


Abstract
The MBT repeat has been recently identified as a key domain capable of methyl–lysine histone recognition. Functional work has pointed to a role for MBT domain-containing proteins in transcriptional repression of developmental control genes such as Hox genes. In this study, L3MBTL2, a human homolog of Drosophila Sfmbt critical for Hox gene silencing, is demonstrated to preferentially recognize lower methylation states of several histone-derived peptides through its fourth MBT repeat. High-resolution crystallographic analysis of the four MBT repeats of this protein reveals its unique asymmetric rhomboid architecture, as well as binding mechanism, which preclude the interaction of the first three MBT repeats with methylated peptides. Structural elucidation of an L3MBTL2–H4K20me1 complex and comparison with other MBT-histone peptide complexes also suggests that an absence of distinct surface contours surrounding the methyl–lysine-binding pocket may underlie the lack of sequence specificity observed for members of this protein family.
INTRODUCTION
Chromatin structure is regulated by chromatin remodeling factors, histone exchange, linker histone association and histone modification. Histone lysine methylation has emerged as a key post-translational modification (PTM) implicated in both gene activation and silencing depending on the site and methylation degree of PTM, however the mechanisms involved are complex and not well understood. To date, seven different histone lysine residues have been identified as functionally relevant sites of methylation (K4, K9, K27, K36 and K79 of histone H3, K20 of histone H4 and K26 of histone H1b). Each of these lysine residues can be mono-, di- or tri-methylated, often with functional consequences. Histone methylation at specific lysine residues brings about various downstream events, which are mediated by different effector proteins. For example, in Drosophila, position effect variegation (PEV) is regulated by Heterochromatin Protein 1 (HP1) (1) and homeotic gene silencing is regulated by polycomb (Pc) (2), whose chromodomain recognizes histone H3 methylated at K9 and K27, respectively (3–6).
To date, several families of protein domains have been shown to recognize various lysine methylation sites with selective methylation state preference. The chromodomains of HP1, Pc, CHD1 and Eaf3 preferentially bind tri-methylated lysine at H3K9, H3K27, H3K4 and H3K36, respectively (7–9). The double Tudor domain of JMJD2A selectively binds tri-methylated H3K4 and H4K20 (10–12), and that of 53BP1 favors mono- or di-methylated H4K20 (13). The ankyrin repeat domains of G9a and GLP preferentially bind mono- or di-methylated H3K9 (14). The PHD finger family of proteins is especially interesting, because various PHD motifs have been shown to recognize histones with different selectivity for the methylation states and sites of lysine (8).
The malignant brain tumor (MBT) repeat is a structural motif of ∼100 amino acids that is conserved from C. elegans to humans and often exists as tandem repeats (15). It was originally identified in the Drosophila tumor-suppressor protein L(3)MBT and mutations in L(3)MBT gene cause malignant transformations of the optic neuroblasts (16). Based on sequence analysis, the MBT repeat shows structural similarity with the chromodomain, Tudor domain and PWWP domain, which are jointly referred to as the Tudor domain ‘Royal Family’ (17). Like Tudor and chromodomain domains, the methyl–lysine-binding ability of the MBT domain was confirmed by protein-array studies (10).
Recent functional studies have suggested important connections between MBT domain-containing proteins and the transcriptional state of chromatin regions. The 3-MBT repeat fragment of human L3MBTL1 can compact nucleosomal arrays within the context of mono- and di-methylated states, but not the tri-methylated state of H4K20 and H1bK26 (18). The association of L3MBTL1 with chromatin is dependent on the presence of H4K20me1 and is abolished by the knockdown of the H4K20 mono-methyltranferase PR-SET7 (18,19). Additionally, the 4-MBT repeats of human SFMBT were shown to be both necessary and sufficient for nuclear matrix association and transcriptional repression as well as binding to histone H3 and H4 (20). In Drosophila, the 2-MBT-repeat protein Scm is a core component in the PRC1 complex, and is essential for the repression of Hox genes (21). Embryos lacking Scm protein show widespread misexpression of Hox genes and die at the end of embryogenesis (21–23)
Crystal structures of MBT repeats have been reported for three proteins: human L3MBTL1 (24–26) and SCML2 (27,28), and Drosophila Scm (23). L3MBTL1 contains three MBT repeats, and the structural studies clearly delineate the specificity of the second MBT repeat towards mono- or di-methylated lysines (24,25,29). SCML2 and Scm both contain two MBT repeats and preferentially bind to mono-methylated lysine residues through their second MBT repeat (23,27). Based on the structural and biophysical characterization, these 3-MBT repeat proteins selectively bind the lower lysine methylation states of histone peptides without obvious sequence selectivity among the histone peptides measured to date.
