

Abstract
Growth factors and their corresponding receptors are commonly overexpressed and/or dysregulated in many cancers including hepatocellular cancer (HCC). Clinical trials indicate that growth factor receptors and their related signalling pathways play important roles in HCC cancer etiology and progression, thus providing rational targets for innovative cancer therapies. A number of strategies including monoclonal antibodies, tyrosine kinase inhibitors (“small molecule inhibitors”) and antisense oligonucleotides have already been evaluated for their potency to inhibit the activity and downstream signalling cascades of these receptors in HCC. First clinical trials have also shown that multi-kinase inhibition is an effective novel treatment strategy in HCC. In this respect sorafenib, an inhibitor of Raf-, VEGF- and PDGF-signalling, is the first multi-kinase inhibitor that has been approved by the FDA for the treatment of advanced HCC. Moreover, the serine-threonine kinase of mammalian target of rapamycin (mTOR) upon which the signalling of several growth factor receptors converge plays a central role in cancer cell proliferation. mTOR inhibition of HCC is currently also being studied in preclinical trials. As HCCs represent hypervascularized neoplasms, inhibition of tumour vessel formation via interfering with the VEGF/VEGFR system is another promising approach in HCC treatment. This review will summarize the current status of the various growth factor receptor-based treatment strategies and in view of the multitude of novel targeted approaches, the rationale for combination therapies for advanced HCC treatment will also be taken into account.
Keywords: Growth factor receptor, Hepatocellular cancer, Small molecule inhibitor, Monoclonal antibody, Innovative cancer treatment, Sorafenib, Bevacizumab, Erlotinib
INTRODUCTION
Hepatocellular carcinoma (HCC) is the fifth most common malignancy worldwide. With an alerting 5 years relative survival rate of about 7% HCC is estimated to cause more than half a million deaths annually. HCC is the third most common cause of cancer deaths. The disease is most prevalent in Eastern and Southeastern Asia, and Middle Africa, with more than half of the patients being reported from China[1]. However, also in the developed countries the incidence of HCC dramatically increased in the past decades, mainly due to the increasing prevalence of chronic hepatitis C[2].
HCC is one of the few cancers with well-defined major risk factors. In Western countries > 80% of HCC develop in livers with cirrhosis mainly due to chronic hepatitis C, alcohol abuse, chronic hepatitis B or hemochromatosis. Especially in developed countries there is increasing concern regarding the epidemic of obesity which is associated with type 2 diabetes and other features of the metabolic syndrome and which frequently leads to non-alcoholic steatohepatitis (NASH). Here NASH may become to be one of the major causes of cirrhosis; diabetes and NASH are risk factors for developing HCC[2–4]. Rarer causes are cirrhosis due to hemochromatosis, autoimmune liver diseases or congenital disorders of metabolism. Cirrhosis in a setting of chronic liver cell injury, with inflammation, hepatocyte necrosis and regeneration, is a particular breeding ground for hepatocyte dedifferentiation and HCC[5]. In developing countries, HCC frequently arises in non-cirrhotic livers, mostly on the basis of congenital infection with the hepatitis B virus which acts as mutagen due to insertion in the human genome and/or on the basis of aflatoxin exposure from contaminated food[2,4].
Unfortunately, the majority of patients suffer from advanced HCC at presentation. Therefore, curative treatment like local ablation, surgical resection or liver transplantation can be achieved in only a minority of HCC patients[6]. Local tumour destruction, chemoembolisation or systemic therapy are the treatment options of advanced HCC. Apart from transarterial chemoembolisation, which improves survival in well-selected patients with unresectable HCC, conventional palliative treatment options do not appear to improve overall outcome[5,6]. A recent meta-analysis of Simonetti and coworkers, who evaluated the results of randomized clinical trials of systemic and regional chemotherapy of HCC patients confirmed the disappointing results and revealed that nonsurgical therapies are more or less ineffective and do not prolong the survival of HCC patients, while further compromising quality of life[7].
Effective palliative treatment is hampered by the fact that advanced HCC represents a tumour entity which is extremely resistant to radiotherapy and conventional chemotherapy[8]. Moreover, the existing conventional chemotherapeutics are more or less non-selective cytotoxic drugs with significant systemic side-effects. Importantly, as most patients with advanced HCC have compromised liver function aggressive medical therapy regimens can not be applied. Thus, usually no effective therapy can be offered to these patients.
Because of the lack of any survival benefit of treatment with conventional drugs, new agents and novel therapeutic strategies are urgently needed to improve palliative treatment, prolong life expectancy and improve quality of life in patients with advanced HCC.
POTENTIAL TARGETS FOR FUTURE HCC THERAPIES
Growth factors and their related receptors are interesting targets for future therapeutic approaches. During foetal life, a large number of growth factors including the epidermal growth factor (EGF), insulin-like growth factors (IGFs), the hepatocyte growth factor (HGF), the vascular endothelial growth factor (VEGF), the fibroblast growth factor (FGF), the platelet-derived growth factor (PDGF) and the transforming growth factors -α and -β (TGF-α, TGF-β) are produced in the liver. In the adult normal liver many of them decline or are even absent. On the other hand adult hepatocytes are able to upregulate the production of particular growth factors like EGF, TGF-α, IGFs and VEGF, when liver regeneration is required after injury or damage[9,10]. This normally transient upregulation is dysregulated in the chronic injured liver leading to sustained mito-oncogenic signalling. Thus, dysregulation of the growth factor production and growth factor receptor signalling of adult hepatocytes plays an important role in hepatocarcinogenesis.
Furthermore, members of the fibroblast growth factor and platelet-derived growth factor families, FGFs and PDGF play important roles in promoting liver fibrosis and HCC growth[11,12]. Like HGF, these growth factors are produced and released from non-hepatocyte sources like activated hepatic stellate cells, myofibroblasts, endothelial cells, Kupffer cells and bile duct epithelia and do also contribute to hepatocarcinogenesis.
GROWTH FACTOR RECEPTOR RELATED SIGNALLING PATHWAYS IN HCC CELLS
In the last decade some of the relevant pathways in cancer biology have been deciphered and there is emerging evidence that particularly growth factor receptors and their related downstream signalling pathways play a pivotal role in the development and maintenance of various cancers including HCC. Among the most critical cellular signalling pathways which support hepatocarcinogenesis are the receptor tyrosine kinase-activated pathways which include the rat sarcoma/rat sarcoma-activated factor (raf) /mitogen activated protein kinase/extracellular regulated kinase kinase/extracellular regulated kinase pathway (Ras/Raf/MEK/ERK), the Janus kinase/signal tranducers and activator of transcription pathway (JAK/STAT)s, and the phosphatidyl inositide 3 kinase/protein kinase B (AKT) /mammilian target of rapamycin pathway (PI3K/AKT/mTOR) (Figure 1).
Figure 1.

Major growth factor receptor signalling pathways in HCC.
Ras/Raf/MEK/ERK pathway
The Ras/Raf/MEK/ERK pathway appears to be one of the most significant cellular signalling sequences in the development and maintenance of hepatocellular cancer. This pathway transduces extracellular signals from ligand-bound tyrosine kinase receptors, such as the epidermal growth factor receptor (EGFR), the insulin-like growth factor receptor (IGFR), the vascular endothelial growth factor receptor (VEGFR) or the platelet-derived growth factor receptor (PDGFR) to the nucleus in a series of specific phosphorylation events, starting with the activation of Ras which in turn activates serine threonine kinases of the Raf-family[1]. Activated Raf phosphorylates MEK 1/2 kinases which finally activate the extracellular regulated kinases ERK 1/2. Once activated, ERK 1/2 translocates to the nucleus where it acts as a regulator of gene expression of various proteins, including those for cell cycle progression, apoptosis resistance, extracellular matrix (ECM) remodeling, cellular motility angiogenesis and drug resistance[13]. Dysregulation of this crucial pathway occurs due to oncogenic transformation of Ras and Raf isoforms, or overexpression and/or overactivation of the Ras and Raf genes. In a recent study overexpression of the Raf-1 gene was shown in 50% of HCC biopsies, while increased activation of the Raf-1 protein was found even in 100% of the n = 30 evaluated HCC biopsies[14]. However, significant contribution of the proliferative Ras pathway to the development of HCC has long been a matter of debate. Previous studies suggested that activation of the Ras pathway might only be important in rodent, but not in human HCC, because of the low incidence of Ras gene mutations found in human HCC, while activation of the Ras cascade also occurred in the presence of wild-type Ras[15]. Recently, the components of the Ras cascade in human HCCs were characterized, demonstrating a downregulation or loss in the expression of specific members of the RAS inhibitor family. Among them the RAS association family 1 gene A (RASSF1A) and its homologue NORE1A in 100% of the n = 35 examined HCC[16]. The inactivation of these inhibitors resulted in a persistent activation of the Ras pathway and the authors suggested that the use of Ras inhibitors may thus be an interesting therapeutic modality for future treatment of HCC.
JAK/STAT pathway
The same holds true for the JAK/STAT pathway which plays an important role in cellular processes like differentiation, proliferation, and apoptosis[17]. STATs are latent in the cytoplasm and become activated through tyrosine phosphorylation which typically occurs through JAKs or growth factor receptor tyrosine kinases. Activated STATs enter the nucleus and serve as transcription factors. As to apoptosis and cell cycle related genes the transcriptional changes induced by STATs are similar to those described for ERK1/2.
In normal cells, ligand-dependent activation of STATs is transient, but in tumours the STAT proteins (in particular STAT-1, -3 and -5) are often constitutively activated[18,19]. This constitutive activation is partly due to inactivation of specific STAT inhibitors, the suppressors of cytokine signalling (SOCS), which normally balance and terminate STAT activity[20,21]. Thus, loss of activation of the STAT inhibitors such as cytokine-inducible SH2-protein (CIS), SOCS1, SOCS2, SOCS3, and SH2-containing phosphatases (SHP1) was shown to account for the constitutive activation of antiapoptotic and mitogenic STAT-3 and -5 in HCC[16].
In terms of both the Ras and the JAK/STAT pathway it may not be the increase in gene mutations of the respective pathway proteins, but the state of activation of these pathways due to an imbalanced interplay of activators and inhibitors, which accounts for the pivotal role of these pathways in HCC.
PI3K/AKT/mTOR pathway
The activated PI3K/AKT/mTOR pathway has only recently emerged as a novel contributor to (HCC) tumour development. PI3K associates with the intracellular domain of several growth factor receptors. Upon activation PI3K triggers the generation of phosphatidylinositol 3,4,5-triphosphate (PIP3) which provokes the subsequent activation of AKT, a serine/threonine kinase which regulates multiple cellular target proteins. Among these proteins is proapoptotic BAD, which becomes inactivated by phosphorylation, and also the mammalian target of rapamycin (mTOR) subfamily of proteins, which become activated by AKT. mTOR proteins regulate the phosphorylation of p70 S6 serine-threonine kinase and the translational repressor protein PHAS-1/4E-BP. Both proteins regulate the translation of proliferation- and angiogenesis-relevant proteins, such as c-myc, cyclin-D1, ornithine decarboxylase, hypoxia-induced factor 1-α, and are indirectly involved in the expression of VEGF[1,22].
In nontransformed cells the PI3K/AKT/mTOR pathway is controlled by the phosphatase and tensin homolog deleted on chromosome ten (PTEN), a tumour suppressor which inhibits this pathway by reversing the PI3K reaction and blocking AKT activation. Mutation or silencing of the PTEN gene leads to activation of the pathway and promotes carcinogenesis. PTEN expression is reduced or absent in almost half of the studied HCCs, and hepatocyte-specific abrogation of PTEN expression in mice results in the development of HCC[23]. Thus, constitutive activation of this pathway can be due to enhanced stimulation of growth factor receptors, like EGFR and IGFRs, but can also result from decreased PTEN expression[1]. In non-HCC tumour models loss of PTEN expression has been demonstrated to negatively influence the sensitivity towards EGFR-TK inhibition by gefitinib[24]. Thus, it will be interesting to evaluate if this also holds true for HCC. If so, PTEN expression can serve as a novel marker for predicting the response to tyrosine kinase inhibition-based treatment strategies in HCC and therapeutic strategies in which normal PTEN expression can be restored, should be an attractive approach for combined therapeutic strategies in HCC treatment in the future.
Moreover, recent work by Boyault and coworkers demonstrated that in specific subgroups of HCC patients, namely those that are infected with low copy number hepatitis B virus (HBV) and concomitant overexpression of genes expressed in fetal liver, as well as those infected with a high copy number of HBV, but concomitant mutations in the catalytic domain of the phosphoinositide-3 kinase (PIK3CA) and the tumor suppressor protein 53 (TP53), there is a frequent upregulation of AKT expression and activation, rendering these patients especially susceptible to therapeutic approaches that inhibit the AKT-pathway[25].
