25 and the transcription factor Foxp3. CD4+Foxp3+ Treg, which arise both in the thymus (15, 16) and in the periphery dependent upon TGF-β (17), are one of the most important elements for preventing autoimmunity (18, 19). The dependence of Treg upon B7-CD28 costimulation is demonstrated by their reduced number in mice lacking both B7 molecules, as well as by a report of exacerbated autoimmune disease in CD28-deficient mice (20). Despite the opposing functions of CTLA-4 and CD28 interacting with the same B7 molecules, optimal CTLA-4 expression and the maintenance of Treg both depend on CD28 costimulation and subsequent IL-2 production, suggesting the balance between CD28-mediated activation and CTLA-4-mediated suppression is critical for normal immune function.
In this study, we describe a new mouse model of spontaneous colitis in transgenic mice that express a chimeric, soluble B7-2 Ig Fc (B7-2 Fc) transgene (Tg). Constitutive expression of the chimeric protein in B7-2 Fc transgenic (B7-2 Fc Tg) mice induced extensive colonic inflammation as well as systemic lymphadeonopathy, indicating the critical role of normal B7 interactions in maintaining immune homeostasis in the mucosae and systemically. Our experimental results provide insights into the mechanism of T cell-mediated intestinal inflammation in these mice, and they indicate that the B7-2 Fc Tg mice are a useful model both for studying the roles of the B7 ligand-dependent functions of CTLA-4 and CD28, as well as for understanding the mechanisms for the initiation and exacerbation of colitis.
Materials and Methods
Mice
To generate B7-2 Fc Tg mice, the chimeric sequence was subcloned into the liver specific apolipoprotein E expression vector. Microinjection, embryo manipulation, and transgene screening were conducted as previously described (21–23). C57BL/6, B7-1−/−B7-2−/−, CD28−/−, Igh-6−/−, TCRβ−/−, IFN-γ−/−, and RAG1−/− mice on the C57BL/6 background were purchased from The Jackson Laboratory and bred to the B7-2 Fc Tg mice at the La Jolla Institute for Allergy and Immunology. Mice used in these studies were backcrossed 3– 6 times to the C57BL/6 background, and all Tg mice used were littermates hemizygous for the transgene. OT-II TCR transgenic mice (24) were a gift of Dr. Stephen P Schoenberger, La Jolla Institute for Allergy and Immunology (La Jolla, CA).
Abs and reagents
mAbs for CD4 (RM4-5), CD8α (53-6.7), CD11b (M1/70), CD19 (1D3), CD25 (PC61), CD40 (3/23), CD43 (1B11), MHC class II Ab (AF6-120.1), B7-1 (16-10A1), B7-2 (GL1), and CTLA-4 (UC10-4F10-11) were purchased from BD Biosciences. Anti-F4/80 (BM8), CD90.2 (Thy-1 allele specific, 53-2.1) and TCRβ (H57-597) were purchased from eBiosciences. Biotinylated Abs against CD8α (53-6.7), CD11b (M1/70), CD11c (N418), B220 (RA3-6B2), TER-119 (TER-119), NK1.1 (PK136), and CD25 (7D4) for CD4+ T cell negative selection were purchased from eBiosciences, except anti-CD25 (BD Biosciences). To purify B7-2 Ig Fc chimeric protein, conditioned medium from CHO cells expressing the extracellular domain of murine B7-2 fused to human IgG1 Fc was concentrated 15-fold using a Pellicon ultrafiltration device (Millipore) fitted with a 30 kD Molecular Weight Cut Off screen channel cassette. Filtered concentrate was bound to protein A-Sepharose at ~10 mg/ml resin, then washed with several volumes 20 mM HEPES, 100 mM NaCl (pH 7.3) before elution with neutral elution buffer (Pierce). Eluted fusion protein was dialyzed vs two 40× volumes of column wash buffer 24 h each, then filtered through 0.2 mm. Purified material was tested for endotoxin using Pyrotell LAL vials (Associates of Cape Cod) and contained 0.2 to 0.5 EU/mg.
Flow cytometric analysis
To isolate cells from the large intestine, tissues from the rectum to the cecum were opened longitudinally, washed with HBSS (Invitrogen), minced thoroughly, and then incubated with RPMI-1620 medium containing 1.5 mg/ml Collagenase type IV (Sigma Aldrich), supplemented with 10% FCS, 2-ME, and penicillin-streptomycin-glutamine (Life Technologies) for 20 –30 min at 37°C. Digested cells were filtered through a metal mesh and resuspended with 35% Percoll, followed by centrifugation at 1700 rpm for 20 min. The cell pellets were further run over a 44 –70% discontinuous Percoll gradient at 2400 rpm for 30 min. Cells recovered from the interface were used as large intestinal cells for immunostaining. Foxp3 was detected with the anti-mouse/rat Foxp3 staining kit (FJK-16s) (eBiosciences), according to the manufacturer’s protocol. For intracellular cytokine staining, ex vivo isolated or cultured cells were restimulated with PMA (2 ng/ml) and ionomycin (0.5 μg/ml) for 4 h, with Brefeldin A added for the last 2 h before staining with the BD Cytofix/Cytoperm Fixation/Permeabilization Solution Kit (BD Biosciences).
Histological scoring
For colitis scoring, three samples of 2–3 mm were obtained from the distal, middle, and proximal portions of the large intestine, and were processed for H&E staining. Samples were coded and scored in a blinded fashion. Each specimen was scored according to a simplified scoring system: 0 = normal without any focal inflammation; 1 = focal infiltration of mononuclear cells without myeloid cell accumulation; 2 = focal inflammatory cell infiltration without affecting villous structure; 3 = focal accumulation of inflammatory cells causing epithelial hyperplasia or disturbed villous structure; 4 = severe diffuse inflammation with epithelial hyperplasia and crypt abscesses; and 5 = apparent defects or ulceration of the epithelial layer in addition to the findings for score 4.
Ig isotype measurements
Ig isotypes in sera were detected using standard capture ELISA according to the manufacturer’s protocol (Southern Biotechnology Associates), with biotinylated anti-Ig Abs and streptavidin-labeled HRP (Jackson Immuno-Reseach Laboratories) for detection.
CD4+ T cell transfer
For transfer colitis experiments, CD4+CD45RBhigh cells (99.9% pure, containing <0.1% CD4+CD25+) were sorted on a FACS Vantage SE with the FACS DiVa option (BD Biosciences) after the preisolation of CD4+ cells using CD4 (L3T4) Microbeads (Miltenyi Biotec) according to the manufacturer’s protocol. Five × 105 sorted cells were resuspended with PBS and injected i.v. to RAG1−/− or B7-2 Fc Tg+ RAG1−/− recipients. In some experiments, 100 μg B7-2 Ig Fc protein was injected i.p. three times a week starting just after the transfer of donor cells.
Attenuation of colitis with CD4+CD25+ Treg
CD4+ cells were negatively selected using a combination of 6 biotinylated mAbs (anti-CD8α, CD11b, CD11c, NK1.1, B220, TER-119) and anti-biotin microbeads (Miltenyi Biotec). CD4+ T cells (92–95% purity) were further separated into CD25+ and CD25− populations using a biotinylated anti-CD25 mAb and anti-biotin microbeads (75– 85 and 96 –98% purity, respectively). One × 106 CD4+CD25+ or CD4+CD25− cells were injected to randomly selected 4-wk-old B7-1−/−B7-2−/− B7-2 Fc Tg+ mice once per week for 4 wk, and the mice were analyzed at ages 9 to 10 wk old.
