

Abstract
We present a high-resolution mass spectrometric (MS) footprinting method enabling identification of contact amino acids in protein–protein complexes. The method is based on comparing surface topologies of a free protein versus its complex with the binding partner using differential accessibility of small chemical group selective modifying reagents. Subsequent MS analysis reveals the individual amino acids selectively shielded from modification in the protein–protein complex. The current report focuses on probing interactions between full-length HIV-1 integrase and its principal cellular partner lens epithelium-derived growth factor. This method has a generic application and is particularly attractive for studying large protein–protein interactions that are less amenable for crystallographic or NMR analysis.
Keywords: Protein–protein interactions, Mass spectrometric footprinting, Surface topology of proteins, Amino acid modifying reagents, MALDI-ToF
1. Introduction
Protein–protein interactions established by HIV-1 integrase (IN) within the preintegration complex (PIC) are essential for productive integration of the viral cDNA into human chromatin. Dimeric and tetrameric forms of IN have been implicated in promoting its two catalytic activities, 3′ processing and strand transfer, respectively [1–4]. IN is comprised of three-domains, the N-terminal domain, catalytic core domain (CCD), and C-terminal domain. While the atomic structures of the individual domains [5–9] and two-domain fragments [10,11] are available, the intra- and inter-protein interfaces formed by full-length multimeric IN within the catalytic complexes are for the most part unknown. Limited protein solubility at relatively low ionic strength, as well as an inherent flexibility of the three-domain protein, has likely contributed to the inability to solve the structure of the full-length HIV-1 enzyme.
Here, we present a mass spectrometric (MS) footprinting method that allows to probe in detail interactions between full-length proteins within complexes that may resist rigorous structural analysis like X-ray crystallography or nuclear magnetic spectroscopy (NMR). The application of this approach for studying nucleic acid–protein interactions consistently revealed biologically essential contact amino acid residues [12–15] and, as documented here, the method can be readily extended to examine protein–protein interfaces. MS footprinting provides a tool to build upon available high-resolution structures of protein subdomains as “pieces of the puzzle” to assemble a plausible molecular model for full-length protein–protein complexes. By example, we provide details of the analysis of HIV-1 IN with its principal cellular binding partner lens epithelium-derived growth factor (LEDGF), which is critically important for effective integration [16–21]. The solution structure of the IN binding domain (IBD) of LEDGF and its co-crystallization with the IN CCD have been reported [22,23]. However, mutagenesis studies had indicated that contacts contributing to the high affinity interaction between full-length IN and LEDGF extended beyond the inter-protein interfaces observed in the co-crystal structure of isolated domains [24]. The MS-based approach detailed here enabled us to reveal novel inter- and intra-protein contacts important for effective formation of the IN–LEDGF complex [25].
2. Description of method
2.1. Overview
The experimental strategy is depicted in Fig. 1. The method exploits differential accessibility of small chemical modifying reagents in free protein versus the protein–protein complex. To identify IN residues essential for high affinity binding with LEDGF, free IN and the IN–LEDGF complex are subjected to treatment by small covalent modifiers. Surface residues in free IN and the complex are susceptible to modification, while the interacting amino acids in the complex are shielded from modification. The concentrations of the modifying reagents are carefully optimized in preliminary experiments to ensure mild modification conditions, under which the integrity of the protein–protein complex is preserved. To recover only the LEDGF bound form of IN from the reaction mixture, we use His-tag LEDGF and tag-free IN proteins. Following the modification reactions, the IN–LEDGF complex and free LEDGF are recovered from the reaction mixture using Ni-nitrilotriacetic acid (Ni-NTA) beads (GE Healthcare). The separate control reaction, which interrogates IN protein in the absence of LEDGF, proceeds without the affinity purification step. Following separation by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS–PAGE), the IN bands are excised and subjected to in-gel proteolysis to generate peptide fragments amenable for MS experiments. Subsequent MS analyses are conducted to compare the modification patterns of IN in its free form and in the complex with LEDGF. The modified peaks persisting in both free IN and the complex indicate accessible, surface-exposed residues, while the modified peaks observed in free IN but effectively diminished in the complex reveal the amino acids specifically shielded via the protein–protein interaction (Fig. 1).
Fig. 1.