Due to the importance of the above-described signalling pathways linked to growth factor receptors in the development and maintenance of (hepatocellular) cancer, several attempts have been undertaken to develop specific inhibitors which either block the communication of growth factors and their cognate receptors using antibodies or growth factor trapping decoy receptors, or by interrupting the transmission of growth factor receptor which signals to the respective downstream signalling cascades by membrane permeable small molecule inhibitors which block the intrinsic receptor tyrosine kinase activity. The following section will provide a concise overview of selected agents which are currently in the development and testing for the targeted treatment of solid tumours and HCC.
EGFR-based strategies
The crucial role of epidermal growth factor receptor (EGFR) in tumour proliferation and its overexpression in several solid tumours have provided the rationale for targeting and interrupting this key signalling network. EGFR blockade through monoclonal antibodies and tyrosine kinase inhibitors has translated into promising evidence of clinical benefit in gastrointestinal tumours, particularly colorectal cancer[26]. EGFR is expressed in a high proportion of HCCs, and EGFR-inhibitors, such as the monoclonal antibody cetuximab or the tyrosine kinase inhibitors gefitinib, erlotinib or ANAPD have been shown to inhibit HCC growth and metastasis formation in vitro[27–31] and in vivo[32]. Recently, Philips and coworkers conducted a phase II trial with erlotinib for advanced HCC and could demonstrate very encouraging results, as they observed good response rates in approx. One third of the treated patients and a prolonged survival with mild and tolerable side effects after treatment with a dose of 150 mg/d[33].
Despite the encouraging findings on the general suitability of anti-EGFR-based-approaches for the treatment of HCC, only few clinical trials have been conducted so far. Most of our current knowledge on the clinical benefit of anti-EGFR-based therapies originates from studies on other tumour entities, such as colorectal cancer, renal cell carcinoma and non-small cell lung cancer (NSCLC). Nevertheless, at present several clinical trials evaluate the efficacy of anti-EGFR-interventions for the treatment for HCC (www.clinical-trials.gov), and based on the data that are available so far there is hope that anti-EGFR mono- or combination therapies will qualify for improving the treatment of advanced HCC in the near future. Currently, another phase II trial is conducted in patients with advanced HCC which evaluates the efficacy of a combination of erlotinib and the anti-angiogenic VEGF-blocking antibody bevacizumab (NIH, NCT00365391). Dual-targeting of the HCC cells and their nutrient supply via the surrounding vasculature may improve the antitumoural effects as compared to monotherapy with either erlotinib or bevacizumab alone[34].
Thus the majority of the currently tested anti-EGFR-based approaches are increasingly combined either with conventional cytostatics or with other targeted-agents[28,29,35,36]. The rationale for applying combination therapies is the existence of multilevel receptor cross-stimulation or of redundant signalling pathways that lead to neoplasia. Blocking only one of these pathways allows others to act as salvage or escape mechanisms for cancer cells. Preclinical evidence of synergistic antitumour activity achievable by combining targeted agents that block multiple signalling pathways has recently emerged[37–41]. The multi-target approach can be accomplished by using either combinations of selective agents or agents which interfere with various targets[42]. Table 1 shows the current status of anti-EGFR-strategies for the treatment of solid tumours including HCC.
Table 1.
Agents for anti-EGFR-based therapy of solid tumours
| Name | Target | Mechanism | Current status |
| Small molecule inhibitors | |||
| Gefitinib (Iressa) | EGFR | Reversibly acting tyrosine kinase inhibitor | Approved for NSCLC with restricted indications |
| PhaseIfor HCC[133] | |||
| Erlotinib (Tarceva) | EGFR | Reversibly acting tyrosine kinase inhibitor | Approved for NSCLC and pancreatic cancer |
| Phase II for hepatocellular cancer[33] | |||
| EKB-569 | EGFR | Irreversibly acting tyrosine kinase inhibitor | Phase I/II for colorectal cancer[134] |
| Phase II for NSCLC[135] | |||
| Lapatinib (Tykerb) | EGFR, erbB2 | Reversibly acting tyrosine kinase inhibitor | Phase III for breast cancer |
| Phase II for HCC[136] | |||
| Canertinib (CI-1033) | Pan-erbB | Irreversibly acting tyrosine kinase inhibitor | Phase II for SCC and ovarian cancer[137] |
| BMS-599626 | EGFR, erbB2 | Reversibly acting tyrosine kinase inhibitor | Phase II for HCC [138] |
| Monoclonal antibodies | |||
| Cetuximab | EGFR | Approved for colorectal cancer | |
| Phase III for head and neck cancer, NSCLC and pancreatic cancer | |||
| Phase II for HCC[139] | |||
| Trastuzumab | erbB2 | Approved for breast cancer | |
| ABX-EGF | EGFR | Phase III for colorectal-, head and neck-, and renal cell cancer | |
| Matuzumab (EMD 72 000) | EGFR | Phase I/II for NSCLC[140], ovarian-[141], pancreatic cancer[142] |
IGF/IGFR-based strategies
There is compelling evidence that both insulin-like growth factors IGF-I and -II and their receptor tyrosine kinase, IGF-1R, are involved in the development and progression of cancer[43–46]. Interaction of IGF-I and -II with the IGF-1R plays a pivotal role in tumourigenesis, proliferation and spread of many cancers, by promoting cell cycle progression, preventing apoptosis, and by regulating and maintaining the tumourigenic phenotype. A wide variety of tumours including HCC show abnormal, or enhanced expression of IGFs and IGF-1R, which has been correlated with disease stage, reduced survival, development of metastases and tumour de-differentiation[47–49]. In men, obesity and diabetes are clearly associated with an increased risk of HCC, and this seems to be due to alterations in the metabolism of endogenous hormones, including sex steroids, insulin and the IGF/IGFR system. Thus, a promising approach of innovative HCC treatment may be the blockade of the IGF/IGFR, but also the mTOR-signalling system, which is functionally upregulated in HCC cells in vitro[50–52] and in vivo[47], and which has been shown to exert strong stimulatory effects on the growth of hepatoma cells[48]. In addition to the increased expression of IGF-1R and IGFs, a simultaneous reduction of IGF binding protein expression (IGFBP) and enhanced proteolytic cleavage of IGFBPs often occurs. Both mechanisms lead to an excessive increase in the amount of bioactive IGF[50,53] which further enhances the mito-oncogenic effects of IGFR-signalling in HCC and other cancer cells. The expression of IGF-1R is very low in normal hepatocytes that are poorly responsive to IGFs, whereas significant expression is found in Kupffer, endothelial and hepatic stellate cells[50].
Several approaches have demonstrated the therapeutic potential of interfering with IGF-1R mediated signalling in vitro and in vivo, including the use of IGF-1R blocking antibodies, IGF-1R antisense oligonucleotides or IGF-1R siRNA[54–57].
Recently, we and others introduced the potent and selective IGF-1R tyrosine kinase inhibitor, NVP-AEW541, as promising novel agent for the therapy of several cancers[58–60], including HCC[61]. The antineoplastic properties of NVP-AEW541 and related compounds such as NVP-ADW742[62] have been demonstrated in preclinical studies on Ewing’s sarcoma-bearing mice[63], fibrosarcoma, breast cancer and muscelosceletal carcinoma[64–66]. Specific IGFR-antibodies have also shown to suppress prostate and breast cancer cell growth in a recent preclinical study[67]. The clinically most advanced anti-IGFR antibody is CP-751,871 which is currently being tested in three phase II trials for advanced breast cancer, NSCLC and prostate cancer (www.clinical-trials.gov). Importantly, IGFR-inhibition appears to be well-tolerated in the preliminary clinical studies conducted so far[63,68,69]. Safety is important, since IGFR-based inhibition has long been regarded as a high risk intervention, because of the high homology of the IGF-1R receptor with the related insulin-receptor, and the fear that especially IGF-1R-TK inhibitors might do also block the insulin receptor which could lead to insulin resistance and overt diabetes[70]. However, the current in vivo studies did not confirm this apprehension, resulting in growing interest in anti-IGFR-based therapies[71].
It is widely accepted that a therapy which inhibits IGF signalling may have to be combined with other therapies to enhance the antiproliferative overall-effect, since crosstalk between the signalling of IGF and other growth factor receptors have already been shown to be able to attenuate the antineoplastic effects of a respective monotherapeutic approach[72]. Thus, we and others could show that IGFR and concomitant EGFR-inhibition or conventional chemotherapy enhances the antineoplastic effect of the respective monotherapies[28,29,61]. Especially, dual-targeting EGFR and IGF-1R is a promising approach for future treatment of HCC. The rationale for this particular combination is derived from observations that in HCC cells the EGFR-system is activated by the IGF/IGFR-system via receptor cross-talk leading to mito-oncogenic EGFR-tyrosine kinase activity[73,74]. Thus inhibition of IGF-2-related signalling leads to sensitization of HCC cells to anti-EGFR-treatment with gefitinib[72], and it was postulated that inhibition of IGF/IGF-1R-signalling may not only enhance the effects of gefitinib treatment, but may also help to overcome resistance to anti-EGFR-based therapy of HCC[75]. Table 2 summarizes the most promising IGF/IGFR-targeted agents which are currently under intense investigation in preclinical and early clinical trials.
Table 2.
Agents for anti-IGF-1R-based cancer treatment
| Name | Target | Mechanism | Current status |
| Small moelcule inhibitors | |||
| INSM-18 | IGF-1R and HER2 | Substrate competitive inhibitor | Phase I[44] |
| NVP-AEW541 | IGF-1R | ATP-competitive inhibitor | Preclinical[63] |
| NVP-ADW742 | IGF-1R | ATP-competitive inhibitor, activation of proapoptotic pathways | Preclinical[62] |
| BMS-536924 | IGF1R and IR | ATP competitive inhibitor, | Preclinical[143] |
| Cyclolignans | IGFR-1R | IGF competitive inhibitor | Preclinical[144] |
| Antibodies | |||
| CP-751, 871 | IGF-1R | IGF1R downregulation | PhaseIfor multiple myeloma |
| Phase II for Breast-[145], lung-[146], and prostate[147] cancer | |||
| A12 | IGF-1R | IGF1R down-regulation, apoptosis, cell cycle arrest | Phase I[148] |
| scFv-Fc | IGF-1R | IGF1R downregulation | Preclinical[149] |
| AVE-1642 | IGF-1R | IGF1R downregulation, cell-cycle arrest, induction of apoptosis | Preclinical[69] |
VEGF/VEGFR-based strategies
VEGF is the key angiogenic factor in tumours. The VEGF gene and protein have been reported to be transcribed, expressed, and secreted by HCC cells[76]. Endothelial cells which line tumour vessels express VEGFR-1 and VEGFR-2 which communicate to stimulate each other in a feedback-loop[77]. Given that VEGF protein expression is related to HCC grade[78] and given that the degree of microvascular density correlates with HCC grade[79,80], it is comprehensible that inhibitors of VEGF signalling are promising therapeutic agents for HCC treatment.
Bevacizumab is a humanized murine monoclonal anti-VEGF antibody which has entered the clinic for treatment of cancer. Standard cytostatic treatment plus bevacizumab significantly increased survival in metastatic colorectal cancer compared to standard treatment alone in a phase III clinical trial[81] which led to its approval for treatment of colorectal cancer 2005. Comparable results were obtained in a recent phase III clinical with bevacizumab for treatment of NSCLC. This study had to be interrupted because of the obvious survival advantage of patients in the bevacizumab arm[82]. Bevacizumab monotherapy is currently being tested in patients with unresectable HCC[83] (National Cancer Institute: NCT00162669). Moreover, a phase II trial is currently being conducted testing the efficacy of bevacuzimab in combination with capcetabine and oxaliplatin in patients with advanced HCC. An intermediate evaluation of this trial is encouraging and shows that this combination is tolerable to patients with advanced HCC and cirrhosis[84]. As described earlier, bevacizumab is also currently tested in combination with the EGFR-tyrosine kinase inhibitor erlotinib (see above).
In addition, several agents that inhibit the tyrosine kinase activity of VEGFR have been synthesized by combinatorial chemistry. Recent clinical studies revealed the suppression of HCC growth by vatalanib (PTK787/ZK 222584), which inhibits the activities of VEGFR-1 and -2 and has shown antineoplastic effects in other solid tumours[85,86]. Another interesting agent is the tyrosine kinase inhibitor sunitinib, which inhibits the VEGFR- as well as the PDGF-βR, c-KIT and FLT-3 tyrosine kinases. Sunitinib has been approved for the treatment of renal cell carcinoma[87]. With restricted indications sunitinib is also approved for the therapy of gastrointestinal stromal tumours (GIST)[88] and is currently tested in phaseIand II trials for HCC (NIH: NCT00361309; NCT00247676).