In vitro CD4+ T cell culture
To test a costimulatory function of soluble B7-2 Ig Fc, CD4+CD25− T cells were isolated from B7-1−/−B7-2−/− spleens by negative selection using the combination of biotinylated mAbs mentioned above and anti-biotin microbeads (Miltenyi Biotec). CD4+CD25− cells selected from B7-1−/−B7-2−/− mice were >97% CD4+CD45RBhigh, containing <0.05% CD4+CD25+ cells (data not shown). Before the culture, 96-well plates were coated with 1.0 μg/ml anti-CD3 mAb diluted in PBS for 3– 4 h at 37°C. One × 105 CFSE-labeled T cells were stimulated in the anti-CD3 coated plate for 4 days in the presence or absence of 60 μg/ml soluble B7-2 Ig Fc or 0.5 μg/ml anti-CD28 (37.51, BD Biosciences). Proliferation and IL-2 secretion in the culture supernatants were analyzed by flow cytometry (CFSE dilution) and ELISA. In some experiments, to stimulate T cells with immobilized B7-2 Ig Fc, 1.0 μg/ml anti-CD3 (145-2C11, BD Biosciences) and 1.0 μg/ml anti-human IgG1 Fc (Jackson ImmunoResearch Laboratories) were coated onto 96-well culture plates. Some wells were further incubated with 10 μg/ml B7-2 Ig Fc for 60 min at room temperature before adding T cells with fresh medium. In coculture, experiments with CD4+CD25+ Treg, for the preparation of APCs, T lmphocytes were depleted from spleen cells using CD90.2 microbeads (Miltenyi Biotec). Selected CD90.2− cells were gamma-irradiated before use. One × 105 CFSE-labeled naive T cells, isolated as mentioned above, and 2.5 × 104 FACS-sorted CD45.1+ CD4+CD25high Treg were stimulated with 1.0 μg/ml anti-CD3 in the presence of 5 × 105 APC. The proliferation index of the CD45.1− CD90.2+ cells was calculated using FlowJo software. For T cell restimulation experiments, OT-II OVA specific T cells were primed for 3 days with 1 μM OVA peptide ISQAVHAAHAEINEAGR (Abgent) in the presence of 5 × 105 irradiated CD90.2− spleen cells as APC. For restimulation, primed T cells were restimulated with 0.1 μM OVA peptide in the presence of spleen cells from B6 RAG1−/− mice as APC. On day 3 of the restimulation culture, cells were harvested and processed for intra-cellular cytokine staining as mentioned above.
Statistical tests
An unpaired t test was used for all statistical comparisons.
Results
Soluble B7-2 Fc transgenic mice develop chronic colitis
Our previous work indicated that reduced costimulation could lead to accelerated colitis induction in the context of the T cell transfer model of colitis, in which the recipients are immune deficient. To determine how the modulation of the amount of B7-mediated costimulation might affect inflammation in the intestine and elsewhere in intact mice, we generated and analyzed Tg mice that express a soluble B7-2 Ig Fc fusion protein under the control of a liver-specific promoter. The constitutively produced soluble B7-2 Ig Fc should interact with its counterreceptors, CD28 and CTLA-4, to modify CD28-mediated co-stimulation and/or the B7 ligand-dependent regulatory function of CTLA-4. B7-2 Ig Fc Tg mice matured normally without early lethality or severe systemic inflammation. Interestingly, however, some B7-2 Ig Fc Tg mice exhibited symptoms of chronic inflammatory bowel disease, such as diarrhea and rectal prolapse, starting between the ages of 5 to 8 wk. Although the severity of the symptoms was variable, many of the diseased B7-2 Ig Fc Tg mice survived long term, carrying the symptoms of chronic colitis. Histopathological analysis demonstrated severe diffuse inflammation throughout the large intestine including the cecum of sick mice (Fig. 1A), as well as a marked enlargement of the spleen (Fig. 1B) and the mesenteric lymph nodes (MLN) (Fig. 1C). Microscopically, the colonic mucosa was hypertrophic and severely damaged by an extensive infiltrate of mononuclear cells and myeloid cells (Fig. 1D). In many cases, the inflammation involved the whole mucosal layer and lamina propria, which often contained enlarged lymphoid aggregates or follicles (Fig. 1E) that were composed mostly of B lymphocytes, as indicated by staining for CD19 (Fig. 1F). The inflammation was restricted mostly to the large intestine, although mild villous swelling was seen at the terminal part of the ileum (Fig. 1G). Later in the chronic phase in some mice, the inflammation progressed toward the submucosal layer, however, no granulomatous changes, a typical finding in human Crohn’s disease, were observed. Because the inflammation was continuous, diffuse, and restricted to the mucosa, the lesions bore a closer resemblance to those typical of ulcerative colitis.
FIGURE 1.
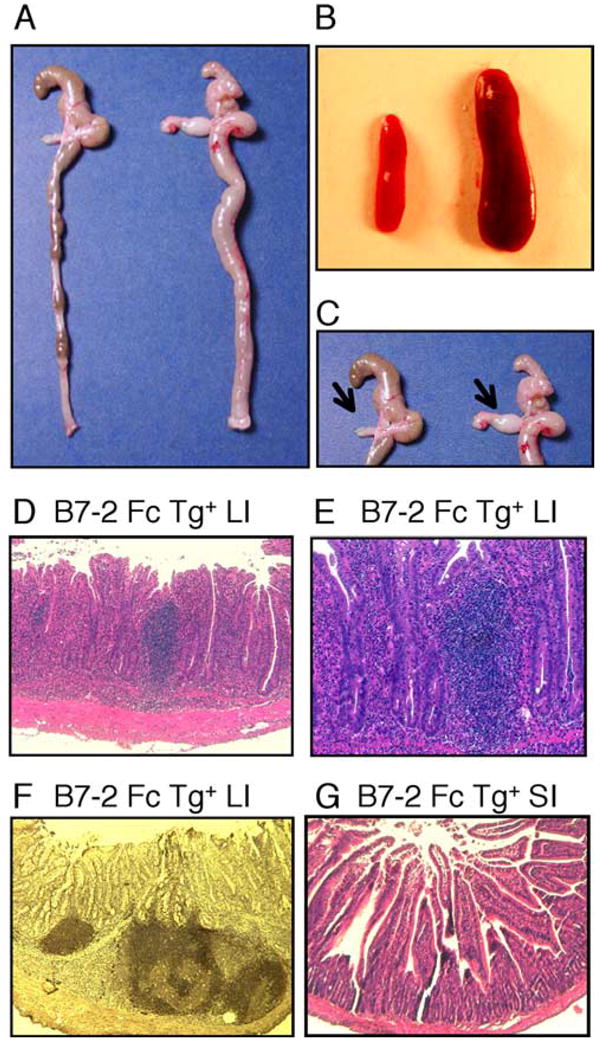
Spontaneous colitis in B7-2 Fc Tg mice. A–C, Diffuse inflammation of the whole large intestine (A), splenomegaly (B), and enlarged MLN (arrows in C) in tissues from a B7-2 Fc Tg+ littermate (right) compared with a B7-2 Fc Tg− mouse (left). D, Severe mucosal inflammation in the large intestine (LI) of the B7-2 Fc Tg+ mouse. E, A characteristic enlarged lymphoid aggregate in the large intestine of a B7-2 Fc Tg+ mouse. F, CD19 staining of the organized lymphoid follicles in the large intestine of the B7-2 Fc Tg+ mouse. G, A mild inflammation in the small intestine (SI) of the B7-2 Fc Tg+ mouse.