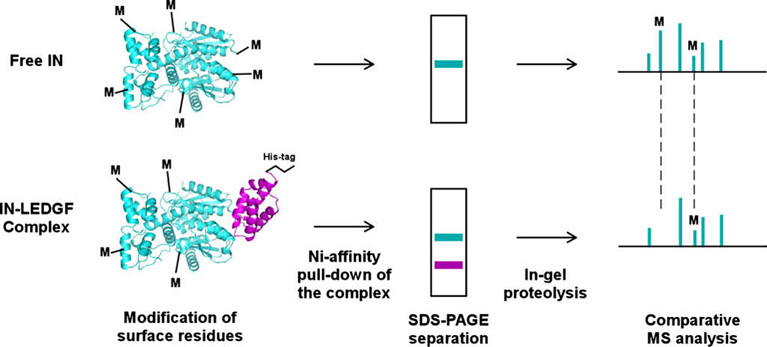
Schematic of the MS-based protein footprinting method. The structures of the IN CCD and its complex with the LEDGF IBD are used for illustration, while the experiments were performed with full-length IN and LEDGF. In parallel experiments free IN and the complex are subjected to treatments by small chemical modifiers (M) and analyzed as described in the text.
2.2. Step-by-step protocol
2.2.1. Modification of surface-exposed Lys residues
-
Recombinant HIV-1 IN and LEDGF proteins are purified following their expression in Escherichia coli using established procedures [24,26]. Prepare and analyze the following two reactions in parallel: free IN and IN–LEDGF complex. Should protein stocks contain amines (for example, Tris), dialyze the preparations against non-amine buffers such as phosphate, Hepes, borate, or carbonate. HIV-1 IN modification reactions are carried out in buffer containing 50 mM Hepes, pH 8.0, 250 mM NaCl, 10 mM MgCl2. Prepare a stock solution of 1 µM IN in one tube, and a stoichiometric mixture of 1 µM IN and 1 µM LEDGF in the other tube. The total volume of each reaction is 25 µl. Incubate the reactions at room temperature for 30 min to allow formation of protein–protein complexes.
Add 2.5 µl of a freshly prepared aqueous solution of 10 mM sulfo-N-hydroxysuccinimide (NHS)-biotin (Pierce) to both reactions. Incubate the reactions at 37 °C for 30 min. Note that these reaction conditions were optimized by examining the effects of varying NHS-biotin concentrations (0.1–10 mM) on the IN–LEDGF interaction using a Ni-NTA pull-down assay [24]. The highest concentration of NHS-biotin that did not disturb IN–LEDGF complex formation was selected for the footprinting analysis.
Quench the reactions by adding 2.5 µl of 3 M Na-acetate, pH 5.0, containing 1 M Lys in its free form. Spin the samples at 1000g for 2 min to remove any precipitation.
For the sample containing free IN, proceed to the Section 2.2.4, while for the IN–LEDGF complex, follow the protocol presented in Section 2.2.3.
2.2.2. Modification of surface-exposed Arg residues
-
Prepare and analyze the following two reactions in parallel: free IN and IN–LEDGF complexes in buffer containing 50 mM Hepes, pH 8.0, 50 mM boric acid, 250 mM NaCl, 10 mM MgCl2. Place 1 µM IN in one tube and mix 1 µM IN with a stoichiometric amount of LEDGF in the other tube. The total volume of each reaction is 25 µl. Incubate the reactions at room temperature for 30 min to allow formation of protein–protein complexes.
Add 2.5 µl of a freshly prepared aqueous solution of 200 mM p-hydroxyphenylglyoxal (HPG) (Pierce) to both reactions. Incubate the reactions in the dark at 37 °C for 60 min. As indicated above for NHS-biotin, these conditions were optimized by examining the effects of varying HPG concentrations (0.1–50 mM) on IN–LEDGF complex formation.
Quench the reactions by adding 2.5 µl of 3 M Na-acetate, pH 5.0, containing 1 M Arg in its free form. Spin the samples at 1000g for 2 min to remove any precipitation.
For the sample containing free IN, proceed to the Section 2.2.4; the next section describes the protocol for analyzing the IN–LEDGF complex.