Another promising approach is the use of dual-targeting tyrosine kinase inhibitors, which inhibit less related tyrosine kinases, such as NVP-AEE788 or zactima (ZD6474) which target both the VEGFRs and the EGFR. In recent in vivo studies of non-HCC tumour models (colon, cholangiocarcinoma, prostate, NSCLC) NVP-AEE788 displayed significant antineoplastic efficacy. These agents can inhibit both tumour cell proliferation and survival by blocking hepatoma EGFR and angiogenesis by inhibiting endothelial VEGFR. These promising recent results warrant further evaluation in clinical trials[89–92]. For zactima successful testing in clinical trials has already been reported for non-HCC tumour entities like NSCLC and thyroid cancer[93,94]. Table 3 summarizes the current status of anti-VEGF/VEGFR-based approaches in the treatment of solid tumours including HCC.
Table 3.
Agents for anti-VEGF/VEGFR-based therapy of solid tumours
| Name | Target | Mechanism | Current status |
| Small molecule inhibitors | |||
| Sunitinib (Sutent) | PDGFR, VEGFR, c-KIT, FLT-3 | Tyrosine kinase inhibitor | Approved for advanced renal cell carcinoma and GIST (with restricted indications)[87,150] |
| PhaseIfor HCC[151] | |||
| Zactima (ZD6474) | VEGFR, EGFR | Tyrosine kinase inhibitor | Phase III for NSCLC[156] |
| Phase II for thyroid cancer[157] | |||
| Vatalanib (PTK787/ZK 222584) | VEGFR, PDGFR, C-KIT | Tyrosine kinase inhibitor | Phase II/III for colorectal cancer[152] |
| PhaseIfor HCC[153] | |||
| Anti-ligand targeting | |||
| Bevacizumab | VEGF | VEGF-neutralizing antibody | Approved for colorectal cancer |
| Phase III for NSCLC[166] | |||
| Phase II for HCC[154] | |||
| VEGF trap | VEGF | Soluble decoy receptor which neutralizes all VEGF A isoforms | PhaseIfor advanced solid tumours[155] |
OTHER MULTI-KINASE AND GROWTH FACTOR RECEPTOR INDEPENDENT INHIBITORS
Multi-kinase inhibition
The novel bi-aryl urea sorafenib is an orally available multi-kinase inhibitor which targets kinases of wild-type B-Raf, mutantV559EB-Raf and C-Raf, thus blocking tumour growth. Furthermore, sorafenib potently inhibits receptor tyrosine kinases involved in angiogenesis, including human vascular endothelial growth factor receptors-2 and -3 (VEGFR-2/-3) and PDGF-βR. The principal mechanism of action of sorafenib is the competitive inhibition of ATP-binding to the catalytic domains of the respective kinases[95]. However, the fact that sorafenib is an oral multi-kinase inhibitor, with effects on several molecular targets in addition to the Raf isoforms makes it difficult to determine which of these targets contributes most to its anti-tumour activity in particular tumour types.
A recent phase II HCC clinical trial, which identified an association between high baseline tumour p-ERK levels and improved response to sorafenib, suggests that inhibition of the Raf/MEK/ERK pathway is central to sorafenib’s mode of anti-tumour action in HCC[96]. If this generally holds true for HCC remains to be determined. In other tumour entities the antineoplastic potency of sorafenib appears to be mainly due to its antiangiogenic activity[97,98].
It is of particular clinical importance to have reliable markers to individually predict the treatment outcome. It has been suggested that rash, which is commonly associated with EGF-pathway inhibition, could be predictive of treatment outcome, and that the onset of rash could be used for optimal dose titration[99]. This might also be effective in treatment with sorafenib, as it is an inhibitor of Raf kinase, which is a downstream effector molecule of the EGFR signalling pathway. A recent report combining data from four phase I trials supported this hypothesis. Patients receiving sorafenib dosed at or close to the recommended dose of 400 mg bid, and experiencing skin toxicity and/or diarrhea, had a significantly increased time to progression compared with patients without such toxicity[100].
Sorafenib inhibits the proliferation of a variety of human cancer cell lines and retards tumour growth in related xenograft models of NSCLC, breast, colon and pancreas carcinoma[13,101,102]. Sorafenib is also active in otherwise fairly therapy resistant cholangiocarcinoma cells. Here, it over-additively enhanced the antineoplastic effects of cytostatics such as doxorubicin or the histone deacetylase inhibitor MS-275 and acts synergistically with IGFR blockade[103,104]. Recent in vitro studies by our group confirmed the synergistic antiproliferative effects of a combination treatment with sorafenib and MS-275 in hepatocellular carcinoma models. Proliferation studies, with either Hep-G2 or Huh-7 cells, resulted in half-maximal growth inhibition at a sorafenib concentration of 1.6 ± 0.3 μmol/L (Hep-G2) and 4.4 ± 0.2 μmol/L (Huh-7), respectively. The IC50 of MS-275 amounted to 1.2 ± 0.1 μmol/L in Hep-G2 cells and 0.9 ± 0.2 μmol/L in Huh-7 cells. Co-application of sub-IC50 concentrations of sorafenib and MS-275 for three days resulted in significant over-additive growth inhibition of Huh-7 cells, while in Hep-G2 cells a rather additive growth inhibitory effect was observed (Figure 2). Our data support the idea of dual-targeting hepatocellular carcinoma cells for enhanced treatment efficacy and show that multi-kinase inhibition plus histone deacetylase inhibition appear to be a promising combination, warranting further elucidation in clinical trials.
Figure 2.
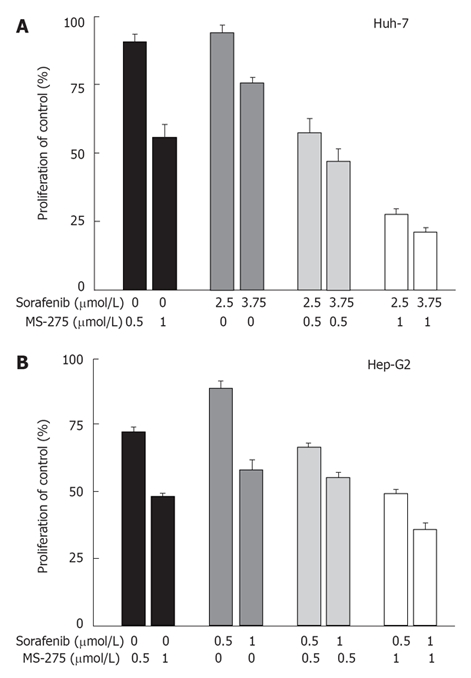
Antiproliferative effects of sorafenib-based combination treatment. A: Huh-7 and B: HEP-G2 cells were treated for 72 h with sub-IC50 concentrations of sorafenib and the histone deactylase inhibitor MS-275. Combination of both agents resulted in synergistic growth inhibition of Huh-7 cells, while rather additive growth inhibitory effects were observed in HepP-G2 cells (mean ± SEM).
A series of clinical studies have tested sorafenib’s antineoplastic potency in cancer patients. Phase I trials showed a favourable safety profile of 400 mg sorafenib administered twice daily for 12 wk in patients with advanced solid tumours (e.g. colon, ovary, breast, pancreas, kidney)[100,105]. Promising antitumour activities of sorafenib were observed in a phase II clinical study of patients with advanced melanoma[106]. Most encouraging results were seen in phase II and III trails of patients with metastatic renal cell carcinoma (RCC) which led to approval in the US for advanced RCC[107].
Sorafenib has also been tested for the treatment of advanced HCC in phase II and III trials. In a phase IItrial on 137 patients with inoperable HCC the continuous oral application of sorafenib 400 mg bid in 4-wk cycles revealed a significant attenuation of HCC growth in 1/3 of the patients[108] resulting in a further evaluation in a randomized double-blinded phase III trial with 602 patients with advanced HCC. An interim evaluation of this international multi-center SHARP-study (Sorafenib HCC Assessment Randomized Protocol) led to discontinuation, as the HCC patients treated with sorafenib achieved a significant survival benefit over the placebo-treated controls. Llovet et al presented the respective data for the Sharp investigators study group at the ASCO meeting in 2007 and showed that the treatment of advanced HCC patients with sorafenib leads to a 44% improvement in the overall survival as compared to the control group. The median overall survival in the sorafenib treated arm was 10.7 mo vs 7.9 mo in the control arm. Moreover, the median time to progression was almost doubled (5.5 mo in the sorafenib arm vs 2.8 mo in the control arm). The authors concluded that the effects of sorafenib treatment are clinically meaningful and establish sorafenib as first-line treatment for patients with advanced HCC[109]. Based on these findings, sorafenib has recently gained accelerated approval by the FDA for the treatment of advanced unresectable HCC.
mTOR inhibition
The natural antibiotic rapamycin is a potent inhibitor of mTOR[110]. Recently, three analogues of rapamycin with superior pharmacokinetic and biological properties have been synthesized and tested in clinical trials for different malignancies. The cell cycle inhibitor-779 (CCI-779, temsirolimus) is a soluble ester analogue. RAD001 (40-O-[2-hydroxyethyl]-rapamycin, everolimus) is an orally bioavailable derivative of rapamycin, and finally AP23573, which is a non-pro-drug analogue of rapamycin. These agents have been tested successfully in early clinical trials for their antineoplastic potency and/or tolerability in various malignancies, such as renal, breast and lung cancers (CCI-779), or are currently being studied in open clinical trials for the treatment of colorectal, endometrial cancer, recurrent or refractory solid tumours, and brain tumours (RAD001, everolimus)[111–113]. AP23573 has been successfully tested in a phase II trial in sarcomas[114] and two phaseIstudies in patients with refractory or advanced solid tumours, showing partial responses and disease stabilisation in individual patients[115].
In vitro as well as preclinical in vivo data of HCC show, that mTOR inhibition by rapamycin and analogues significantly reduces HCC growth and improves survival primarily via antiangiogenic effects[116]. A Phase I/II trial evaluating everolimus for advanced HCC is currently starting to recruit patients[117] (NIH, NCT00390195). Moreover, the use of rapamycin and analogues for combination treatment together with conventional cytostatic drugs such as doxorubicin or vinblastine has been demonstrated to additively or even synergistically enhance the antineoplastic potency of the respective monotherapeutic HCC treatment with either doxorubicin or vinblastine alone[118–120].
Taken together, the in vitro and preclinical in vivo data as well as the clinical trials conducted so far show that mTOR inhibitors, including the rapamycin analogues CCI-779, RAD001 and AP23573, are promising combination agents for future cancer therapy. They are well tolerated and can produce stable disease or even substantial responses in relapsed or conventional therapy resistant solid tumours[115].
Proteasome inhibition
Another interesting therapeutic approach for innovative cancer treatment is the inhibition of the 26S proteasome, which is a large protease that is present in both the nucelus and the cytoplasm of eukaryotic cells and functions as an identifier and destructor of proteins branded for destruction by the ubiquitin system. The so called ubiquitin-proteasome pathway (UPP) is the major non-lysosomal proteolytic system in eukaryotic cells and triggers degradation of proteins involved in cell cycle progression, apoptosis, nuclear factor kappaB (NF-κB) activation, and angiogenesis. UPP also degrades mutant, damaged, and misfolded proteins[121]. Since these signalling pathways are critical for cell survival and proliferation, especially in cancer cells, the inhibition of the proteasome has emerged as an attractive target for cancer therapy.
Bortezomib (Velcade™) is a proteasome inhibitor, which blocks multi-ubiquitinated protein degradation by reversibly and competitively inhibiting the active site threonine residue of the 26S proteasome[122]. Antineoplastic activity of bortezomib has already been shown in several in vitro and in vivo studies[104,123,124]. Bortezomib is the first proteasome inhibitor which has been approved for cancer therapy and is in use for the treatment of advanced multiple myeloma[121]. Based on the results of a phase II trial on bortzezomib in the treatment of mantle cell lymphoma (ML) the FDA recently granted approval to bortezomib for the treatment of patients with ML (www.cancer.gov)[125]. Other cancers, including neuroendocrine tumours, RCC, NSCLC, or metastatic sarcomas have also been evaluated in recent phase II clinical trials. In some of these studies a significant antineoplastic effect of monotherapy with bortezomib was observed, while in some other studies no or only marginal responses to single treatment with bortezomib were found[126–128]. However, in these cases it was recommended to investigate the role of bortezomib in combination with other antitumoural drugs. The rationale for using bortezomib in combination treatment regimes is that bortezomib's mode of action is mainly based on the inhibition of the NF-κB pathway, which has been shown to exert chemosensitizing effects when administered together with other antitumoural drugs. Combination treatment studies with encouraging results have been reported for lung cancer and lymphoma[129–131]. A phase I/II trial of bortezomib in patients with unresectable HCC was recently reported to result in disease stabilization in some patients and the treatment was generally well tolerated. In this study it was also suggested to especially focus on combination treatment strategies using bortezomib together with HCC-relevant cytostatics such as doxorubicin[132]. We have recently conducted an in vitro evaluation of bortezomib-based treatment of HCC cells. Our findings underline the suitability of bortezomib for the treatment of HCC-alone or in combination with sorafenib. In Huh-7 and Hep-G2 cells nanomolar concentrations of bortezomib induced a marked growth inhibition after three days of treatment. Moreover, the combination of bortezomib and sub-IC50 concentrations of sorafenib resulted in additive growth inhibition in both hepatocellular Huh-7 and hepatoma Hep-G2 cells (Figure 3). Thus, our data support the idea of dual-targeting hepatocellular carcinoma cells for enhanced treatment efficacy using bortezomib as a combinatory drug. Our data strengthen the conception of multi-kinase inhibition plus bortezomib to be a promising combination for future HCC treatment, warranting further elucidation in clinical trials. Table 4 summarizes the current status of multi-kinase inhibitors and growth factor independent inhibitors for the treatment of solid tumours.