B cell and myeloid expansions in B7-2 Ig Fc Tg mice
Flow cytometric analyses revealed a marked expansion of CD19+ B cells especially in the peripheral lymph nodes (PLN) and MLN of the diseased B7-2 Fc Tg (average of 58% among CD45+ cells) (Fig. 2A). In contrast, the percentage of CD11b+ cells was greatly increased in the spleen. The CD11b+ cells consisted of both F4/80+ macrophages and F4/80− and Gr-1+ (data not shown) granulocytes (right panel of Fig. 2A). The migration of both macrophages and granulocytes also was characteristic of the inflamed large intestine. The absolute number of cells in spleen and MLN, was significantly increased in the diseased B7-2 Fc Tg+ mice, especially for CD11b+ cells. This increase was evident not only compared with the B7-2 Fc Tg− littermates, but also to those B7-2 Fc Tg+ mice that did not bear colitis (Fig. 2B), indicating that these cellular increases were secondary to the development of inflammation. The expanded B cell population exhibited an activated phenotype, including higher expression of MHC class II Ab molecules, B7-1, B7-2, and CD40, as shown for splenic CD19+CD11b− cells (Fig. 2C). Serum Ig was significantly increased for all the subclasses analyzed (Fig. 2D). In accordance with the histology indicating enlarged lymphoid aggregates and follicles, flow cytometric analyses also demonstrated that B cells accumulated in the large intestine (Fig. 2A). These data suggested that modification of T cell-APC interactions by the soluble B7-2 Fc fusion protein altered systemic immunity causing spontaneous expansions of B and myeloid cells in addition to an organ-based inflammation in the large intestine.
FIGURE 2.

Systemic expansion and activation of CD11b+ cells and CD19+ cells in B7-2 Fc Tg mice. A, A representative surface staining for CD11b together with CD19 (left panel) and F4/80 (right panel). Numbers show the percentage of the population gated as a square among total cells from each organ. Cells were isolated from spleen, MLN, PLN, and large intestine of B7-2 Fc Tg+ (left of each panel) and Tg− (right) littermates. B, Total cell numbers in spleen (top left), CD11b+ spleen cells (top right), total MLN cells (bottom left), and CD19+ cells in MLN (bottom right). Bars represent the averages from diseased B7-2 Fc Tg+ (left, n = 7), disease-free B7-2 Fc Tg+ (middle, n = 6), and B7-2 Fc Tg− littermates (right, n = 10). Statistical analyses were performed using a t test. (*, p < 0.05; ns, not significant) (C) I-Ab, B7-1, B7-2 and CD40 staining of splenic B cells (CD19+CD11b−) from diseased B7-2 Fc Tg+ (solid line) and disease-free B7-2 Fc Tg− (gray filled) mice. Stainings with isotype control for diseased B7-2 Fc Tg+ were shown as broken lines. D, Increased serum Ig in B7-2 Fc Tg mice (both diseased and disease-free). The concentrations of serum Ig subclasses as well as total Ig are shown. Each triangle and square represents individual B7-2 Fc Tg+ and Tg− mouse, respectively. The differences were significant (p < 0.05) in all the subclasses (shown as *).
Altered activation of CD4+ T cells in B7-2 Ig Fc Tg mice
Because the B7-2 Ig Fc should bind to two receptors, CD28 and CTLA-4, mostly expressed by T cells, we hypothesized that an excess amount of soluble B7-2 Ig Fc in the transgenic mice would modulate positive and/or negative costimulatory signals. Alteration of the activation state of T cells likely would then elicit dysregulated immune responses, including the expansions of B cells and myeloid cells that we observed. Consistent with this idea, despite a pronounced expansion of B cells in the lymphoid organs, T lymphocytes were predominantly accumulated in the large intestine of the diseased B7-2 Fc Tg mice, as determined by the ratio of CD19+ to TCRβ+ lymphocytes among the gated CD45+ cells (Fig. 3A). Although the percentage of T cells was decreased in the lymphoid organs, the absolute T cell number was not significantly changed when increases in the total cell number were accounted for (data not shown). The ratio of CD8+ to CD4+ T cells was increased in the lymphoid organs of younger B7-2 Fc Tg mice, as shown in representative data from an 8-wk-old diseased Tg mouse (Fig. 3B, left panel). This increased CD8/CD4 ratio was not evident, however, when we analyzed older, chronically diseased, Tg mice (Fig. 3B, right panel). Despite these dynamic changes in LN T cell populations, increases in CD4+ T cells in the large intestine were consistently observed in the diseased Tg mice, regardless of the age or phase of the disease. We speculate that the relative increase of CD8+ T cells compared with CD4+ T cells in the lymphoid organs at early stages may have been caused by an abnormal activation and enhanced migration of CD4+ T cells to the large intestine. In accordance with this idea, CD4+ T cells were strongly activated in lymphoid organs and the large intestine in the early phases of the disease, as indicated by staining for the activation-induced 130kDa glycoform of CD43 (Fig. 3C). These data suggested a critical role for activated and migrating CD4+ T cells in the induction of a spontaneous inflammation in the large intestine.
FIGURE 3.

Accumulation of activated CD4+ T cells in B7-2 Fc Tg mice. A, Ratio of B cells (CD19+) to T cells (TCR+ in the spleen, MLN, PLN, and large intestine). Bars represent ratios in the diseased B7-2 Fc Tg+ (filled) and Tg− (open) littermates (four mice each).*, Significant (p < 0.05) differences. B, Accumulation of CD4+ T cells in the large intestine of the B7-2 Fc Tg mice. CD4 and CD8 staining for gated TCRβ+ cells from 8- (left) and 14-wk-old (right) diseased B7-2 Fc Tg+ mice and disease-free Tg− littermates (left and right of each panel, respectively) (representative data of four mice analyzed in each group). C, Representative CD43 (1B11) staining for the CD4+ (left) and CD8+ (right) TCRβ+ cells isolated from a diseased B7-2 Fc Tg+ mouse (bold lines) and a disease-free B7-2 Fc Tg− littermate (gray filled). Stainings for the cells isolated from spleen, MLN, PLN, and large intestine are shown.
Colitis induction in Tg mice requires CD28 costimulation
To confirm the idea that the B7-2 Ig Fc chimera induced a spontaneous colitis by altering the activation and regulation of T cells, we bred the B7-2 Fc Tg mice onto the CD28−/−, TCRβ−/− or B cell deficient (Igh-6−/−) genetic backgrounds. We assessed colitis by histological scoring when the mice were between 8 to 10 wk old. As shown in Table I, none of the TCRβ−/− B7-2 Fc Tg mice exhibited any symptoms of chronic colitis, or any other inflammatory disease. Furthermore, the CD28−/− B7-2 Fc Tg mice also did not develop colitis. These data clearly indicated that spontaneous colitis in the B7-2 Fc Tg mice is T cell mediated and requires CD28 costimulation. B cells were not required, however, as 4 of 12 Igh-6−/− B7-2 Fc Tg mice developed colitis, although the disease penetrance was slightly lower than that of Igh-6 wild type B7-2 Fc Tg mice. Despite a massive cell infiltration in the large intestine, the enlarged lymphoid follicles and aggregates that were seen in the Igh-6 wild type B7-2 Fc Tg mice were not seen in the inflamed B7-2 Fc Tg mice that lack B lymphocytes (data not shown).
Table I.
Histological scores and disease penetrance of B7-2 Fc Tg micea
| Histological Scores | |||
|---|---|---|---|
| No Colitis | Mild to Moderate | Severe | |
| Strain | (0–1) | (2–3) | (4–5) |
| B7-2 Fc Tg+/− | 5/13 (39%) | 3/13 (23%) | 5/13 (39%) |
| B7-2 Fc Tg+/− TCRβ−/− | 10/10 (100%) | 0/10 (0%) | 0/10 (0%) |
| B7-2 Fc Tg+− Igh-6−/− | 8/12 (67%) | 1/12 (8.3%) | 3/12 (25%) |
| B7-2 Fc Tg+/− CD28−/− | 10/10 (100%) | 0/10 (0%) | 0/10 (0%) |
Maximal score from distal, middle, and proximal part of large intestine was used.