2.2.3. Affinity pull-down of protein–protein complexes
-
In separate eppendorf centrifuge tubes, prepare 20 µl of Ni-NTA resin for each reaction. For this, wash the beads three times with 400 µl of pull-down buffer: 50 mM Hepes, pH 7.1, 150 mM NaCl, 2 mM MgCl2, 35 mM imidazole, 0.1% w/v Nonidet P40. Following the washes, spin the mixtures at 1000g for 1 min, discard the supernatants, and use the beads in the next step.
Add 400 µl pull-down buffer to the NHS-biotin (Section 2.2.1) and HPG (Section 2.2.2) treated IN–LEDGF complexes, and apply the mixtures onto the pre-washed Ni-NTA beads. Incubate the reactions on a shaker for 1 h at room temperature.
Centrifuge the mixtures at 1000g for 2 min, remove the supernatants, and wash the resin twice using 650 µl pull-down buffer each time. Discard the pull-down buffers and add 16 µl of an aqueous solution of 50 mM dithiothreitol (DTT) and 50 mM EDTA to the beads.
2.2.4. Capping Cys residues, SDS–PAGE separation of interacting proteins, and in-gel proteolysis
From this step forward, treat all samples including free IN and IN–LEDGF complexes identically.
-
Add 2 µl 20% SDS and 0.2 µl 1 M DTT to each sample, and incubate the reactions for 20 min at 70 °C to unfold the proteins.
Cap Cys residues by adding 2.5 µl of a freshly prepared aqueous solution of 1 M iodoacetamide and incubating the reactions for 45 min at room temperature. Terminate the capping reactions by adding 2.5 µl 1 M DTT.
Add SDS–PAGE loading buffer (Invitrogen) to the samples and heat the mixtures at 95 °C for 10 min. Perform SDS–PAGE separation of the proteins.
Following electrophoresis, stain the gel with Microwave Blue stain (Protiga). Once the proteins are clearly visible, transfer the gel to a clean glass plate.
Excise the IN bands, slice each band into about four pieces with a clean scalpel, and transfer the slices to a 1.5 ml tube. Add 500 µl 50 mM ammonium bicarbonate and 500 µl 100% acetonitrile to each Eppendorf tube, and shake them at 200 rpm for at least 4 h to destain.
Carefully remove the destain solution without touching the gel slices. Add 1 ml of 50 mM ammonium bicarbonate. Vortex for 1 min, spin briefly, and discard the solution. Repeat twice.
Add 200 µl pure acetonitrile to each sample. Shake the samples for 15 min or until the gel slices are white and shrunken. Spin the samples and carefully remove the acetonitrile. Lyophilize the gel slices in a SpeedVac at 45 °C for 15 min or until dry.
While drying the samples, prepare the trypsin solution. First, prepare a stock solution (0.2 µg/µl) of modified sequencing grade trypsin (Roche) in 10 mM HCl. Immediately prior to use, dilute the stock solution 20-fold with 50 mM ammonium bicarbonate, pH 8.0. Add 50 µl trypsin solution to each dried gel slice. Allow ~15 min for the gel slices to soak up the solution. If needed add 50 mM ammonium bicarbonate, pH 8.0 to fully cover the gel slices. Spin briefly and place the samples on a shaker for overnight at room temperature.
The next day, add 150 µl pure acetonitrile to each digestion, and immediately vortex the samples for 5 min.
Centrifuge the tubes at 1000g for 2 min, and carefully remove 180 µl supernatant without touching the gel slice. Transfer the solution into 500 µl micro-centrifuge tubes.
Dry the peptide mixtures completely using vacuum desiccation at medium heat (45 °C).
Add 10 µl HPLC grade water to each sample. At this stage, the samples are ready for MS analysis.
2.2.5. Mass spectrometric analysis
-
Prepare the matrix solution: dissolve α-cyano-4-hydroxycinnamic acid (Sigma) to 5 mg/ml in 75% (v/v) acetonitrile. Thoroughly vortex the mixture and centrifuge at 10,000g for 5 min.
Apply 0.7 µl of each sample onto a MALDI plate, and immediately mix with 1.7 µl matrix solution by pipetting the mixture up and down several times. Air dry the samples at room temperature (~15 min). These samples can now be analyzed by a MALDI-ToF instrument.