Figure 3.

Antiproliferative effects of combination treatment with bortezomib and sorafenib in hepatocelluar carcinoma cells. A: Huh-7 and B: Hep-G2 cells were treated for 72 h with sub-IC50 concentrations of sorafenib and the proteasome inhibitor bortezomib. Combination of both agents led to additive growth inhibition both in Huh-7 as well as in Hep-G2 cells (mean ± SEM).
Table 4.
Agents of multi-kinase- and growth factor independent inhibition for the therapy of solid tumours
| Name | Target | Mechanism | Current status |
| Sorafenib | c-Raf-1, | Tyrosine kinase inhibitor | Approved for advanced RCC |
| B-Raf, VEGFR, PDGFR | Phase III for advanced HCC[108] | ||
| Phase II for melanoma[106], breast cancer[158] and NSCLC[159] | |||
| Phase Ifor advanced solid tumours[160] | |||
| Everolimus (RAD001) | mTOR | Tyrosine kinase inhibitor | Phase II for colorectal cancer[165] |
| PhaseI/II for advanced HCC[117,116] | |||
| PhaseIfor endometrial and brain tumours[111,112] | |||
| Temsirolimus (CCI-779) | mTOR | Tyrosine kinase inhibitor | Phase II for metastatic breast cancer[161], advanced RCC[162], and mantle cell lymphoma[163] |
| PhaseIfor advanced solid tumours (e.g. colorectal, ovarian, lung cancer)[164] | |||
| AP23573 | mTOR | Tyrosine kinase inhibitor | Phase II for sarcomas of soft tissue and bone[114] |
| PhaseIfor advanced solid tumours | |||
| Bortezomib (Velcade) | Proetasome | Proteasome inhibitor | Approved for multiple myeloma and mantle cell lymphoma[125] |
| Phase II for colorectal cancer[166], neuroendocrine tumours[128], sarcoma[126], RCC[127], and NSCLC[129] | |||
| PhaseI/II for unresectable HCC[132] | |||
| PhaseIfor advanced solid tumours[167] |
CONCLUSION
The concept of targeted-therapies which specifically inhibit growth factor receptors and their related signalling pathways emerged to be a promising approach for the innovative and effective medical treatment of various cancers, including hepatocellular carcinoma. Thus, advanced HCC is no longer a tumour disease without specific medical treatment options. The recent findings and clinical trials clearly demonstrate that especially combination treatments inhibiting more than just one signalling pathway will be particularly efficient, as it leaves less mechanisms of escape for the tumour cells.
In addition, there are several other promising new drugs which are currently being tested or which should be investigated in future HCC trials. In this respect combinations with drugs such as multi-kinase inhibitors are particularly intriguing. Thus in the future agents like the multi-kinase inhibitor sorafenib will likely be combined with growth factor receptor inhibitors, proteasome inhibitors, HDAC inhibitors or cytostatics as to effectively control advanced HCC. The advantage of such novel combination therapies is their higher efficacy at lowered toxicity as compared to monotherapeutic approaches. The novel combination treatments will offer new chances for drug therapy even in HCC patients with underlying cirrhosis. Fortunately, most of the new drugs can be taken orally.
Footnotes
S- Editor Ma N L- Editor Rippe RA E- Editor Liu Y
References
- 1.Avila MA, Berasain C, Sangro B, Prieto J. New therapies for hepatocellular carcinoma. Oncogene. 2006;25:3866–3884. doi: 10.1038/sj.onc.1209550. [DOI] [PubMed] [Google Scholar]
- 2.El-Serag HB, Davila JA, Petersen NJ, McGlynn KA. The continuing increase in the incidence of hepatocellular carcinoma in the United States: an update. Ann Intern Med. 2003;139:817–823. doi: 10.7326/0003-4819-139-10-200311180-00009. [DOI] [PubMed] [Google Scholar]
- 3.Angulo P, Lindor KD. Non-alcoholic fatty liver disease. J Gastroenterol Hepatol. 2002;17 Suppl:S186–S190. doi: 10.1046/j.1440-1746.17.s1.10.x. [DOI] [PubMed] [Google Scholar]
- 4.McGlynn KA, Tsao L, Hsing AW, Devesa SS, Fraumeni JF Jr. International trends and patterns of primary liver cancer. Int J Cancer. 2001;94:290–296. doi: 10.1002/ijc.1456. [DOI] [PubMed] [Google Scholar]
- 5.Bruix J, Hessheimer AJ, Forner A, Boix L, Vilana R, Llovet JM. New aspects of diagnosis and therapy of hepatocellular carcinoma. Oncogene. 2006;25:3848–3856. doi: 10.1038/sj.onc.1209548. [DOI] [PubMed] [Google Scholar]
- 6.Llovet JM, Burroughs A, Bruix J. Hepatocellular carcinoma. Lancet. 2003;362:1907–1917. doi: 10.1016/S0140-6736(03)14964-1. [DOI] [PubMed] [Google Scholar]
- 7.Simonetti RG, Liberati A, Angiolini C, Pagliaro L. Treatment of hepatocellular carcinoma: a systematic review of randomized controlled trials. Ann Oncol. 1997;8:117–136. doi: 10.1023/a:1008285123736. [DOI] [PubMed] [Google Scholar]
- 8.Zollner G, Wagner M, Fickert P, Silbert D, Fuchsbichler A, Zatloukal K, Denk H, Trauner M. Hepatobiliary transporter expression in human hepatocellular carcinoma. Liver Int. 2005;25:367–379. doi: 10.1111/j.1478-3231.2005.01033.x. [DOI] [PubMed] [Google Scholar]
- 9.Duncan SA. Mechanisms controlling early development of the liver. Mech Dev. 2003;120:19–33. doi: 10.1016/s0925-4773(02)00328-3. [DOI] [PubMed] [Google Scholar]
- 10.Strain AJ, Diehl AM. Liver growth and repair. London: Chapman & Hall Ltd; 1998. pp. 28–44. [Google Scholar]
- 11.Campbell JS, Hughes SD, Gilbertson DG, Palmer TE, Holdren MS, Haran AC, Odell MM, Bauer RL, Ren HP, Haugen HS, et al. Platelet-derived growth factor C induces liver fibrosis, steatosis, and hepatocellular carcinoma. Proc Natl Acad Sci USA. 2005;102:3389–3394. doi: 10.1073/pnas.0409722102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ogasawara S, Yano H, Iemura A, Hisaka T, Kojiro M. Expressions of basic fibroblast growth factor and its receptors and their relationship to proliferation of human hepatocellular carcinoma cell lines. Hepatology. 1996;24:198–205. doi: 10.1053/jhep.1996.v24.pm0008707262. [DOI] [PubMed] [Google Scholar]
- 13.Sridhar SS, Hedley D, Siu LL. Raf kinase as a target for anticancer therapeutics. Mol Cancer Ther. 2005;4:677–685. doi: 10.1158/1535-7163.MCT-04-0297. [DOI] [PubMed] [Google Scholar]
- 14.Hwang YH, Choi JY, Kim S, Chung ES, Kim T, Koh SS, Lee B, Bae SH, Kim J, Park YM. Over-expression of c-raf-1 proto-oncogene in liver cirrhosis and hepatocellular carcinoma. Hepatol Res. 2004;29:113–121. doi: 10.1016/j.hepres.2004.02.009. [DOI] [PubMed] [Google Scholar]
- 15.Grisham JW. Interspecies comparison of liver carcinogenesis: implications for cancer risk assessment. Carcinogenesis. 1997;18:59–81. doi: 10.1093/carcin/18.1.59. [DOI] [PubMed] [Google Scholar]
- 16.Calvisi DF, Ladu S, Gorden A, Farina M, Conner EA, Lee JS, Factor VM, Thorgeirsson SS. Ubiquitous activation of Ras and Jak/Stat pathways in human HCC. Gastroenterology. 2006;130:1117–1128. doi: 10.1053/j.gastro.2006.01.006. [DOI] [PubMed] [Google Scholar]
- 17.Bromberg J, Darnell JE Jr. The role of STATs in transcriptional control and their impact on cellular function. Oncogene. 2000;19:2468–2473. doi: 10.1038/sj.onc.1203476. [DOI] [PubMed] [Google Scholar]
- 18.Bowman T, Garcia R, Turkson J, Jove R. STATs in oncogenesis. Oncogene. 2000;19:2474–2488. doi: 10.1038/sj.onc.1203527. [DOI] [PubMed] [Google Scholar]
- 19.Calo V, Migliavacca M, Bazan V, Macaluso M, Buscemi M, Gebbia N, Russo A. STAT proteins: from normal control of cellular events to tumorigenesis. J Cell Physiol. 2003;197:157–168. doi: 10.1002/jcp.10364. [DOI] [PubMed] [Google Scholar]
- 20.Nagai H, Kim YS, Lee KT, Chu MY, Konishi N, Fujimoto J, Baba M, Matsubara K, Emi M. Inactivation of SSI-1, a JAK/STAT inhibitor, in human hepatocellular carcinomas, as revealed by two-dimensional electrophoresis. J Hepatol. 2001;34:416–421. doi: 10.1016/s0168-8278(00)00038-6. [DOI] [PubMed] [Google Scholar]
- 21.Yoshikawa H, Matsubara K, Qian GS, Jackson P, Groopman JD, Manning JE, Harris CC, Herman JG. SOCS-1, a negative regulator of the JAK/STAT pathway, is silenced by methylation in human hepatocellular carcinoma and shows growth-suppression activity. Nat Genet. 2001;28:29–35. doi: 10.1038/ng0501-29. [DOI] [PubMed] [Google Scholar]
- 22.Adjei AA, Hidalgo M. Intracellular signal transduction pathway proteins as targets for cancer therapy. J Clin Oncol. 2005;23:5386–5403. doi: 10.1200/JCO.2005.23.648. [DOI] [PubMed] [Google Scholar]
- 23.Horie Y, Suzuki A, Kataoka E, Sasaki T, Hamada K, Sasaki J, Mizuno K, Hasegawa G, Kishimoto H, Iizuka M, et al. Hepatocyte-specific Pten deficiency results in steatohepatitis and hepatocellular carcinomas. J Clin Invest. 2004;113:1774–1783. doi: 10.1172/JCI20513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bianco R, Shin I, Ritter CA, Yakes FM, Basso A, Rosen N, Tsurutani J, Dennis PA, Mills GB, Arteaga CL. Loss of PTEN/MMAC1/TEP in EGF receptor-expressing tumor cells counteracts the antitumor action of EGFR tyrosine kinase inhibitors. Oncogene. 2003;22:2812–2822. doi: 10.1038/sj.onc.1206388. [DOI] [PubMed] [Google Scholar]
- 25.Boyault S, Rickman DS, de Reynies A, Balabaud C, Rebouissou S, Jeannot E, Herault A, Saric J, Belghiti J, Franco D, et al. Transcriptome classification of HCC is related to gene alterations and to new therapeutic targets. Hepatology. 2007;45:42–52. doi: 10.1002/hep.21467. [DOI] [PubMed] [Google Scholar]
- 26.Sangro B, Mazzollini G, Prieto J. Future therapies for hepatocellular carcinoma. Eur J Gastroenterol Hepatol. 2005;17:515–521. doi: 10.1097/00042737-200505000-00007. [DOI] [PubMed] [Google Scholar]
- 27.Hopfner M, Sutter AP, Huether A, Schuppan D, Zeitz M, Scherubl H. Targeting the epidermal growth factor receptor by gefitinib for treatment of hepatocellular carcinoma. J Hepatol. 2004;41:1008–1016. doi: 10.1016/j.jhep.2004.08.024. [DOI] [PubMed] [Google Scholar]
- 28.Huether A, Hopfner M, Sutter AP, Schuppan D, Scherubl H. Erlotinib induces cell cycle arrest and apoptosis in hepatocellular cancer cells and enhances chemosensitivity towards cytostatics. J Hepatol. 2005;43:661–669. doi: 10.1016/j.jhep.2005.02.040. [DOI] [PubMed] [Google Scholar]