Lack of endogenous B7 molecules exacerbates B7-2 Ig Fc-induced colitis
We considered the possibility that additional costimulatory signals potentially provided by the B7-2 Fc fusion protein might be driving super normal T cell activation in the Tg mice, leading to colitis. If this were the case, then disease should be ameliorated when the B7-2 Fc Tg mice were crossed to mice that lack endogenous expression of both B7-1 and B7-2 (hereafter B7− mice). Surprisingly, however, the B7− B7-2 Fc Tg mice developed a more severe colitis, with a higher disease penetrance. As shown in Fig. 4A, all of the eight B7− B7-2 Fc Tg mice analyzed developed very severe colitis, as determined by higher histological scores and an ~2-fold increase in CD11b+ cells in the spleen and CD19+ cells in the MLN, when compared with diseased, B7 wild type B7-2 Fc Tg mice (Fig. 4B). These data indicate that excessive CD28-mediated costimulation is unlikely to be the main factor leading to colitis. However, because colitis pathogenesis required CD28-mediated costimulation (Table I), it is apparent that the B7-2 Fc protein must be capable of providing at least some costimulation in vivo.
FIGURE 4.

Increased disease severity in B7− B7-2 Fc Tg mice. A, Histological scores of colitis in B7+ (left, circles) and B7− (right, squares) B7-2 Fc Tg+ mice (filled) compared with Tg− littermates (open). (*, p < 0.05 between B7+ and B7− B7-2 Fc Tg+). B, Number of CD11b+ cells in the spleen (left) and CD19+ cells in the MLN (right) of diseased B7+ B7-2 Fc Tg+ mice (●), B7− B7-2 Fc Tg+ mice (■) and B7− B7-2 Fc Tg− littermates (□). C, Serum IgG1, IgG2a and IgM concentrations from the B7-2 Fc Tg+ (filled) and B7-2 Fc Tg− (open) littermates that are B7+ (circles) and B7− (squares).*, Significant (p < 0.05) differences.
The amount of serum Ig exhibited a distinct pattern in the B7− B7-2 Fc Tg mice. IgM was significantly elevated in the B7− B7-2 Fc Tg mice (n = 9), even compared with diseased B7 wild type B7-2 Fc Tg mice (n = 13) (average 377.7 compared with 214.9 μg/ml, p = 0.042). Serum IgG1 and IgG2a concentrations were quite low (27.2 and 28.7 μg/ml, respectively) in B7− B7-2 Fc Tg mice due to impaired costimulation, although they were still higher than in healthy B7− mice lacking the B7-2 transgene (10.9 and 3.9 μg/ml, respectively) (Fig. 4C). These data indicate that the in vivo positive effects of the B7-2 Fc fusion protein on CD28 mediated T cell costimulation in the absence of endogenous B7 expression were relatively weak, as they were not sufficient to restore B cell class switching, although they could trigger T cell mediated colitis.
The B7-2 Ig Fc has a minimal affect on Treg function in vitro
One important role of B7 costimulation is to maintain the peripheral Treg population (20). As we expected from the data showing a minimal effect on B cell class switching by the B7-2 Ig Fc, Treg were severely reduced in diseased B7− B7-2 Fc Tg mice in spleen, MLN and PLN (Fig. 5, A and B).
FIGURE 5.

A reduced percentage of Treg contributes to the severe colitis in B7− B7-2 Fc Tg mice. A, Representative stainings for CD25+ Foxp3+ Treg in the spleen (top) and MLN (bottom) from the B7+ (left) and B7− (right) background B7-2 Fc Tg+ (left in each) and B7-2 Fc Tg− (right in each) mice. B, Percentages of Foxp3+ cells averaged from four mice each from the B7+ or B7− background, diseased B7-2 Fc Tg+ or B7-2 Fc Tg− mice.*, Significant (p < 0.05) differences. C, In vitro coculture experiment showing the suppression of T cell proliferation by Treg in the presence or absence of the B7-2 Ig Fc. CFSE dilutions of responder CD4+CD45RBhigh cells (gated as CD45.1−) cocultured without Treg (gray area), with Treg (solid line), and with Treg in the presence of B7-2 Ig Fc (dotted line) are shown. Numbers indicate the proliferation indexes. D, Injection of CD4+CD25+ cells but not CD4+CD25− cells attenuated the severity of colitis in the B7− B7-2 Fc Tg mice. Averaged histological scores from three parts of the large intestines of B7− B7-2 Fc Tg+ mice injected with CD4+CD25− (n = 4) or CD4+CD25+ (n = 4) cells. (*, p < 0.05).
To determine whether the B7-2 Ig Fc inhibited the function of Treg, we performed conventional cocultures of naive T lymphocytes and Treg. In this experiment, CFSE-labeled, sorted CD4+CD45RBhigh cells were cultured with T cell depleted irradiated spleen cells as APC, in the presence or absence of sorted, CD45 congenic CD4+CD25high cells. The T cells were anti-CD3 stimulated, and the proliferation indexes were calculated from the CFSE dilution (Fig. 5C). The data indicate that the addition of the B7-2 Ig Fc did not affect the in vitro suppressive function of Tregs (proliferation indices with Treg = 2.48 and 2.41, with or without the B7-2 Ig Fc, respectively, compared with 3.83 without Treg).
As shown above, the B7− B7-2 Fc Tg mice had a decreased percentage of Treg compared with their B7 wild type B7-2 Fc Tg counterparts, likely due to an amount of B7-mediated costimulation that was not sufficient for Treg homeostasis. To determine whether a reduction in Treg could be an important mechanism for the enhanced colitis pathogenesis in B7− B7-2 Fc Tg mice, we injected CD4+CD25+ cells once a week for 4 wk, starting when the mice were 4 wk old. Repeated injections of CD4+CD25+ cells were conducted, because an efficient expansion of Treg was not expected in the B7− B7-2 Fc Tg mice. As shown in Fig. 5D, injections of CD4+CD25+ cells, but not CD4+CD25− cells, attenuated the disease severity to the levels in the B7 wild type B7-2 Fc Tg mice.
The B7-2 Ig Fc acts on naive CD4+ T cells
To address the question if the B7-2 Ig Fc enhances CD4+ T cell mediated colitis induction independently of Treg, we tested the effect of the B7-2 Ig Fc in the CD4+CD45RBhigh T cell transfer model of colitis. In this model, transfer of naive, CD4+ CD45RBhigh T cells in the absence of Treg induces chronic colitis in immune deficient RAG1−/− mice by 4 to 8 wk post transfer. Interestingly, injection of a soluble B7-2 Ig Fc fusion protein to the recipients after CD4+CD45RBhigh T cell transfer dramatically accelerated colitis induction, assessed by weight loss (Fig. 6A) and severe diarrhea. These data were confirmed by a similar acceleration of disease induction in RAG1−/− recipients that express the B7-2 Fc transgene (B7-2 Fc Tg+). In accordance with the T cell dependence of spontaneous colitis in B7-2 Fc Tg mice, none of the B7-2 Fc Tg+ RAG1−/− developed colitis in the absence of donor CD4+CD45RBhigh T cells (data not shown). Only 6 days after T cell transfer, however, there was a massive migration of mononuclear and myeloid cells in the B7-2 Fc Tg+ RAG1−/− recipients (Fig. 6B), while only a few mononuclear cells were observed in the control B7-2 Fc Tg− RAG1−/− recipients. These data indicated that the B7-2 Ig Fc could accelerate colitis mediated by naive CD4+ T cells in the absence of the other arms of the adaptive immune system, although these mice showed no signs of disease in the absence of CD4+ T cells.
FIGURE 6.

The soluble B7-2 Ig Fc accelerates colitis independently of Treg. A, CD4+CD45RBhigh cells were injected to RAG1−/− (circles) or B7-2 Fc Tg+ RAG1−/− (squares) recipients. In a separate group of four RAG1−/− recipients, the B7-2 Ig Fc (100 μg) was injected three times a week for 2 wk (○). The average relative body weight of each group is shown. B, Representative histology (×40 magnification) of the large intestine 6 days post transfer of CD4+CD45RBhigh cells into B7-2 Fc Tg− RAG1−/− (left) and B7-2 Fc Tg+ RAG1−/− (right) recipients. Arrows represent infiltration of mononuclear cells into the mucosa.