Drosophila Sfmbt, a 4-MBT repeat-containing protein, has been reported to bind mono- or di-methylated H3K9 and H4K20 peptides (30). Drosophila Sfmbt knockout experiments revealed that Drosophila Sfmbt is a novel PcG protein and is critical for HOX gene silencing (30). The human genome encodes four proteins containing four MBT repeats. Among these 4-MBT repeat proteins, the MBT repeat domain of L3MBTL2 has the highest sequence homology with that of Drosophila Sfmbt (Supplementary Figure 1). Despite the nomenclature, the MBT repeat domain of L3MBTL2 has 48% identity with that of Drosophila Sfmbt, while both SFMBT1 and SFMBT2 MBT domains have only 30% identity with that of Drosophila Sfmbt. Therefore, like Drosophila Sfmbt, L3MBTL2 may potentially bind methylated histones and play a role in transcriptional repression. Binding data presented in this work indeed reveals that L3MBTL2 selectively binds lower methylation degrees of histone peptides. In order to gain insight into how L3MBTL2 binds methyl–lysine histones, we have also determined the structure of a 4-MBT repeat domain of human L3MBTL2.
MATERIALS AND METHODS
Protein expression and purification
The human L3MBTL2 protein (residues 170–625) was subcloned into pET28a-MHL vector and transformed in Escherichia coli BL21 (DE3) Codon plus RIL (Stratagene). Cells were grown and harvested as described in (31) in Luria–Bertanin media. The cell pellets were resuspended in a buffer solution containing 50 mM Tris 8.0, 250 mM NaCl, 5% glycerol and lysed by sonication. The supernatant fraction obtained by centrifugation at 16 000 rpm for 1 h was passed through a HiTrap Ni column (GE Healthcare, Piscataway, NJ), which was then washed and eluted with 20 column volumes of buffer in a gradient change to 20 mM Tris–HCl (pH 8.0), 250 mM NaCl, 500 mM imidazole. HiTrap Q HP column (GE Healthcare, Piscataway, NJ) and Superdex 75 gel-filtration column (GE Healthcare, Piscataway, NJ) were used for further purification. The protein was concentrated to 10 mg/ml in a buffer containing 20 mM Tris–HCl, pH 8.0, 0.2 M NaCl, 1 mM EDTA and 1 mM DTT.
Isothermal titration calorimetry
Isothermal titration calorimetry measurements were performed in duplicate at 25°C, using a VP-ITC microcalorimeter (MicroCal Inc.). Experiments were performed by injecting 10 µl of peptide solution (2–4 mM) into a sample cell containing 30–70 µM L3MBTL2 in 25 mM Tris–HCl, pH 7.5, 200 mM NaCl and 2 mM β-mercaptoethanol. Unmodified, mono-methylated, di-methylated and tri-methylated peptides were dissolved and dialyzed into the same buffer as L3MBTL2. Tyrosine-containing peptide concentrations were estimated with absorbance spectroscopy using the extinction coefficient, ε280 = 1280 M−1 cm−1. Otherwise, peptide concentrations were estimated from the mass. A total of 25 injections were performed with a spacing of 180 s and a reference power of 13 µcal/s. Binding isotherms were plotted and analyzed using Origin Software (MicroCal Inc.). The ITC measurements were fit to a one-site binding model.
Protein crystallization
Crystals of all complexes were obtained at 18°C by vapor diffusion of hanging drops of 1 µl of 10 mg ml−1 protein solution with 1 mM final concentration of peptides (which is about 5× molar excess peptide by directly adding peptide to protein solution) mixed with 1 µl of a reservoir solution. The reservoir solution was 10%–17% PEG3350, 0.1 M tri-sodium citrate (pH 5.0–5.9), 0.1 M ammonium acetate or 0.1 M ammonium phosphate, 10 mM DTT. Crystals of the L3MBTL2 fragment itself were also obtained in the same conditions. Before flash-cooling crystals in liquid nitrogen, crystals were soaked in a cryoprotectant of paratone.