- 29.Huether A, Hopfner M, Baradari V, Schuppan D, Scherubl H. EGFR blockade by cetuximab alone or as combination therapy for growth control of hepatocellular cancer. Biochem Pharmacol. 2005;70:1568–1578. doi: 10.1016/j.bcp.2005.09.007. [DOI] [PubMed] [Google Scholar]
- 30.Ueda S, Basaki Y, Yoshie M, Ogawa K, Sakisaka S, Kuwano M, Ono M. PTEN/Akt signaling through epidermal growth factor receptor is prerequisite for angiogenesis by hepatocellular carcinoma cells that is susceptible to inhibition by gefitinib. Cancer Res. 2006;66:5346–5353. doi: 10.1158/0008-5472.CAN-05-3684. [DOI] [PubMed] [Google Scholar]
- 31.Liu Y, Poon RT, Shao W, Sun X, Chen H, Kok TW, Fan ST. Blockage of epidermal growth factor receptor by quinazoline tyrosine kinase inhibitors suppresses growth of human hepatocellular carcinoma. Cancer Lett. 2007;248:32–40. doi: 10.1016/j.canlet.2006.05.018. [DOI] [PubMed] [Google Scholar]
- 32.Matsuo M, Sakurai H, Saiki I. ZD1839, a selective epidermal growth factor receptor tyrosine kinase inhibitor, shows antimetastatic activity using a hepatocellular carcinoma model. Mol Cancer Ther. 2003;2:557–561. [PubMed] [Google Scholar]
- 33.Philip PA, Mahoney MR, Allmer C, Thomas J, Pitot HC, Kim G, Donehower RC, Fitch T, Picus J, Erlichman C. Phase II study of Erlotinib (OSI-774) in patients with advanced hepatocellular cancer. J Clin Oncol. 2005;23:6657–6663. doi: 10.1200/JCO.2005.14.696. [DOI] [PubMed] [Google Scholar]
- 34.Greten TF. Molecular therapy for HCC? Z Gastroenterol. 2006;44:205–206. doi: 10.1055/s-2005-859031. [DOI] [PubMed] [Google Scholar]
- 35.Bourhis J, Rivera F, Mesia R, Awada A, Geoffrois L, Borel C, Humblet Y, Lopez-Pousa A, Hitt R, Vega Villegas ME, et al. Phase I/II study of cetuximab in combination with cisplatin or carboplatin and fluorouracil in patients with recurrent or metastatic squamous cell carcinoma of the head and neck. J Clin Oncol. 2006;24:2866–2872. doi: 10.1200/JCO.2005.04.3547. [DOI] [PubMed] [Google Scholar]
- 36.Moore MJ, Goldstein D, Hamm J, Kotecha J, Gallinger S, Au HJ. Ding K. Christy-Bittel J, Parulekar W. Erlotinib improves survival when added to gemcitabine in patients with advanced pancreatic cancer. A phase III trial of the National Cancer Institute of Canada Clinical Trials Group [NCIC-CTG] J Clin Oncol. 2005;ASCO Annual Meeting Proceedings; 23 Suppl 16::LBA1. [Google Scholar]
- 37.Ciardiello F, Troiani T, Bianco R, Orditura M, Morgillo F, Martinelli E, Morelli MP, Cascone T, Tortora G. Interaction between the epidermal growth factor receptor (EGFR) and the vascular endothelial growth factor (VEGF) pathways: a rational approach for multi-target anticancer therapy. Ann Oncol. 2006;17:vii109–vii114. doi: 10.1093/annonc/mdl962. [DOI] [PubMed] [Google Scholar]
- 38.Tortora G, Caputo R, Damiano V, Melisi D, Bianco R, Fontanini G, Veneziani BM, De Placido S, Bianco AR, Ciardiello F. Combination of a selective cyclooxygenase-2 inhibitor with epidermal growth factor receptor tyrosine kinase inhibitor ZD1839 and protein kinase A antisense causes cooperative antitumor and antiangiogenic effect. Clin Cancer Res. 2003;9:1566–1572. [PubMed] [Google Scholar]
- 39.Ganslmayer M, Ocker M, Kraemer G, Zopf S, Hahn EG, Schuppan D, Herold C. The combination of tamoxifen and 9cis retinoic acid exerts overadditive anti-tumoral efficacy in rat hepatocellular carcinoma. J Hepatol. 2004;40:952–956. doi: 10.1016/j.jhep.2004.02.004. [DOI] [PubMed] [Google Scholar]
- 40.Ganslmayer M, Ocker M, Zopf S, Leitner S, Hahn EG, Schuppan D, Herold C. A quadruple therapy synergistically blocks proliferation and promotes apoptosis of hepatoma cells. Oncol Rep. 2004;11:943–950. [PubMed] [Google Scholar]
- 41.Herold C, Ganslmayer M, Ocker M, Blauberger S, Zopf S, Hahn EG, Schuppan D. Overadditive anti-proliferative and pro-apoptotic effects of a combination therapy on colorectal carcinoma cells. Int J Oncol. 2003;23:751–756. [PubMed] [Google Scholar]
- 42.Maione P, Gridelli C, Troiani T, Ciardiello F. Combining targeted therapies and drugs with multiple targets in the treatment of NSCLC. Oncologist. 2006;11:274–284. doi: 10.1634/theoncologist.11-3-274. [DOI] [PubMed] [Google Scholar]
- 43.Sachdev D, Yee D. Disrupting insulin-like growth factor signaling as a potential cancer therapy. Mol Cancer Ther. 2007;6:1–12. doi: 10.1158/1535-7163.MCT-06-0080. [DOI] [PubMed] [Google Scholar]
- 44.Hofmann F, Garcia-Echeverria C. Blocking the insulin-like growth factor-I receptor as a strategy for targeting cancer. Drug Discov Today. 2005;10:1041–1047. doi: 10.1016/S1359-6446(05)03512-9. [DOI] [PubMed] [Google Scholar]
- 45.Wang Y, Sun Y. Insulin-like growth factor receptor-1 as an anti-cancer target: blocking transformation and inducing apoptosis. Curr Cancer Drug Targets. 2002;2:191–207. doi: 10.2174/1568009023333863. [DOI] [PubMed] [Google Scholar]
- 46.Wang Z, Ruan YB, Guan Y, Liu SH. Expression of IGF-II in early experimental hepatocellular carcinomas and its significance in early diagnosis. World J Gastroenterol. 2003;9:267–270. doi: 10.3748/wjg.v9.i2.267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sedlaczek N, Hasilik A, Neuhaus P, Schuppan D, Herbst H. Focal overexpression of insulin-like growth factor 2 by hepatocytes and cholangiocytes in viral liver cirrhosis. Br J Cancer. 2003;88:733–739. doi: 10.1038/sj.bjc.6600777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Scharf JG, Braulke T. The role of the IGF axis in hepatocar-cinogenesis. Horm Metab Res. 2003;35:685–693. doi: 10.1055/s-2004-814151. [DOI] [PubMed] [Google Scholar]
- 49.Yao X, Hu JF, Daniels M, Yien H, Lu H, Sharan H, Zhou X, Zeng Z, Li T, Yang Y, et al. A novel orthotopic tumor model to study growth factors and oncogenes in hepatocarcinogenesis. Clin Cancer Res. 2003;9:2719–2726. [PubMed] [Google Scholar]
- 50.Alexia C, Fallot G, Lasfer M, Schweizer-Groyer G, Groyer A. An evaluation of the role of insulin-like growth factors (IGF) and of type-I IGF receptor signalling in hepatocarcinogenesis and in the resistance of hepatocarcinoma cells against drug-induced apoptosis. Biochem Pharmacol. 2004;68:1003–1015. doi: 10.1016/j.bcp.2004.05.029. [DOI] [PubMed] [Google Scholar]
- 51.Tsai TF, Yauk YK, Chou CK, Ting LP, Chang C, Hu CP, Han SH, Su TS. Evidence of autocrine regulation in human hepatoma cell lines. Biochem Biophys Res Commun. 1988;153:39–45. doi: 10.1016/s0006-291x(88)81186-0. [DOI] [PubMed] [Google Scholar]
- 52.Scharf JG, Schmidt-Sandte W, Pahernik SA, Ramadori G, Braulke T, Hartmann H. Characterization of the insulin-like growth factor axis in a human hepatoma cell line (PLC) Carcinogenesis. 1998;19:2121–2128. doi: 10.1093/carcin/19.12.2121. [DOI] [PubMed] [Google Scholar]
- 53.Yu H, Rohan T. Role of the insulin-like growth factor family in cancer development and progression. J Natl Cancer Inst. 2000;92:1472–1489. doi: 10.1093/jnci/92.18.1472. [DOI] [PubMed] [Google Scholar]
- 54.Scotlandi K, Benini S, Nanni P, Lollini PL, Nicoletti G, Landuzzi L, Serra M, Manara MC, Picci P, Baldini N. Blockage of insulin-like growth factor-I receptor inhibits the growth of Ewing's sarcoma in athymic mice. Cancer Res. 1998;58:4127–4131. [PubMed] [Google Scholar]
- 55.Shapiro DN, Jones BG, Shapiro LH, Dias P, Houghton PJ. Antisense-mediated reduction in insulin-like growth factor-I receptor expression suppresses the malignant phenotype of a human alveolar rhabdomyosarcoma. J Clin Invest. 1994;94:1235–1242. doi: 10.1172/JCI117441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Salisbury AJ, Macaulay VM. Development of molecular agents for IGF receptor targeting. Horm Metab Res. 2003;35:843–849. doi: 10.1055/s-2004-814158. [DOI] [PubMed] [Google Scholar]
- 57.Ellouk-Achard S, Djenabi S, De Oliveira GA, Desauty G, Duc HT, Zohair M, Trojan J, Claude JR, Sarasin A, Lafarge-Frayssinet C. Induction of apoptosis in rat hepatocarcinoma cells by expression of IGF-I antisense c-DNA. J Hepatol. 1998;29:807–818. doi: 10.1016/s0168-8278(98)80263-8. [DOI] [PubMed] [Google Scholar]
- 58.Tanno B, Mancini C, Vitali R, Mancuso M, McDowell HP, Dominici C, Raschella G. Down-regulation of insulin-like growth factor I receptor activity by NVP-AEW541 has an antitumor effect on neuroblastoma cells in vitro and in vivo. Clin Cancer Res. 2006;12:6772–6780. doi: 10.1158/1078-0432.CCR-06-1479. [DOI] [PubMed] [Google Scholar]
- 59.Hopfner M, Baradari V, Huether A, Schofl C, Scherubl H. The insulin-like growth factor receptor 1 is a promising target for novel treatment approaches in neuroendocrine gastrointestinal tumours. Endocr Relat Cancer. 2006;13:135–149. doi: 10.1677/erc.1.01090. [DOI] [PubMed] [Google Scholar]
- 60.Hopfner M, Sutter AP, Huether A, Baradari V, Scherubl H. Tyrosine kinase of insulin-like growth factor receptor as target for novel treatment and prevention strategies of colorectal cancer. World J Gastroenterol. 2006;12:5635–5643. doi: 10.3748/wjg.v12.i35.5635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hopfner M, Huether A, Sutter AP, Baradari V, Schuppan D, Scherubl H. Blockade of IGF-1 receptor tyrosine kinase has antineoplastic effects in hepatocellular carcinoma cells. Biochem Pharmacol. 2006;71:1435–1448. doi: 10.1016/j.bcp.2006.02.006. [DOI] [PubMed] [Google Scholar]
- 62.Warshamana-Greene GS, Litz J, Buchdunger E, Garcia-Echeverria C, Hofmann F, Krystal GW. The insulin-like growth factor-I receptor kinase inhibitor, NVP-ADW742, sensitizes small cell lung cancer cell lines to the effects of chemotherapy. Clin Cancer Res. 2005;11:1563–1571. doi: 10.1158/1078-0432.CCR-04-1544. [DOI] [PubMed] [Google Scholar]
- 63.Manara MC, Landuzzi L, Nanni P, Nicoletti G, Zambelli D, Lollini PL, Nanni C, Hofmann F, Garcia-Echeverria C, Picci P, et al. Preclinical in vivo study of new insulin-like growth factor-I receptor--specific inhibitor in Ewing's sarcoma. Clin Cancer Res. 2007;13:1322–1330. doi: 10.1158/1078-0432.CCR-06-1518. [DOI] [PubMed] [Google Scholar]