CTLA-4 blockade by the B7-2 Ig Fc enhances T cell differentiation
To further analyze the function of the B7-2 Ig Fc protein in T cell activation, we stimulated naive CD4+ T cells with plate-bound anti-CD3 mAb in the presence or absence of the soluble B7-2 Ig Fc. Unexpectedly, there was no effect of the soluble chimera on T cell proliferation (Fig. 7A) and IL-2 production (Fig. 7B). A similar experiment using OT-II OVA specific T cells cultured with OVA peptide and APC also gave no costimulatory effect by the soluble B7-2 Ig Fc protein. Furthermore, the soluble B7-2 Ig Fc did not restore costimulation in the culture with B7− APC (data not shown). These data indicate that the weak costimulatory activity of the B7-2 Ig Fc is not sufficient to activate T cells in vitro during priming when added in a soluble form. To confirm an interaction between the B7-2 Ig Fc and CD28 causing costimulation, we immobilized the B7-2 Ig Fc together with anti-CD3 mAb in tissue culture wells. To have the same density of the anti-CD3 mAb in the presence or absence of the B7-2 Ig Fc, we coated the plate first with anti-CD3 mAb together with an anti-human IgG1 Fc mAb to capture the fusion protein. Some of the wells were further incubated with the B7-2 Ig Fc before adding T cells. Although it was not effective when soluble, when immobilized, the B7-2 Ig Fc clearly enhanced T cell proliferation and IL-2 production (Fig. 7, C and D).
FIGURE 7.

Weak agonistic and antagonistic properties of the B7-2 Ig Fc chimera. A and B, Soluble B7-2 Ig Fc. CFSE-labeled naive CD4+ T cells were stimulated for 4 days with 1.0 μg/ml plate-bound anti-CD3 mAb. A, CFSE dilutions in the presence (solid line) or absence (gray-filled) of 60 μg/ml soluble B7-2 Ig Fc (top) or 0.5 μg/ml soluble anti-CD28 mAb (bottom) are shown. B, IL-2 concentration in the supernatant was measured from cultures in Fig. 7A. C and D, Plate-bound B7-2 Ig Fc. CFSE-labeled naive CD4+ T cells were stimulated with plate-bound anti-CD3 or anti-CD3 with B7-2 Ig Fc for 4 days. C, CFSE dilution in the presence (solid line) or absence (gray-filled) of immobilized B7-2 Ig Fc is shown. These data are representative of two independent experiments. D, IL-2 concentration in the supernatant from a culture in the presence (×) or absence (●) of the immobilized B7-2 Ig Fc. E, The B7-2 Ig Fc inhibits Ab binding to CTLA-4 but not to CD28. Pre-activated CD4+ T cells (for CTLA-4) or naive CD4+ T cells (for CD28) were incubated with 100 μg/ml B7-2 Ig Fc (solid line) for 10 min before staining with PE-conjugated anti-CTLA-4 (top) or anti-CD28 (bottom) Ab. Cells preincubated with 100 μg/ml unconjugated anti-CTLA-4 (for CTLA-4) and naive CD4+ T cells isolated from CD28 deficient mice were used as negative controls, respectively (shown as broken lines). CTLA-4 or CD28 expression without pre-incubation is shown by gray-filled area. F, OT-II OVA specific CD4+ T cells were primed and restimulated in the presence of 0.1 μM OVA. B7-2 Ig Fc (left), anti-CTLA-4 (middle), or neither of them (right) was added in both primary and secondary cultures. Intracellular IFN-γ staining after a brief activation with PMA; ionomycin in the presence of Brefeldin A is shown. Data for all experiments in the figure are representative of at least two experiments.
We hypothesized that CTLA-4 blockade by the B7-2 Ig Fc could be at least part of the mechanism for dysregulated CD4+ T cell responses and colitis. Preincubation of activated CD4+ T cells with a B7-2 Ig Fc completely inhibited surface staining with a PE-labeled anti-CTLA-4 mAb, although it did not block staining with an anti-CD28 mAb (Fig. 7E). These data indicated that the B7-2 Ig Fc could bind to CTLA-4 and compete for the binding site of this blocking mAb, but do not by themselves demonstrate functional role for this interaction.
It has been reported that CTLA-4 induces anergy and inhibits T cell differentiation. We therefore conducted a culture experiment to test whether the soluble B7-2 Ig Fc could facilitate T cell differentiation, although it has only limited effect on priming. After priming with irradiated spleen cells, OT-II T cells were rested and restimulated with total spleen cells from RAG1−/− mice for 3 days. After a third stimulation with PMA and ionomycin for 4 h, T cells that were cultured during priming and restimulation either with the B7-2 Ig Fc or an anti-CTLA-4 blocking Ab were more capable of producing IFN-γ detected by intracellular cytokine staining (Fig. 7F). These data demonstrated that the B7-2 Ig Fc efficiently bound to CTLA-4 and caused a similar effect on T cell differentiation to generate IFN-γ producing cells as a CTLA-4 blocking Ab.
The role of IFN-γ in colitis induction
Effector cytokines produced by differentiated T cells are important for the induction of T cell mediated inflammatory diseases, including colitis. Intracellular staining of the ex vivo restimulated CD4+TCRβ+ cells for IFN-γ, IL-4, IL-17, IL-10 (Fig. 8A), IL-5, IL-13, TNF-α, and IL-2 (data not shown) revealed that a significantly higher percentage of CD4+TCRβ+ cells in the spleen, MLN, and large intestine of the diseased B7-2 Fc Tg were capable of producing IFN-γ, compared with those from the transgenic negative littermates. Increases in other proinflammatory cytokines were less consistent, and there was no evidence for increased Th2 cytokine production. In accordance with these data, B7-2 Fc Tg mice that were crossed onto IFN-γ deficient background (IFN-γ−/− B7-2 Fc Tg) did not develop colitis. None of the 18 IFN-γ−/− B7-2 Fc Tg exhibited any symptom of colitis, which was confirmed by histological scores for colonic specimens (Fig. 8B, top). Interestingly, however, IFN-γ−/− B7-2 Fc Tg that also are deficient for B7-1 and B7-2 developed severe colitis, as shown in Fig. 8C. Interestingly, this colitis exhibited different histological features represented by a vigorous accumulation of mononuclear cells in the submucosal area as a prominent feature. These data indicate that while IFN-γ is important for the induction of colonic inflammation, this requirement is diminished if the host lacks the elements needed for normal immune regulation, including Tregs. Therefore endogenous B7 molecules are critical factors for the maintenance of Treg and the integrity of immune tolerance.
FIGURE 8.

IFN-γ dependent and independent colitis induction in the B7-2 Fc Tg mice. A, Cells were isolated from spleen, MLN and LI (as indicated) from 8-wk-old diseased B7-2 Fc Tg+ mice (top) or disease-free B7-2 Fc Tg− littermates. Intracellular stainings of CD4+TCRβ+ cells for IFN-γ/IL-17 (left) and IL-4/IL-10 (right) after ex vivo restimulation are shown. Data are representative from eight diseased B7-2 Fc Tg+ and four B7-2 Fc Tg− mice. B, Averaged histological scores from B7+ (top) or B7− (bottom) background IFN-γ−/− B7-2 Fc Tg+ mice. C, Representative H&E stainings from a middle part of large intestine of B7+ (left) and B7− (right) background IFN-γ−/− B7-2 Fc Tg+ mice. Scale bars showing 1 mm are placed at the left bottom corner of each figure.
Discussion
In this study, we characterized a new model for mouse colitis that exhibited some symptoms similar to human IBD. The constitutive production of a soluble mouse B7-2-human IgG1 Fc fusion protein by liver cells induced chronic colitis and systemic lymphadenopathy. The inflammation observed was a diffuse colitis that involved the entire large intestine, from the cecum to the rectum, but that was mostly restricted to the large intestine. Histological analysis demonstrated a very severe mucosal inflammation with crypt abscesses and enlarged lymphoid follicles and aggregates. Although these features are often observed in human ulcerative colitis, we are not proposing that this model provides a completely faithful representation of human ulcerative colitis.