Structure determination
X-ray diffraction data were collected at 100 K at APS BEAMLINE 19-ID at Argonne National Laboratory. Data were processed using the HKL software package. The apo-structure of L3MBT MBT repeat domain has been solved by molecular replacement in Phaser (32) using the L3MBTL1 3MBT structure [1OZ2 (26)] as a search model. Models were placed for two copies each of repeats 3 and 4, then the structure was completed by using ARP/wARP (33) and phenix.autobuild (34) to build automatically into maps averaged in DM (35). The graphics program COOT (36) was used for manual model building and visualization. Crystal diffraction data and refinement statistics for both the apo-form and complex structures are displayed in Table 1. The atomic coordinates of the refined models have been deposited into the PDB, and the entries have been assigned the accession codes 3CEY and 3F70.
Table 1.
Crystallography data and refinement statistics of L3MBTL2
| 4MBT | 4MBT–H4K20me1 | |
|---|---|---|
| Data collection | ||
| Space group | P212121 | P212121 |
| Cell dimensions | ||
| a, b, c (Å) | 55.64, 55.94, 329.09, | 55.93, 56.31, 331.09 |
| α, β, γ (°) | 90, 90, 90 | 90, 90, 90 |
| Resolution (Å) | 2.20 | 2.15 |
| Rmerge | 7.4 (30.6) | 8.0 (65.8) |
| I/σI | 10.5 (1.4) | 8.9 (1.5) |
| Completeness (%) | 87.6 (46.2) | 98.7 (90.4) |
| Redundancy | 5.9 (1.8) | 6.7 (3.8) |
| Refinement | ||
| Number of reflections | 44600 | 54054 |
| Rwork/Rfree | 21.0/25.2 | 20.9/27.1 |
| Number of atoms/B factor (Å2) | ||
| Protein | 6182/35.7 | 6423/32.5 |
| Peptide | NA | 20/58.1 |
| Water | 166/55.1 | 366/45.1 |
| R.m.s. deviations | ||
| Bond lengths (Å) | 0.027 | 0.021 |
| Bond angles (°) | 1.85 | 1.88 |
RESULTS AND DISCUSSION
L3MBTL2, like L3MBTL1, selectively binds histone peptides with mono- or di-methylated lysine
The human L3MBTL2 protein comprises 705 amino acids, and contains one atypical zinc finger of unknown function in the N-terminus, and 4-MBT repeats which could potentially bind lysine-methylated histones, like human L3MBTL1, SCML2 and Drosophila Scm. Structural studies of these proteins in complex with histone peptides showed that all these 3 MBT repeat proteins use a semi-aromatic pocket to recognize mono- or di-methylated lysine. This semi-aromatic binding pocket is formed by three aromatic residues and a negatively charged residue. Sequence alignment results show that the fourth MBT repeat of L3MBTL2 also contains these four conserved residues, which we predicted would form the potential binding pocket to accommodate methyl–lysine histones (Supplementary Figure 2). To investigate if L3MBTL2 is also able to recognize methyl–lysine histone peptides, we purified a fragment of human L3MBTL2 encompassing all four MBT repeats (residues 170–625, hereafter referred to as 4MBT), and measured its binding affinity for histone H4K20 bearing various lysine methylation states using isothermal titration calorimetry (ITC). We confirmed that the 4 MBT protein selectively binds mono- and di-methylated histone peptides in a monovalent mode, and does not exhibit detectable binding to any unmodified or tri-methylated peptides (Figure 1). Furthermore, the ITC-binding data also show that 4MBT binds mono-methylated or di-methylated H3K4, H3K9 and H3K27 containing peptides with similar affinities (Supplementary Figure 3). Based on the 4MBT-histone-binding studies, it appears that L3MBTL2, like L3MBTL1, selectively binds lower-methylated lysine histones and does not have sequence selectivity among the four sequences tested. Nevertheless, 4MBT binds mono- and di-methylated lysine centrally located in a peptide more tightly than free mono-methylated lysine (Supplementary Figure 3). To shed light onto the structural basis for the selectivity for mono- or di-methylated lysines over unmodified or tri-methylated lysine histones, we determined the crystal structure of 4MBT in complex with a histone H4 peptide with K20 mono-methylated (H4K20me1).
Figure 1.

Isothermal titration calorimetry data for binding of unmodified and different methylated H4K20 peptides (AKRHRK20VLRDN in sequence) to the 4-MBT-repeat domain (4MBT) of L3MBTL2. ND: No detectable binding.