- 64.Arteaga CL, Kitten LJ, Coronado EB, Jacobs S, Kull FC Jr, Allred DC, Osborne CK. Blockade of the type I somatomedin receptor inhibits growth of human breast cancer cells in athymic mice. J Clin Invest. 1989;84:1418–1423. doi: 10.1172/JCI114315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Garcia-Echeverria C, Pearson MA, Marti A, Meyer T, Mestan J, Zimmermann J, Gao J, Brueggen J, Capraro HG, Cozens R, et al. In vivo antitumor activity of NVP-AEW541-A novel, potent, and selective inhibitor of the IGF-IR kinase. Cancer Cell. 2004;5:231–239. doi: 10.1016/s1535-6108(04)00051-0. [DOI] [PubMed] [Google Scholar]
- 66.Scotlandi K, Manara MC, Nicoletti G, Lollini PL, Lukas S, Benini S, Croci S, Perdichizzi S, Zambelli D, Serra M, et al. Antitumor activity of the insulin-like growth factor-I receptor kinase inhibitor NVP-AEW541 in musculoskeletal tumors. Cancer Res. 2005;65:3868–3876. doi: 10.1158/0008-5472.CAN-04-3192. [DOI] [PubMed] [Google Scholar]
- 67.Feng Y, Zhu Z, Xiao X, Choudhry V, Barrett JC, Dimitrov DS. Novel human monoclonal antibodies to insulin-like growth factor (IGF)-II that potently inhibit the IGF receptor type I signal transduction function. Mol Cancer Ther. 2006;5:114–120. doi: 10.1158/1535-7163.MCT-05-0252. [DOI] [PubMed] [Google Scholar]
- 68.Hofmann F, Brueggen J, Capraro HG, Cozens R, Evans DB, Fabbro D, Ferrari S, Furet P, Garcia-Echeverria C, Geiger T, Porta DG, Liebetanz J, Maira SM, Marti A, Martiny-Baron G, Mestan J, Meyer T, Ruetz S, Stoltz B, Zimmermann J, Peterson MA. In vitro and in vivo profiling of selective and potent IGF-IR kinase inhibitors. Proc AACR. 2003;44:3798. [Google Scholar]
- 69.Burtrum D, Zhu Z, Lu D, Anderson DM, Prewett M, Pereira DS, Bassi R, Abdullah R, Hooper AT, Koo H, et al. A fully human monoclonal antibody to the insulin-like growth factor I receptor blocks ligand-dependent signaling and inhibits human tumor growth in vivo. Cancer Res. 2003;63:8912–8921. [PubMed] [Google Scholar]
- 70.Garber K. IGF-1: old growth factor shines as new drug target. J Natl Cancer Inst. 2005;97:790–792. doi: 10.1093/jnci/97.11.790. [DOI] [PubMed] [Google Scholar]
- 71.Leary A, Johnston SR. Small molecule signal transduction inhibitors for the treatment of solid tumors. Cancer Invest. 2007;25:347–365. doi: 10.1080/07357900701259694. [DOI] [PubMed] [Google Scholar]
- 72.Desbois-Mouthon C, Cacheux W, Blivet-Van Eggelpoel MJ, Barbu V, Fartoux L, Poupon R, Housset C, Rosmorduc O. Impact of IGF-1R/EGFR cross-talks on hepatoma cell sensitivity to gefitinib. Int J Cancer. 2006;119:2557–2566. doi: 10.1002/ijc.22221. [DOI] [PubMed] [Google Scholar]
- 73.Gilmore AP, Valentijn AJ, Wang P, Ranger AM, Bundred N, O'Hare MJ, Wakeling A, Korsmeyer SJ, Streuli CH. Activation of BAD by therapeutic inhibition of epidermal growth factor receptor and transactivation by insulin-like growth factor receptor. J Biol Chem. 2002;277:27643–27650. doi: 10.1074/jbc.M108863200. [DOI] [PubMed] [Google Scholar]
- 74.Huether A, Hopfner M, Sutter AP, Baradari V, Schuppan D, Scherubl H. Signaling pathways involved in the inhibition of epidermal growth factor receptor by erlotinib in hepatocellular cancer. World J Gastroenterol. 2006;12:5160–5167. doi: 10.3748/wjg.v12.i32.5160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Jones HE, Gee JM, Hutcheson IR, Knowlden JM, Barrow D, Nicholson RI. Growth factor receptor interplay and resistance in cancer. Endocr Relat Cancer. 2006;13 Suppl 1:S45–S51. doi: 10.1677/erc.1.01275. [DOI] [PubMed] [Google Scholar]
- 76.Thomas MB, Abbruzzese JL. Opportunities for targeted therapies in hepatocellular carcinoma. J Clin Oncol. 2005;23:8093–8108. doi: 10.1200/JCO.2004.00.1537. [DOI] [PubMed] [Google Scholar]
- 77.LeCouter J, Moritz DR, Li B, Phillips GL, Liang XH, Gerber HP, Hillan KJ, Ferrara N. Angiogenesis-independent endothelial protection of liver: role of VEGFR-1. Science. 2003;299:890–893. doi: 10.1126/science.1079562. [DOI] [PubMed] [Google Scholar]
- 78.Yamaguchi R, Yano H, Iemura A, Ogasawara S, Haramaki M, Kojiro M. Expression of vascular endothelial growth factor in human hepatocellular carcinoma. Hepatology. 1998;28:68–77. doi: 10.1002/hep.510280111. [DOI] [PubMed] [Google Scholar]
- 79.Poon RT, Fan ST, Wong J. Clinical significance of angiogenesis in gastrointestinal cancers: a target for novel prognostic and therapeutic approaches. Ann Surg. 2003;238:9–28. doi: 10.1097/01.sla.0000075047.47175.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Jinno K, Tanimizu M, Hyodo I, Nishikawa Y, Hosokawa Y, Doi T, Endo H, Yamashita T, Okada Y. Circulating vascular endothelial growth factor (VEGF) is a possible tumor marker for metastasis in human hepatocellular carcinoma. J Gastroenterol. 1998;33:376–382. doi: 10.1007/s005350050099. [DOI] [PubMed] [Google Scholar]
- 81.Hurwitz H, Fehrenbacher L, Novotny W, Cartwright T, Hainsworth J, Heim W, Berlin J, Baron A, Griffing S, Holmgren E, et al. Bevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic colorectal cancer. N Engl J Med. 2004;350:2335–2342. doi: 10.1056/NEJMoa032691. [DOI] [PubMed] [Google Scholar]
- 82.Sandler A, Gray R, Perry MC, Brahmer J, Schiller JH, Dowlati A, Lilenbaum R, Johnson DH. Paclitaxel-carboplatin alone or with bevacizumab for non-small-cell lung cancer. N Engl J Med. 2006;355:2542–2550. doi: 10.1056/NEJMoa061884. [DOI] [PubMed] [Google Scholar]
- 83.Semela D, Dufour JF. Angiogenesis and hepatocellular carcinoma. J Hepatol. 2004;41:864–880. doi: 10.1016/j.jhep.2004.09.006. [DOI] [PubMed] [Google Scholar]
- 84.Hewitt MR, Sun W, Haller DG, Mykulowycz G, Tuite CM, Rosen MA, Soulen MC, Caparro M, Giantonio BJ, Olthoff KM. A phase II trial of combination of capecitabine, oxaliplatin with bevacizumab in treatment of advanced hepatocellular carcinoma (HCC): Preliminary safety analysis. J Clin Oncol. 2006;ASCO Annual Meeting Proceedings; 24 Suppl 1::4098. [Google Scholar]
- 85.Liu Y, Poon RT, Li Q, Kok TW, Lau C, Fan ST. Both antiangiogenesis- and angiogenesis-independent effects are responsible for hepatocellular carcinoma growth arrest by tyrosine kinase inhibitor PTK787/ZK222584. Cancer Res. 2005;65:3691–3699. doi: 10.1158/0008-5472.CAN-04-3462. [DOI] [PubMed] [Google Scholar]
- 86.Steeghs N, Nortier JW, Gelderblom H. Small molecule tyrosine kinase inhibitors in the treatment of solid tumors: an update of recent developments. Ann Surg Oncol. 2007;14:942–953. doi: 10.1245/s10434-006-9227-1. [DOI] [PubMed] [Google Scholar]
- 87.Motzer RJ, Bukowski RM. Targeted therapy for metastatic renal cell carcinoma. J Clin Oncol. 2006;24:5601–5608. doi: 10.1200/JCO.2006.08.5415. [DOI] [PubMed] [Google Scholar]
- 88.Goodman VL, Rock EP, Dagher R, Ramchandani RP, Abraham S, Gobburu JV, Booth BP, Verbois SL, Morse DE, Liang CY, et al. Approval summary: sunitinib for the treatment of imatinib refractory or intolerant gastrointestinal stromal tumors and advanced renal cell carcinoma. Clin Cancer Res. 2007;13:1367–1373. doi: 10.1158/1078-0432.CCR-06-2328. [DOI] [PubMed] [Google Scholar]
- 89.Wiedmann M, Feisthammel J, Bluthner T, Tannapfel A, Kamenz T, Kluge A, Mossner J, Caca K. Novel targeted approaches to treating biliary tract cancer: the dual epidermal growth factor receptor and ErbB-2 tyrosine kinase inhibitor NVP-AEE788 is more efficient than the epidermal growth factor receptor inhibitors gefitinib and erlotinib. Anticancer Drugs. 2006;17:783–795. doi: 10.1097/01.cad.0000217433.48870.37. [DOI] [PubMed] [Google Scholar]
- 90.Younes MN, Park YW, Yazici YD, Gu M, Santillan AA, Nong X, Kim S, Jasser SA, El-Naggar AK, Myers JN. Concomitant inhibition of epidermal growth factor and vascular endothelial growth factor receptor tyrosine kinases reduces growth and metastasis of human salivary adenoid cystic carcinoma in an orthotopic nude mouse model. Mol Cancer Ther. 2006;5:2696–2705. doi: 10.1158/1535-7163.MCT-05-0228. [DOI] [PubMed] [Google Scholar]
- 91.Busby JE, Kim SJ, Yazici S, Nakamura T, Kim JS, He J, Maya M, Wang X, Do KA, Fan D, et al. Therapy of multidrug resistant human prostate tumors in the prostate of nude mice by simultaneous targeting of the epidermal growth factor receptor and vascular endothelial growth factor receptor on tumor-associated endothelial cells. Prostate. 2006;66:1788–1798. doi: 10.1002/pros.20519. [DOI] [PubMed] [Google Scholar]
- 92.Heymach JV. ZD6474--clinical experience to date. Br J Cancer. 2005;92 Suppl 1:S14–S20. doi: 10.1038/sj.bjc.6602604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Natale RB, Bodkin D, Govindan R, Sleckman B, Rizvi N, Capo A, Germonpré P, Stockman P, Kennedy S, Ranson M. ZD6474 versus gefitinib in patients with advanced NSCLC: Final results from a two-part, double-blind, randomized phase II trial. J Clin Oncol. 2006;ASCO annual meeting proceedings; 24 Suppl 18::7000. doi: 10.1200/JCO.2008.18.6015. [DOI] [PubMed] [Google Scholar]
- 94.Wells S, You YN, Lakhani V, Hou J, Langmuir P, Headley D. Skinner M, Morse M, Burch W, Schlumberger M. A phase II trial of ZD6474 in patients with hereditary metastatic medullary thyroid cancer. J Clin Oncol. 2006;ASCO Annual Meeting Proceedings; 24 Suppl 18::5533. [Google Scholar]
- 95.Wilhelm S, Chien DS. BAY 43-9006: preclinical data. Curr Pharm Des. 2002;8:2255–2257. doi: 10.2174/1381612023393026. [DOI] [PubMed] [Google Scholar]
- 96.Gollob JA, Wilhelm S, Carter C, Kelley SL. Role of Raf kinase in cancer: therapeutic potential of targeting the Raf/MEK/ERK signal transduction pathway. Semin Oncol. 2006;33:392–406. doi: 10.1053/j.seminoncol.2006.04.002. [DOI] [PubMed] [Google Scholar]
- 97.Wilhelm SM, Carter C, Tang L, Wilkie D, McNabola A, Rong H, Chen C, Zhang X, Vincent P, McHugh M, et al. BAY 43-9006 exhibits broad spectrum oral antitumor activity and targets the RAF/MEK/ERK pathway and receptor tyrosine kinases involved in tumor progression and angiogenesis. Cancer Res. 2004;64:7099–7109. doi: 10.1158/0008-5472.CAN-04-1443. [DOI] [PubMed] [Google Scholar]