In the B7-2 Fc Tg mice, there were indications of systemic immune activation, including an increased number of myeloid cells in the spleen, a marked expansion and activation of B cells in the lymphoid organs, and elevated serum Ig. The increased B cell activation was caused by abnormal T cell activation and differentiation. Although B cell activation and Ig may have contributed to disease severity, B lymphocytes were dispensable for colitis induction. On the contrary, the absence of colitis in the TCRβ−/− and CD28−/− B7-2 Fc Tg mice, and the effects of the B7-2 Fc in the transfer model, clearly indicated that the colitis induction in the B7-2 Fc Tg mice required CD4 T cell activation in the presence of B7-CD28 costimulation. We cannot exclude the possibility that other factors contribute to colitis pathogenesis, including interaction of the Fc portion of the transgene with Fc receptors expressed innate immune cells. Clearly, however, this cannot by itself cause disease in the absence of CD4+ T cell activation.
Because B7-2 binds to both CD28 and CTLA-4, which play opposite but interdependent roles in T cell activation, we examined three hypotheses to understand the mechanism for dysregulated CD4+ T cell activation, differentiation and eventually, inflammation. The first hypothesis was that a B7-2 Ig Fc might give an additional and excess costimulatory signal to T cells through CD28, which might enhance T cell activation and survival. The strongest argument against this is the finding of increased disease severity and penetrance in B7− B7-2 Fc Tg mice. This led us to consider whether the B7-2 Fc protein had any agonistic activity. The presence of colitis in the B7− B7-2 Fc Tg mice, in contrast to the absence of disease in the CD28−/− B7-2 Fc Tg mice, clearly demonstrated an agonistic function of the B7-2 Ig Fc in vivo. Our in vitro experiments also were consistent with an agonistic function, but only when the B7-2 Ig Fc was immobilized on APC. We speculate that the fusion protein could associate with APC by a relatively weak affinity between the human IgG1 Fc and mouse Fc receptors. However, the costimulatory signal generated by the B7-2 Ig Fc in vivo in the B7− B7-2 Fc Tg mice was not sufficient even for the basal level functions of B7 costimulation, required for maintaining B cell Ig class switching (25) and a normal population of Treg (20). In summary, the evidence suggests that the fusion protein has weak costimulatory activity, but the dissociation between the total intensity of CD28 costimulation and the disease severity, comparing B7-2 Fc Tg mice on the B7− and B7+ backgrounds, indicated that an excess of B7-CD28 costimulation was not the main mechanism for the induction of colitis.
Because both B7-1 and B7-2 bind to CTLA-4 with a much higher affinity than they do to CD28 (26, 27), it would be reasonable to propose that the soluble B7-2 Ig Fc has a greater affect on CTLA-4 function than on CD28 function. The B7-2 Ig Fc bound to CTLA-4, as demonstrated by the inhibition of CTLA-4 Ab staining. Therefore it is possible that the B7-2 Ig Fc acted primarily by attenuating the function of Treg, by blocking constitutively expressed CTLA-4 (28, 29). There is abundant evidence that CD4+Foxp3+ Treg are essential for the maintenance of peripheral tolerance. The role of CTLA-4 in the function of Treg is controversial. In one study, it was reported that constitutively expressed CTLA-4 is required for the function of Treg, because injections of anti-CTLA-4 in the recipients accelerated colitis induction in the CD4+CD45RBhigh transfer model, but only in the presence of Treg (29). However, in a more recent study, Treg isolated from the CTLA-4 deficient mice successfully prevented colitis in the same model (30).
Regardless of the role of CTLA-4 in Treg function, our CD4+CD45RBhigh transfer experiment, either with coinjections of the B7-2 Ig Fc, or in the B7-2 Fc Tg RAG1−/− recipients, clearly demonstrated that the B7-2 Ig Fc can enhance colitis induction even in the absence of Treg. It should be noted that disease developed in these recipients as early as 1 wk post transfer, well before we have observed the generation of adaptive Treg, or Foxp3+ cells, from the naive CD4 T cell population (data not shown). Furthermore, in the B7-2 Fc Tg B7+ mice, the CD4+Foxp3+ Treg number was increased significantly, with an equivalent level of Foxp3 expression, compared with wild-type littermates. Because the expression level of Foxp3 is directly correlated with the regulatory function of Treg (19), it was expected that the Treg in the B7-2 Fc Tg B7+ mice would be functionally normal. In accordance with this, the addition of B7-2 Ig Fc did not affect the regulatory capacity of Treg in a coculture system. In summary, these data led us to conclude that Treg are quantitatively and qualitatively normal in the B7-2 Fc Tg B7+ mice, and therefore that impaired Treg function is not the main cause of colitis induction.
The last hypothesis we examined was that the B7-2 Ig Fc might enhance naive T cell differentiation, by affecting the T cell autonomous regulatory function of CTLA-4. A number of studies demonstrated that the engagement of CTLA-4 molecule suppresses IL-2 expression and also restricts T cell clonal expansion by limiting cell cycle progression (13, 31–33), which forms a checkpoint before T cells undergo differentiation into effector cells. The results from in vitro experiments demonstrated that the addition of anti-CTLA-4 blocking mAb, and to a similar extent, the addition of the B7-2 Ig Fc, significantly enhanced the induction of IFN-γ producing cells after secondary culture with RAG1−/− spleen cells. These data, together with the data indicating that the soluble B7-2 Ig Fc does not give excess costimulation to T cells, suggested that the blocking of CLTA-4 by the B7-2 Ig Fc could be a principal mechanism for colitis induction. In the context of colitis models, the importance of the cell autonomous regulation of activated T cells by CTLA-4 has recently been demonstrated in the CD4 T cell transfer model (8). The importance of CTLA-4 expression in colitis prevention is further demonstrated by the findings in cancer patients that developed intestinal inflammation following treatment with CTLA-4 blocking Abs.
The physical form of the B7-2 ectodomain in solution might be a key mechanism for explaining the blocking rather than agonistic function of the fusion protein when interacting with CTLA-4. Both B-1 and B7-2 are known to exhibit a complex dimer interface that is critical for a stable ligand-receptor interaction and signaling (34). However, in solution, the receptor binding domain of human B7-2 is exclusively monomeric (35), although it still binds tightly to monomeric and homodimeric CTLA-4. This is in contrast to B7-1, which is in a rapid equilibrium between monomer and dimer (36). We speculate that an excess amount of the soluble form of the B7-2 Ig Fc in the Tg mice, with the B7-2 ectodomain essentially in monomeric form at the ends of the Ig Fc arms, may inhibit the binding between dimeric endogenous B7 molecules and CTLA-4, which would block the inhibitory signal to activated T cells.
Recent studies demonstrated that the ligation of B7 molecules by CTLA-4 could constitute a reverse signaling pathway by which B7 molecules signal to APC to induce immune suppression through the induction of IDO (37). IDO is a tryptophan-degrading enzyme that could possibly suppresses T cell proliferation by causing a deprivation of the essential amino acid tryptophan. However, IDO deficient mice do not exhibit an evident autoimmune phenotype, including no evidence for colitis (data not shown). Therefore, while the reverse signaling mechanism could contribute to pathogenesis, the impaired T cell autonomous regulation of proliferation and differentiation is likely to be the major factor responsible for colitis.
There are several explanations for the surprising finding that the B7-2 Fc Tg mice deficient for endogenous B7 molecules exhibited a much more severe disease phenotype. First, although we do not have evidence indicating that the B7-2 Ig Fc affects the regulatory function of Treg, the number of Treg is severely reduced compared with Tg mice on the wild type background, probably because the homeostasis of Treg is dependent upon the normal level of B7-CD28 costimulation. Consistent with the importance of reduced Treg number in the B7− B7-2 Fc Tg mice, injections of Treg could diminish disease severity in these mice. Second, the level of CTLA-4 on cells other than Treg should be lower in the B7− B7-2 Fc Tg mice, because the maximal level of CTLA-4 expression requires full B7-CD28 costimulation (8, 38). Therefore, blocking of the suppressive function of CTLA-4 by the B7-2 Ig Fc should be more efficient in the B7− B7-2 Fc Tg mice. Lastly, as mentioned above, ligation of endogenous B7 molecules leading to the induction of IDO could also be a disease-limiting factor, although by itself IDO deficiency does not cause colitis.