Use a MALDI-ToF instrument equipped with a curved field reflectron feature (Kratos Analytical Instruments, Manchester, U.K.) in the reflectron mode to analyze the samples.
The assignment of the peptide peaks is accomplished by comparing monoisotopic mass/charge values for the detected peaks with the theoretical profile of IN tryptic fragments, which is generated by the MS-Digest program of the ProteinProspector search engine (http://prospector.ucsf.edu/). The experimental mass/charge measurement for a given peptide has to be within ±0.1% of the corresponding theoretical value. Mass increments of 226 and 132 Da are considered for assigning NHS-biotin and HPG modified peptides, respectively. Note that modified Lys and Arg residues are not readily amenable to trypsin hydrolysis. Therefore, the modified peptides are very likely to contain one or more missed cleavage sites, with the corresponding number of modified groups attached to Lys and/or Arg residues.
To verify the amino acid sequence, a select peptide peak is subjected to post source decay analysis by activating the respective feature in the MALDI-ToF instrument. The obtained results are analyzed using the MS-Seq program of the Protein Prospector search engine.
For quantitative comparison of modifications in free protein and protein–protein complexes, use at least two unmodified prote-olytic peptide peaks as controls.
3. Concluding remarks
Protein-protein interactions play a vital role in numerous biological processes. Like HIV-1 IN, high-resolution structural data are available for many individual protein domains, while biologically-relevant large multi-protein complexes may be less amenable to crystallographic and NMR analysis. Our MS footprinting approach, which has unveiled important protein–protein interactions that occur during HIV-1 integration [25], can be readily adopted for other protein-based interaction systems. Our methodology importantly allows the examination of protein–protein interactions using limited amounts of starting materials. While amine buffers such as Tris should be avoided in the reactions with NHS-biotin, the modification buffers could include any of a wide variety of components that contribute to stability and solubility of large protein–protein complexes. Preliminary functional analyses should precede the footprinting experiments to determine the optimal modification conditions under which the integrity of the complexes is preserved. The above Lys and Arg footprinting could be augmented by probing other residues. For example, commercial reagents are available for surface topology analysis of Cys, His, Tyr, and Trp residues [27].
Acknowledgment
This study has been supported by NIH grants AI062520, AI077341 and CA100730 (to M.K.).
References
- 1.Faure A, Calmels C, Desjobert C, Castroviejo M, Caumont-Sarcos A, Tarrago-Litvak L, Litvak S, Parissi V. Nucleic Acids Res. 2005;33:977–986. doi: 10.1093/nar/gki241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Guiot E, Carayon K, Delelis O, Simon F, Tauc P, Zubin E, Gottikh M, Mouscadet JF, Brochon JC, Deprez E. J. Biol. Chem. 2006;281:22707–22719. doi: 10.1074/jbc.M602198200. [DOI] [PubMed] [Google Scholar]
- 3.Li M, Mizuuchi M, Burke TR, Jr, Craigie R. EMBO J. 2006;25:1295–1304. doi: 10.1038/sj.emboj.7601005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bosserman MA, O’Quinn DF, Wong I. Biochemistry. 2007;46:11231–11239. doi: 10.1021/bi700197a. [DOI] [PubMed] [Google Scholar]
- 5.Cai M, Zheng R, Caffrey M, Craigie R, Clore GM, Gronenborn AM. Nat. Struct. Biol. 1997;4:567–577. doi: 10.1038/nsb0797-567. [DOI] [PubMed] [Google Scholar]
- 6.Dyda F, Hickman AB, Jenkins TM, Engelman A, Craigie R, Davies DR. Science. 1994;266:1981–1986. doi: 10.1126/science.7801124. [DOI] [PubMed] [Google Scholar]