- 98.Liu L, Cao Y, Chen C, Zhang X, McNabola A, Wilkie D, Wilhelm S, Lynch M, Carter C. Sorafenib blocks the RAF/MEK/ERK pathway, inhibits tumor angiogenesis, and induces tumor cell apoptosis in hepatocellular carcinoma model PLC/PRF/5. Cancer Res. 2006;66:11851–11858. doi: 10.1158/0008-5472.CAN-06-1377. [DOI] [PubMed] [Google Scholar]
- 99.Perez-Soler R. Can rash associated with HER1/EGFR inhibition be used as a marker of treatment outcome? Oncology (Williston Park) 2003;17:23–28. [PubMed] [Google Scholar]
- 100.Strumberg D, Awada A, Hirte H, Clark JW, Seeber S, Piccart P, Hofstra E, Voliotis D, Christensen O, Brueckner A, et al. Pooled safety analysis of BAY 43-9006 (sorafenib) monotherapy in patients with advanced solid tumours: Is rash associated with treatment outcome? Eur J Cancer. 2006;42:548–556. doi: 10.1016/j.ejca.2005.11.014. [DOI] [PubMed] [Google Scholar]
- 101.Beeram M, Patnaik A, Rowinsky EK. Raf: a strategic target for therapeutic development against cancer. J Clin Oncol. 2005;23:6771–6790. doi: 10.1200/JCO.2005.08.036. [DOI] [PubMed] [Google Scholar]
- 102.Gridelli C, Maione P, Del Gaizo F, Colantuoni G, Guerriero C, Ferrara C, Nicolella D, Comunale D, De Vita A, Rossi A. Sorafenib and sunitinib in the treatment of advanced non-small cell lung cancer. Oncologist. 2007;12:191–200. doi: 10.1634/theoncologist.12-2-191. [DOI] [PubMed] [Google Scholar]
- 103.Huether A, Hopfner M, Baradari V, Schuppan D, Scherubl H. Sorafenib alone or as combination therapy for growth control of cholangiocarcinoma. Biochem Pharmacol. 2007;73:1308–1317. doi: 10.1016/j.bcp.2006.12.031. [DOI] [PubMed] [Google Scholar]
- 104.Baradari V, Hopfner M, Huether A, Schuppan D, Scherubl H. Histone deacetylase inhibitor MS-275 alone or combined with bortezomib or sorafenib exhibits strong antiproliferative action in human cholangiocarcinoma cells. World J Gastroenterol. 2007;13:4458–4466. doi: 10.3748/wjg.v13.i33.4458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Hotte SJ, Hirte HW. BAY 43-9006: early clinical data in patients with advanced solid malignancies. Curr Pharm Des. 2002;8:2249–2253. doi: 10.2174/1381612023393053. [DOI] [PubMed] [Google Scholar]
- 106.Ahmad T, Eisen T. Kinase inhibition with BAY 43-9006 in renal cell carcinoma. Clin Cancer Res. 2004;10:6388S–6392S. doi: 10.1158/1078-0432.CCR-040028. [DOI] [PubMed] [Google Scholar]
- 107.Escudier B, Eisen T, Stadler WM, Szczylik C, Oudard S, Siebels M, Negrier S, Chevreau C, Solska E, Desai AA, et al. Sorafenib in advanced clear-cell renal-cell carcinoma. N Engl J Med. 2007;356:125–134. doi: 10.1056/NEJMoa060655. [DOI] [PubMed] [Google Scholar]
- 108.Abou-Alfa GK, Schwartz L, Ricci S, Amadori D, Santoro A, Figer A, De Greve J, Douillard JY, Lathia C, Schwartz B, et al. Phase II study of sorafenib in patients with advanced hepatocellular carcinoma. J Clin Oncol. 2006;24:4293–4300. doi: 10.1200/JCO.2005.01.3441. [DOI] [PubMed] [Google Scholar]
- 109.Llovet J, Ricci S, Mazzaferro S, Hilgard P, Raoul J, Zeuzem S, Poulin-Costello M, Moscovici M, Voliotis D, Bruix J For the SHARP Investigators Study Group. Sorafenib improves survival in advanced Hepatocellular Carcinoma (HCC): Results of a Phase III randomized placebo-controlled trial (SHARP trial) J Clin Oncol. 2007;ASCO annual meeting proceedings; 25 Suppl 18::LBA1. [Google Scholar]
- 110.Tsang CK, Qi H, Liu LF, Zheng XF. Targeting mammalian target of rapamycin (mTOR) for health and diseases. Drug Discov Today. 2007;12:112–124. doi: 10.1016/j.drudis.2006.12.008. [DOI] [PubMed] [Google Scholar]
- 111.Dudkin L, Dilling MB, Cheshire PJ, Harwood FC, Hollingshead M, Arbuck SG, Travis R, Sausville EA, Houghton PJ. Biochemical correlates of mTOR inhibition by the rapamycin ester CCI-779 and tumor growth inhibition. Clin Cancer Res. 2001;7:1758–1764. [PubMed] [Google Scholar]
- 112.Easton JB, Houghton PJ. mTOR and cancer therapy. Oncogene. 2006;25:6436–6446. doi: 10.1038/sj.onc.1209886. [DOI] [PubMed] [Google Scholar]
- 113.Wullschleger S, Loewith R, Hall MN. TOR signaling in growth and metabolism. Cell. 2006;124:471–484. doi: 10.1016/j.cell.2006.01.016. [DOI] [PubMed] [Google Scholar]
- 114.Okuno S. Mammalian target of rapamycin inhibitors in sarcomas. Curr Opin Oncol. 2006;18:360–362. doi: 10.1097/01.cco.0000228742.72165.cf. [DOI] [PubMed] [Google Scholar]
- 115.Smolewski P. Recent developments in targeting the mammalian target of rapamycin (mTOR) kinase pathway. Anticancer Drugs. 2006;17:487–494. doi: 10.1097/00001813-200606000-00001. [DOI] [PubMed] [Google Scholar]
- 116.Sahin F, Kannangai R, Adegbola O, Wang J, Su G, Torbenson M. mTOR and P70 S6 kinase expression in primary liver neoplasms. Clin Cancer Res. 2004;10:8421–8425. doi: 10.1158/1078-0432.CCR-04-0941. [DOI] [PubMed] [Google Scholar]
- 117.Rizell M, Lindner P. Inhibition of mTOR suppresses experimental liver tumours. Anticancer Res. 2005;25:789–793. [PubMed] [Google Scholar]
- 118.Semela D, Piguet AC, Kolev M, Schmitter K, Hlushchuk R, Djonov V, Stoupis C, Dufour JF. Vascular remodeling and antitumoral effects of mTOR inhibition in a rat model of hepatocellular carcinoma. J Hepatol. 2007;46:840–848. doi: 10.1016/j.jhep.2006.11.021. [DOI] [PubMed] [Google Scholar]
- 119.Sieghart W, Fuereder T, Schmid K, Cejka D, Werzowa J, Wrba F, Wang X, Gruber D, Rasoul-Rockenschaub S, Peck-Radosavljevic M, et al. Mammalian target of rapamycin pathway activity in hepatocellular carcinomas of patients undergoing liver transplantation. Transplantation. 2007;83:425–432. doi: 10.1097/01.tp.0000252780.42104.95. [DOI] [PubMed] [Google Scholar]
- 120.Ribatti D, Nico B, Mangieri D, Longo V, Sansonno D, Vacca A, Dammacco F. In vivo inhibition of human hepatocellular carcinoma related angiogenesis by vinblastine and rapamycin. Histol Histopathol. 2007;22:285–289. doi: 10.14670/HH-22.285. [DOI] [PubMed] [Google Scholar]
- 121.Roccaro AM, Hideshima T, Richardson PG, Russo D, Ribatti D, Vacca A, Dammacco F, Anderson KC. Bortezomib as an antitumor agent. Curr Pharm Biotechnol. 2006;7:441–448. doi: 10.2174/138920106779116865. [DOI] [PubMed] [Google Scholar]
- 122.Mitsiades CS, Mitsiades N, Hideshima T, Richardson PG, Anderson KC. Proteasome inhibitors as therapeutics. Essays Biochem. 2005;41:205–218. doi: 10.1042/EB0410205. [DOI] [PubMed] [Google Scholar]
- 123.Schwartz R, Davidson T. Pharmacology, pharmacokinetics, and practical applications of bortezomib. Oncology (Williston Park) 2004;18:14–21. [PubMed] [Google Scholar]
- 124.Brignole C, Marimpietri D, Pastorino F, Nico B, Di Paolo D, Cioni M, Piccardi F, Cilli M, Pezzolo A, Corrias MV, et al. Effect of bortezomib on human neuroblastoma cell growth, apoptosis, and angiogenesis. J Natl Cancer Inst. 2006;98:1142–1157. doi: 10.1093/jnci/djj309. [DOI] [PubMed] [Google Scholar]
- 125.Fisher RI, Bernstein SH, Kahl BS, Djulbegovic B, Robertson MJ, de Vos S, Epner E, Krishnan A, Leonard JP, Lonial S, et al. Multicenter phase II study of bortezomib in patients with relapsed or refractory mantle cell lymphoma. J Clin Oncol. 2006;24:4867–4874. doi: 10.1200/JCO.2006.07.9665. [DOI] [PubMed] [Google Scholar]
- 126.Maki RG, Kraft AS, Scheu K, Yamada J, Wadler S, Antonescu CR, Wright JJ, Schwartz GK. A multicenter Phase II study of bortezomib in recurrent or metastatic sarcomas. Cancer. 2005;103:1431–1438. doi: 10.1002/cncr.20968. [DOI] [PubMed] [Google Scholar]
- 127.Kondagunta GV, Drucker B, Schwartz L, Bacik J, Marion S, Russo P, Mazumdar M, Motzer RJ. Phase II trial of bortezomib for patients with advanced renal cell carcinoma. J Clin Oncol. 2004;22:3720–3725. doi: 10.1200/JCO.2004.10.155. [DOI] [PubMed] [Google Scholar]
- 128.Shah MH, Young D, Kindler HL, Webb I, Kleiber B, Wright J, Grever M. Phase II study of the proteasome inhibitor bortezomib (PS-341) in patients with metastatic neuroendocrine tumors. Clin Cancer Res. 2004;10:6111–6118. doi: 10.1158/1078-0432.CCR-04-0422. [DOI] [PubMed] [Google Scholar]
- 129.A Randomized, Multicenter, Open-Label, Phase 2 Study of VELCADE Alone or VELCADE Plus Docetaxel in Previously Treated Patients With Advanced Non-Small Cell Lung Cancer. ClinicalTrials.gov identifier NCT00051974; Available from: URL: http://clinicaltrials.gov/show/ NCT00051974. [Google Scholar]
- 130.Fanucchi MP, Fossella FV, Belt R, Natale R, Fidias P, Carbone DP, Govindan R, Raez LE, Robert F, Ribeiro M, et al. Randomized phase II study of bortezomib alone and bortezomib in combination with docetaxel in previously treated advanced non-small-cell lung cancer. J Clin Oncol. 2006;24:5025–5033. doi: 10.1200/JCO.2006.06.1853. [DOI] [PubMed] [Google Scholar]
- 131.O'Connor OA. Marked clinical activity of the proteasome inhibitor bortezomib in patients with follicular and mantle-cell lymphoma. Clin Lymphoma Myeloma. 2005;6:191–199. doi: 10.3816/CLM.2005.n.046. [DOI] [PubMed] [Google Scholar]
- 132.Hegewisch-Becker S, Sterneck M, Schubert U, Rogiers X, Guerciolini R, Pierce JE, Hossfeld DK. Phase I/II trial of botrezomib in patients with unresectable hepatocellular carcinoma. J Clin Oncol. 2004;ASCO annual meeting proceedings22 Suppl 15: :4089. [Google Scholar]
- 133.A Pilot Study of Adjuvant Therapy of Gefitinib (Iressa, ZD1839) in Patients With Resectable Hepatocellular Carcinoma. ClinicalTrials.gov identifier NCT00228501; Available from: URL: http://clinicaltrials.gov/show/NCT00228501. [Google Scholar]
- 134.A Phase 2 Study of EKB-569 in Subjects With Advanced Non-Small Cell Lung Cancer. ClinicalTrials.gov identifier NCT00067548; Available from: URL: http://clinicaltrials.gov/show/ NCT00067548. [Google Scholar]
- 135.Study Evaluating EKB-569 in Advanced Colorectal Cancer. ClinicalTrials.gov identifier NCT00072748; Available from: URL: http://clinicaltrials.gov/show/ NCT00072748. [Google Scholar]
- 136.A Phase II Study of Efficacy and Tolerability of GW572016 in Patients With Advanced Hepatocellular and Biliary Carcinomas. ClinicalTrials.gov identifier NCT00107536; Available from: URL: http://clinicaltrials.gov/show/ NCT00107536. [Google Scholar]