Although colitis in B7 wild-type B7-2 Fc Tg mice required IFN-γ, disease that developed in the IFN-γ−/− B7− B7-2 Fc Tg mice indicated that the requirement for particular proinflammatory cytokines critically depends upon the expression of costimulatory molecules and regulatory elements in the host. In the absence of IFN-γ in the B7− mice, despite severe inflammation and a markedly enlarged colon, mucosal mononuclear cell infiltration and hyperplasia were decreased. The comparative analysis of inflammation in the presence or absence of IFN-γ should allow us to dissect the roles of IFN-γ in the cascade of events leading to chronic inflammation in the intestine.
In conclusion, the chronic colitis spontaneously induced the B7-2 Fc Tg mice provides a new and very useful model for dissecting the mechanisms of inflammatory disease induction in the intestine. Advantages of this model are that disease occurs spontaneously in the absence of any genetic deficiency, but also, disease can be studied in an acute situation following T cell transfer. In this model, disease penetrance and severity are determined by the balance between progressive factors, such as proinflammatory cytokines, and disease limiting factors, such as Treg. In the B7 wild-type B7-2 Fc Tg mice that exhibited an intermediate disease phenotype and penetrance, endogenous elements controlled disease manifestation and severity. The lack of a progressive factor, such as IFN-γ, abolished the disease. In contrast, the lack of endogenous B7 molecules exaggerated the disease, perhaps due to a deficiency in Treg. Although the significance of soluble B7 molecules in the context of normal physiology has not been determined, a soluble, truncated form of B7-2, capable of binding to both CD28 and CTLA-4, can be detected in human serum (39). Further studies of the amount and variability of these soluble molecules could help in understanding the pathogenesis of some forms of IBD.
Footnotes
This work was supported by National Institutes of Health Grant PO1 DK46763 (to M.K.) and a Career Development Award from the Crohn’s and Colitis Foundation of America (to G.K.).
Abbreviations used in this paper: IBD, inflammatory bowel disease; Treg, regulatory T cell; Tg, transgene; MLN, mesenteric lymph node; PLN, peripheral lymph node.
Disclosures
G. Kim, O. Turovskaya, M. Levin, and M. Kronenberg have no conflicts of interest, while authors F. Byrne, J. Whoriskey, T. Horen, and J. McCabe are employees of Amgen, Inc.
References
- 1.Powrie F, Leach MW, Mauze S, Caddle LB, Coffman RL. Phenotypically distinct subsets of CD4+ T cells induce or protect from chronic intestinal inflammation in C. B-17 scid mice. Int Immunol. 1993;5:1461–1471. doi: 10.1093/intimm/5.11.1461. [DOI] [PubMed] [Google Scholar]
- 2.Jenkins MK, Taylor PS, Norton SD, Urdahl KB. CD28 delivers a costimulatory signal involved in antigen-specific IL-2 production by human T cells. J Immunol. 1991;147:2461–2466. [PubMed] [Google Scholar]
- 3.Lindsten T, June CH, Ledbetter JA, Stella G, Thompson CB. Regulation of lymphokine messenger RNA stability by a surface-mediated T cell activation pathway. Science. 1989;244:339–343. doi: 10.1126/science.2540528. [DOI] [PubMed] [Google Scholar]
- 4.Fraser JD, Irving BA, Crabtree GR, Weiss A. Regulation of interleukin-2 gene enhancer activity by the T cell accessory molecule CD28. Science. 1991;251:313–316. doi: 10.1126/science.1846244. [DOI] [PubMed] [Google Scholar]
- 5.Chang TT, Jabs C, Sobel RA, Kuchroo VK, Sharpe AH. Studies in B7-deficient mice reveal a critical role for B7 costimulation in both induction and effector phases of experimental autoimmune encephalomyelitis. J Exp Med. 1999;190:733–740. doi: 10.1084/jem.190.5.733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lenschow DJ, Ho SC, Sattar H, Rhee L, Gray G, Nabavi N, Herold KC, Bluestone JA. Differential effects of anti-B7-1 and anti-B7-2 monoclonal antibody treatment on the development of diabetes in the nonobese diabetic mouse. J Exp Med. 1995;181:1145–1155. doi: 10.1084/jem.181.3.1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Knoerzer DB, Karr RW, Schwartz BD, Mengle-Gaw LJ. Collagen-induced arthritis in the BB rat: prevention of disease by treatment with CTLA-4-Ig. J Clin Invest. 1995;96:987–993. doi: 10.1172/JCI118146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kim G, Levin M, Schoenberger SP, Sharpe A, Kronenberg M. Paradoxical effect of reduced costimulation in T cell-mediated colitis. J Immunol. 2007;178:5563–5570. doi: 10.4049/jimmunol.178.9.5563. [DOI] [PubMed] [Google Scholar]
- 9.Liu Z, Geboes K, Hellings P, Maerten P, Heremans H, Vandenberghe P, Boon L, van Kooten P, Rutgeerts P, Ceuppens JL. B7 interactions with CD28 and CTLA-4 control tolerance or induction of mucosal inflammation in chronic experimental colitis. J Immunol. 2001;167:1830–1838. doi: 10.4049/jimmunol.167.3.1830. [DOI] [PubMed] [Google Scholar]
- 10.Morrissey PJ, Charrier K, Braddy S, Liggitt D, Watson JD. CD4+ T cells that express high levels of CD45RB induce wasting disease when transferred into congenic severe combined immunodeficient mice: disease development is prevented by cotransfer of purified CD4+ T cells. J Exp Med. 1993;178:237–244. doi: 10.1084/jem.178.1.237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Waterhouse P, Penninger JM, Timms E, Wakeham A, Shahinian A, Lee KP, Thompson CB, Griesser H, Mak TW. Lymphoproliferative disorders with early lethality in mice deficient in Ctla-4. Science. 1995;270:985–988. doi: 10.1126/science.270.5238.985. [DOI] [PubMed] [Google Scholar]
- 12.Tivol EA, Borriello F, Schweitzer AN, Lynch WP, Bluestone JA, Sharpe AH. Loss of CTLA-4 leads to massive lymphoproliferation and fatal multiorgan tissue destruction, revealing a critical negative regulatory role of CTLA-4. Immunity. 1995;3:541–547. doi: 10.1016/1074-7613(95)90125-6. [DOI] [PubMed] [Google Scholar]
- 13.Chambers CA, Krummel MF, Boitel B, Hurwitz A, Sullivan TJ, Fournier S, Cassell D, Brunner M, Allison JP. The role of CTLA-4 in the regulation and initiation of T-cell responses. Immunol Rev. 1996;153:27–46. doi: 10.1111/j.1600-065x.1996.tb00919.x. [DOI] [PubMed] [Google Scholar]
- 14.Phan GQ, Yang JC, Sherry RM, Hwu P, Topalian SL, Schwartzentruber DJ, Restifo NP, Haworth LR, Seipp CA, Freezer LJ, et al. Cancer regression and autoimmunity induced by cytotoxic T lymphocyte-associated antigen 4 blockade in patients with metastatic melanoma. Proc Natl Acad Sci USA. 2003;100:8372–8377. doi: 10.1073/pnas.1533209100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hori S, Nomura T, Sakaguchi S. Control of regulatory T cell development by the transcription factor Foxp3. Science. 2003;299:1057–1061. doi: 10.1126/science.1079490. [DOI] [PubMed] [Google Scholar]