- 7.Goldgur Y, Dyda F, Hickman AB, Jenkins TM, Craigie R, Davies DR. Proc. Natl. Acad. Sci. USA. 1998;95:9150–9154. doi: 10.1073/pnas.95.16.9150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Eijkelenboom AP, Lutzke RA, Boelens R, Plasterk RH, Kaptein R, Hard K. Nat. Struct. Biol. 1995;2:807–810. doi: 10.1038/nsb0995-807. [DOI] [PubMed] [Google Scholar]
- 9.Lodi PJ, Ernst JA, Kuszewski J, Hickman AB, Engelman A, Craigie R, Clore GM, Gronenborn AM. Biochemistry. 1995;34:9826–9833. doi: 10.1021/bi00031a002. [DOI] [PubMed] [Google Scholar]
- 10.Chen JC, Krucinski J, Miercke LJ, Finer-Moore JS, Tang AH, Leavitt AD, Stroud RM. Proc. Natl. Acad. Sci. USA. 2000;97:8233–8238. doi: 10.1073/pnas.150220297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang JY, Ling H, Yang W, Craigie R. EMBO J. 2001;20:7333–7343. doi: 10.1093/emboj/20.24.7333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhao Z, McKee CJ, Kessl JJ, Santos WL, Daigle JE, Engelman A, Verdine G, Kvaratskhelia M. J. Biol. Chem. 2008;283:5632–5641. doi: 10.1074/jbc.M705241200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shkriabai N, Datta SA, Zhao Z, Hess S, Rein A, Kvaratskhelia M. Biochemistry. 2006;45:4077–4083. doi: 10.1021/bi052308e. [DOI] [PubMed] [Google Scholar]
- 14.Kvaratskhelia M, Miller JT, Budihas SR, Pannell LK, Le Grice SF. Proc. Natl. Acad. Sci. USA. 2002;99:15988–15993. doi: 10.1073/pnas.252550199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shell SM, Hess S, Kvaratskhelia M, Zou Y. Biochemistry. 2005;44:971–978. doi: 10.1021/bi048208a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.De Rijck J, Vandekerckhove L, Gijsbers R, Hombrouck A, Hendrix J, Vercammen J, Engelborghs Y, Christ F, Debyser Z. J. Virol. 2006;80:11498–11509. doi: 10.1128/JVI.00801-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Vandekerckhove L, Christ F, Van Maele B, De Rijck J, Gijsbers R, Van den Haute C, Witvrouw M, Debyser Z. J. Virol. 2006;80:1886–1896. doi: 10.1128/JVI.80.4.1886-1896.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hombrouck A, De Rijck J, Hendrix J, Vandekerckhove L, Voet A, De Maeyer M, Witvrouw M, Engelborghs Y, Christ F, Gijsbers R, Debyser Z. PLoS Pathog. 2007;3:e47. doi: 10.1371/journal.ppat.0030047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Llano M, Saenz DT, Meehan A, Wongthida P, Peretz M, Walker WH, Teo W, Poeschla EM. Science. 2006;314:461–464. doi: 10.1126/science.1132319. [DOI] [PubMed] [Google Scholar]
- 20.Shun MC, Raghavendra NK, Vandegraaff N, Daigle JE, Hughes S, Kellam P, Cherepanov P, Engelman A. Genes Dev. 2007;21:1767–1778. doi: 10.1101/gad.1565107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Marshall HM, Ronen K, Berry C, Llano M, Sutherland H, Saenz D, Bickmore W, Poeschla E, Bushman FD. PLoS ONE. 2007;2:e1340. doi: 10.1371/journal.pone.0001340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cherepanov P, Sun ZY, Rahman S, Maertens G, Wagner G, Engelman A. Nat. Struct. Mol. Biol. 2005;12:526–532. doi: 10.1038/nsmb937. [DOI] [PubMed] [Google Scholar]
- 23.Cherepanov P, Ambrosio AL, Rahman S, Ellenberger T, Engelman A. Proc. Natl. Acad. Sci. USA. 2005;102:17308–17313. doi: 10.1073/pnas.0506924102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Maertens G, Cherepanov P, Pluymers W, Busschots K, De Clercq E, Debyser Z, Engelborghs Y. J. Biol. Chem. 2003;278:33528–33539. doi: 10.1074/jbc.M303594200. [DOI] [PubMed] [Google Scholar]
- 25.McKee CJ, Kessl JJ, Shkriabai N, Dar MJ, Engelman A, Kvaratskhelia M. J. Biol. Chem. 2008 doi: 10.1074/jbc.M805843200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cherepanov P. Nucleic Acids Res. 2007;35:113–124. doi: 10.1093/nar/gkl885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lundblad RL. Techniques in Protein Modification. Boca Raton, FL: CRC Press; 1995. [Google Scholar]