- 137.Campos S, Hamid O, Seiden MV, Oza A, Plante M, Potkul RK, Lenehan PF, Kaldjian EP, Varterasian ML, Jordan C, et al. Multicenter, randomized phase II trial of oral CI-1033 for previously treated advanced ovarian cancer. J Clin Oncol. 2005;23:5597–5604. doi: 10.1200/JCO.2005.08.091. [DOI] [PubMed] [Google Scholar]
- 138.A Randomized 2-Arm, Open Label, Phase II Study of BMS-582664, Administered Orally at A Dose of 800 mg Daily or Doxorubicin Administered Intravenously at a Dose of 60 mg/m2 Every 3 Weeks in Patients With Unresectable, Locally Advanced or Metastatic Hepatocellular Carcinoma. ClinicalTrials.gov identifier NCT00355238; Available from: URL: http://clinicaltrials.gov/show/ NCT00355238. [Google Scholar]
- 139.Cetuximab in Patients With Unresectable or Metastatic Hepatocellular Carcinoma. ClinicalTrials.gov identifier NCT00162669; Available from: URL: http://clinicaltrials.gov/show/ NCT00162669. [Google Scholar]
- 140.Kollmannsberger C, Schittenhelm M, Honecker F, Tillner J, Weber D, Oechsle K, Kanz L, Bokemeyer C. A phase I study of the humanized monoclonal anti-epidermal growth factor receptor (EGFR) antibody EMD 72000 (matuzumab) in combination with paclitaxel in patients with EGFR-positive advanced non-small-cell lung cancer (NSCLC) Ann Oncol. 2006;17:1007–1013. doi: 10.1093/annonc/mdl042. [DOI] [PubMed] [Google Scholar]
- 141.Seiden MV, Burris HA, Matulonis U, Hall JB, Armstrong DK, Speyer J, Weber JD, Muggia F. A phase II trial of EMD72000 (matuzumab), a humanized anti-EGFR monoclonal antibody, in patients with platinum-resistant ovarian and primary peritoneal malignancies. Gynecol Oncol. 2007;104:727–731. doi: 10.1016/j.ygyno.2006.10.019. [DOI] [PubMed] [Google Scholar]
- 142.Graeven U, Kremer B, Sudhoff T, Killing B, Rojo F, Weber D, Tillner J, Unal C, Schmiegel W. Phase I study of the humanised anti-EGFR monoclonal antibody matuzumab (EMD 72000) combined with gemcitabine in advanced pancreatic cancer. Br J Cancer. 2006;94:1293–1299. doi: 10.1038/sj.bjc.6603083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Wittman M, Carboni J, Attar R, Balasubramanian B, Balimane P, Brassil P, Beaulieu F, Chang C, Clarke W, Dell J, et al. Discovery of a (1H-benzoimidazol-2-yl)-1H-pyridin-2-one (BMS-536924) inhibitor of insulin-like growth factor I receptor kinase with in vivo antitumor activity. J Med Chem. 2005;48:5639–5643. doi: 10.1021/jm050392q. [DOI] [PubMed] [Google Scholar]
- 144.Girnita A, Girnita L, del Prete F, Bartolazzi A, Larsson O, Axelson M. Cyclolignans as inhibitors of the insulin-like growth factor-1 receptor and malignant cell growth. Cancer Res. 2004;64:236–242. doi: 10.1158/0008-5472.can-03-2522. [DOI] [PubMed] [Google Scholar]
- 145.Two-Arm Randomized Open Label Phase 2 Study Of CP-751,871 In Combination With Exemestane Versus Exemestane Alone As First Line Treatment For Postmenopausal Patients With Hormone Receptor Positive Advanced Breast Cancer. ClinicalTrials.gov identifier NCT00372996; Available from: URL: http://clinicaltrials.gov/show/ NCT00372996. [Google Scholar]
- 146.A Phase 1b Dose Escalation/Phase 2 Randomized, Non-Comparative, Multiple Center, Open Label Study Of Cp 751,871 In Combination With Paclitaxel And Carboplatin And Of Paclitaxel And Carboplatin Alone As First Line Treatment For Advanced Non-Small Cell Lung Cancer. ClinicalTrials.gov identifier NCT00147537; Available from: URL: http://clinicaltrials.gov/show/ NCT00147537. [Google Scholar]
- 147.A Phase 2, Randomized, Non-Comparative, Two-Arm Open Label, Multiple-Center Study Of Cp-751,871 In Combination With Docetaxel/Prednisone In Chemotherapy- Naive (Arm A) And Docetaxel/Prednisone Refractory (Arm B) Patients With Hormone Insensitive Prostate Cancer. ClinicalTrials.gov identifier NCT00313781; Available from: URL: http://clinicaltrials.gov/show/ NCT00313781. [Google Scholar]
- 148.Wu JD, Haugk K, Coleman I, Woodke L, Vessella R, Nelson P, Montgomery RB, Ludwig DL, Plymate SR. Combined in vivo effect of A12, a type 1 insulin-like growth factor receptor antibody, and docetaxel against prostate cancer tumors. Clin Cancer Res. 2006;12:6153–6160. doi: 10.1158/1078-0432.CCR-06-0443. [DOI] [PubMed] [Google Scholar]
- 149.Sachdev D, Singh R, Fujita-Yamaguchi Y, Yee D. Down-regulation of insulin receptor by antibodies against the type I insulin-like growth factor receptor: implications for anti-insulin-like growth factor therapy in breast cancer. Cancer Res. 2006;66:2391–2402. doi: 10.1158/0008-5472.CAN-05-3126. [DOI] [PubMed] [Google Scholar]
- 150.Motzer RJ, Michaelson MD, Redman BG, Hudes GR, Wilding G, Figlin RA, Ginsberg MS, Kim ST, Baum CM, DePrimo SE, et al. Activity of SU11248, a multitargeted inhibitor of vascular endothelial growth factor receptor and platelet-derived growth factor receptor, in patients with metastatic renal cell carcinoma. J Clin Oncol. 2006;24:16–24. doi: 10.1200/JCO.2005.02.2574. [DOI] [PubMed] [Google Scholar]
- 151.SU011248 in Advanced Hepatocellular Carcinoma. ClinicalTrials.gov identifier NCT00361309; Available from: URL: http://clinicaltrials.gov/show/ NCT00361309. [Google Scholar]
- 152.Scott EN, Meinhardt G, Jacques C, Laurent D, Thomas AL. Vatalanib: the clinical development of a tyrosine kinase inhibitor of angiogenesis in solid tumours. Expert Opin Investig Drugs. 2007;16:367–379. doi: 10.1517/13543784.16.3.367. [DOI] [PubMed] [Google Scholar]
- 153.Koch I, Baron A, Roberts S, Junker U, Palacay-Ramona M, Masson E, Kay A, Wiedenmann B, Laurent D, Cebon J. Influence of Hepatic Dysfunction on Safety, Tolerability, and Pharmacokinetics of PTK787/ZK 222584 in Patients with Unresectable Hepatocellular Carcinoma (HCC) J Clin Oncol. 2005;23 Suppl 16:4134. [Google Scholar]
- 154.Bevacizumab in Advanced Hepatocellular Carcinoma. ClinicalTrials.gov identifier NCT00162669; Available from: URL: http://clinicaltrials.gov/show/ NCT00162669. [Google Scholar]
- 155.Dupont J, Rothenberg ML, Spriggs DR, Cedarbaum JM, Furfine ES, Cohen DP, Dancy I, Lee HS, Cooper W, Lockhart AC. Safety and pharmacokinetics of intravenous VEGF Trap in a phase I clinical trial of patients with advanced solid tumors. J Clin Oncol. 2005;23 Suppl 16:3029. [Google Scholar]
- 156.Herbst RS, Heymach JV, O’Reilly MS, Onn A, Ryan AJ. Vandetanib (ZD6474): an orally available receptor tyrosine kinase inhibitor that selectively targets pathways critical for tumor growth and angiogenesis. Expert Opin Investig Drugs. 2007;16:239–249. doi: 10.1517/13543784.16.2.239. [DOI] [PubMed] [Google Scholar]
- 157.An International, Phase II, Randomized, Double-Blinded, Placebo-Controlled, Multi-Center Study to Assess the Efficacy of ZD6474 (ZACTIMA?) Versus Placebo in Subjects With Unresectable, Locally Advanced or Metastatic Medullary Thyroid Cancer. ClinicalTrials.gov identifier NCT00410761; Available from: URL: http://clinicaltrials.gov/show/ NCT00410761. [Google Scholar]
- 158.Loibl S, Bianchi G, Zamagni C, Ardizzoni A, Raab G, Siena S, Wolf C, Westermeier T, Bergamini L, Gianni L, et al. Sorafenib (BAY 43-9006) in patients with metastatic breast cancer - a Phase II multicentre open trial. Proceedings of the 27. German Tumor conference; March 22-26; Berlin, Germany. Düsseldorf. pp. German Medical Science, 2006: PO–032. [Google Scholar]
- 159.Sorafenib in Treating Patients With Recurrent or Progressive Stage IV Non-Small Cell Lung Cancer. ClinicalTrials. gov identifier NCT00100763; Available from: URL: http://clinicaltrials.gov/show/NCT00100763. [Google Scholar]
- 160.Strumberg D, Richly H, Hilger RA, Schleucher N, Korfee S, Tewes M, Faghih M, Brendel E, Voliotis D, Haase CG, et al. Phase I clinical and pharmacokinetic study of the Novel Raf kinase and vascular endothelial growth factor receptor inhibitor BAY 43-9006 in patients with advanced refractory solid tumors. J Clin Oncol. 2005;23:965–972. doi: 10.1200/JCO.2005.06.124. [DOI] [PubMed] [Google Scholar]
- 161.Chan S, Scheulen ME, Johnston S, Mross K, Cardoso F, Dittrich C, Eiermann W, Hess D, Morant R, Semiglazov V, et al. Phase II study of temsirolimus (CCI-779), a novel inhibitor of mTOR, in heavily pretreated patients with locally advanced or metastatic breast cancer. J Clin Oncol. 2005;23:5314–5322. doi: 10.1200/JCO.2005.66.130. [DOI] [PubMed] [Google Scholar]
- 162.Atkins MB, Hidalgo M, Stadler WM, Logan TF, Dutcher JP, Hudes GR, Park Y, Liou SH, Marshall B, Boni JP, et al. Randomized phase II study of multiple dose levels of CCI-779, a novel mammalian target of rapamycin kinase inhibitor, in patients with advanced refractory renal cell carcinoma. J Clin Oncol. 2004;22:909–918. doi: 10.1200/JCO.2004.08.185. [DOI] [PubMed] [Google Scholar]
- 163.Witzig TE, Geyer SM, Ghobrial I, Inwards DJ, Fonseca R, Kurtin P, Ansell SM, Luyun R, Flynn PJ, Morton RF, et al. Phase II trial of single-agent temsirolimus (CCI-779) for relapsed mantle cell lymphoma. J Clin Oncol. 2005;23:5347–5356. doi: 10.1200/JCO.2005.13.466. [DOI] [PubMed] [Google Scholar]
- 164.Hidalgo M, Buckner JC, Erlichman C, Pollack MS, Boni JP, Dukart G, Marshall B, Speicher L, Moore L, Rowinsky EK. A phase I and pharmacokinetic study of temsirolimus (CCI-779) administered intravenously daily for 5 days every 2 weeks to patients with advanced cancer. Clin Cancer Res. 2006;12:5755–5763. doi: 10.1158/1078-0432.CCR-06-0118. [DOI] [PubMed] [Google Scholar]
- 165.Phase II Trial of RAD001 in Patients With Refractory Colorectal Cancer. ClinicalTrials.gov identifier NCT00337545; Available from: URL: http://clinicaltrials.gov/show/ NCT00337545. [Google Scholar]
- 166.Giaccone G. The potential of antiangiogenic therapy in non-small cell lung cancer. Clin Cancer Res. 2007;13:1961–1970. doi: 10.1158/1078-0432.CCR-06-2186. [DOI] [PubMed] [Google Scholar]
- 167.Mackay H, Hedley D, Major P, Townsley C, Mackenzie M, Vincent M, Degendorfer P, Tsao MS, Nicklee T, Birle D, et al. A phase II trial with pharmacodynamic endpoints of the proteasome inhibitor bortezomib in patients with metastatic colorectal cancer. Clin Cancer Res. 2005;11:5526–5533. doi: 10.1158/1078-0432.CCR-05-0081. [DOI] [PubMed] [Google Scholar]