- 16.Fontenot JD, Gavin MA, Rudensky AY. Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nat Immunol. 2003;4:330–336. doi: 10.1038/ni904. [DOI] [PubMed] [Google Scholar]
- 17.Chen W, Jin W, Hardegen N, Lei KJ, Li L, Marinos N, McGrady G, Wahl SM. Conversion of peripheral CD4+CD25− naive T cells to CD4+CD25+ regulatory T cells by TGF-β induction of transcription factor Foxp3. J Exp Med. 2003;198:1875–1886. doi: 10.1084/jem.20030152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Asano M, Toda M, Sakaguchi N, Sakaguchi S. Autoimmune disease as a consequence of developmental abnormality of a T cell subpopulation. J Exp Med. 1996;184:387–396. doi: 10.1084/jem.184.2.387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kim JM, Rasmussen JP, Rudensky AY. Regulatory T cells prevent catastrophic autoimmunity throughout the lifespan of mice. Nat Immunol. 2007;8:191–197. doi: 10.1038/ni1428. [DOI] [PubMed] [Google Scholar]
- 20.Salomon B, Lenschow DJ, Rhee L, Ashourian N, Singh B, Sharpe A, Bluestone JA. B7/CD28 costimulation is essential for the homeostasis of the CD4+CD25+ immunoregulatory T cells that control autoimmune diabetes. Immunity. 2000;12:431–440. doi: 10.1016/s1074-7613(00)80195-8. [DOI] [PubMed] [Google Scholar]
- 21.Simonet WS, Hughes TM, Nguyen HQ, Trebasky LD, Danilenko DM, Medlock ES. Long-term impaired neutrophil migration in mice overexpressing human interleukin-8. J Clin Invest. 1994;94:1310–1319. doi: 10.1172/JCI117450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Simonet WS, Lacey DL, Dunstan CR, Kelley M, Chang MS, Luthy R, Nguyen HQ, Wooden S, Bennett L, Boone T, et al. Osteoprotegerin: a novel secreted protein involved in the regulation of bone density. Cell. 1997;89:309–319. doi: 10.1016/s0092-8674(00)80209-3. [DOI] [PubMed] [Google Scholar]
- 23.Brinster RL, Chen HY, Trumbauer ME, Yagle MK, Palmiter RD. Factors affecting the efficiency of introducing foreign DNA into mice by microinjecting eggs. Proc Natl Acad Sci USA. 1985;82:4438–4442. doi: 10.1073/pnas.82.13.4438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Barnden MJ, Allison J, Heath WR, Carbone FR. Defective TCR expression in transgenic mice constructed using cDNA-based α- and β-chain genes under the control of heterologous regulatory elements. Immunol Cell Biol. 1998;76:34–40. doi: 10.1046/j.1440-1711.1998.00709.x. [DOI] [PubMed] [Google Scholar]
- 25.Borriello F, Sethna MP, Boyd SD, Schweitzer AN, Tivol EA, Jacoby D, Strom TB, Simpson EM, Freeman GJ, Sharpe AH. B7-1 and B7-2 have overlapping, critical roles in immunoglobulin class switching and germinal center formation. Immunity. 1997;6:303–313. doi: 10.1016/s1074-7613(00)80333-7. [DOI] [PubMed] [Google Scholar]
- 26.Linsley PS, Greene JL, Brady W, Bajorath J, Ledbetter JA, Peach R. Human B7-1 (CD80) and B7-2 (CD86) bind with similar avidities but distinct kinetics to CD28 and CTLA-4 receptors. Immunity. 1994;1:793–801. doi: 10.1016/s1074-7613(94)80021-9. [DOI] [PubMed] [Google Scholar]
- 27.Greene JL, Leytze GM, Emswiler J, Peach R, Bajorath J, Cosand W, Linsley PS. Covalent dimerization of CD28/CTLA-4 and oligomerization of CD80/CD86 regulate T cell costimulatory interactions. J Biol Chem. 1996;271:26762–26771. doi: 10.1074/jbc.271.43.26762. [DOI] [PubMed] [Google Scholar]
- 28.Takahashi T, Tagami T, Yamazaki S, Uede T, Shimizu J, Sakaguchi N, Mak TW, Sakaguchi S. Immunologic self-tolerance maintained by CD25+CD4+ regulatory T cells constitutively expressing cytotoxic T lymphocyte-associated antigen 4. J Exp Med. 2000;192:303–310. doi: 10.1084/jem.192.2.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Read S, Malmstrom V, Powrie F. Cytotoxic T lymphocyte-associated antigen 4 plays an essential role in the function of CD25+CD4+ regulatory cells that control intestinal inflammation. J Exp Med. 2000;192:295–302. doi: 10.1084/jem.192.2.295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Read S, Greenwald R, Izcue A, Robinson N, Mandelbrot D, Francisco L, Sharpe AH, Powrie F. Blockade of CTLA-4 on CD4+CD25+ regulatory T cells abrogates their function in vivo. J Immunol. 2006;177:4376–4383. doi: 10.4049/jimmunol.177.7.4376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Walunas TL, Lenschow DJ, Bakker CY, Linsley PS, Freeman GJ, Green JM, Thompson CB, Bluestone JA. CTLA-4 can function as a negative regulator of T cell activation. Immunity. 1994;1:405–413. doi: 10.1016/1074-7613(94)90071-x. [DOI] [PubMed] [Google Scholar]
- 32.Doyle AM, Mullen AC, Villarino AV, Hutchins AS, High FA, Lee HW, Thompson CB, Reiner SL. Induction of cytotoxic T lymphocyte antigen 4 (CTLA-4) restricts clonal expansion of helper T cells. J Exp Med. 2001;194:893–902. doi: 10.1084/jem.194.7.893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Greenwald RJ, V, Boussiotis A, Lorsbach RB, Abbas AK, Sharpe AH. CTLA-4 regulates induction of anergy in vivo. Immunity. 2001;14:145–155. doi: 10.1016/s1074-7613(01)00097-8. [DOI] [PubMed] [Google Scholar]
- 34.Ostrov DA, Shi W, Schwartz JC, Almo SC, Nathenson SG. Structure of murine CTLA-4 and its role in modulating T cell responsiveness. Science. 2000;290:816–819. doi: 10.1126/science.290.5492.816. [DOI] [PubMed] [Google Scholar]
- 35.Zhang X, Schwartz JC, Almo SC, Nathenson SG. Expression, refolding, purification, molecular characterization, crystallization, and preliminary X-ray analysis of the receptor binding domain of human B7-2. Protein Expr Purif. 2002;25:105–113. doi: 10.1006/prep.2002.1616. [DOI] [PubMed] [Google Scholar]
- 36.Zhang X, Schwartz JC, Almo SC, Nathenson SG. Crystal structure of the receptor-binding domain of human B7-2: insights into organization and signaling. Proc Natl Acad Sci USA. 2003;100:2586–2591. doi: 10.1073/pnas.252771499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Grohmann U, Orabona C, Fallarino F, Vacca C, Calcinaro F, Falorni A, Candeloro P, Belladonna ML, Bianchi R, Fioretti MC, Puccetti P. CTLA-4-Ig regulates tryptophan catabolism in vivo. Nat Immunol. 2002;3:1097–1101. doi: 10.1038/ni846. [DOI] [PubMed] [Google Scholar]
- 38.Alegre ML, Noel PJ, Eisfelder BJ, Chuang E, Clark MR, Reiner SL, Thompson CB. Regulation of surface and intracellular expression of CTLA4 on mouse T cells. J Immunol. 1996;157:4762–4770. [PubMed] [Google Scholar]
- 39.Jeannin P, Magistrelli G, Aubry JP, Caron G, Gauchat JF, Renno T, Herbault N, Goetsch L, Blaecke A, Dietrich PY, et al. Soluble CD86 is a costimulatory molecule for human T lymphocytes. Immunity. 2000;13:303–312. doi: 10.1016/s1074-7613(00)00030-3. [DOI] [PubMed] [Google Scholar]