Overall structure of L3MBTL2
The crystal structure of the 4MBT domain of L3MBTL2 was solved by molecular replacement using L3MBTL1 [1OZ2 (26)] as a search model. The structure of the L3MBTL2–H4K20me1 complex was solved using the apo-L3MBTL2 structure as a search model. The crystal diffraction data and refinement statistics for both structures are summarized in Table 1. These two structures are almost identical except for extra electron density for the bound peptide in the L3MBTL2–H4K20me1 complex structure. The overall structure of 4MBT is shown in Figure 2A. The crystal structure of 4MBT shows that 4MBT exhibits an irregular rhombus architecture (Figure 2A and Supplementary Figure 4). Consistent with previously reported structures, each MBT repeat consists of an extended ‘arm’ and a globular β subunit core with the N-terminal arm of each MBT repeat packing against the β subunit core of its preceding repeat. A model of the assembly of 4-MBT repeats had been proposed based on the 2-MBT protein SCML2, which predicted that 4-MBT repeats would form a ring-like structure with the N-terminal arm packing against the C-terminal barrel and the other arm packing against the preceding barrel (28). Surprisingly, the 4-MBT repeats do not assume a four-blade propeller architecture. Instead, the last 3-MBT repeats form a three-blade propeller-shaped architecture similar to the packing of L3MBTL1 (Figure 3), and a narrow channel runs through the middle of the propeller-like structure, which is filled with water molecules. Nonetheless, the arm of the first repeat packs against the core of the fourth repeat, as predicted (28).
Figure 2.

Only the fourth MBT repeat of L3MBTL2 binds H4K20me1. (A) Overall structure of 4MBT bound to an H4K20me1 peptide (AKRHRK20VLRDN). MBT1, MBT2, MBT3 and MBT4 are shown in blue, yellow, green and purple, respectively. The K20me1 lysine is shown in a stick model. (B) K20me1 recognition by the fourth repeat MBT4. The lysine-binding pocket residues and K20me1 are shown in a stick model. The electron density map 2Fo–Fc is contoured at 1 σ. The electron density map is calculated with the peptide omitted.
Figure 3.
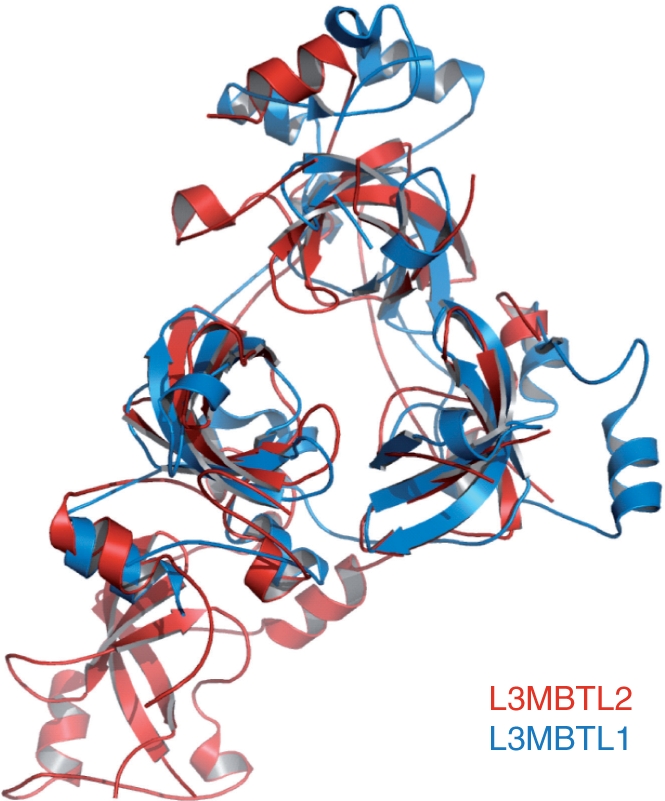
Superposition of the crystal structures of L3MBTL1 and L3MBTL2 MBT domains. L3MBTL1 is colored in blue and L3MBTL2 is colored in red.
Structural basis for binding of the fourth MBT repeat to H4K20me1 and its preference for lower lysine methylation marks
The 4MBT–H4K20me1(AKRHRK20VLRDN) complex shows that only the fourth MBT repeat (MBT4) bound to the methyl–lysine histone, confirming our prediction based on sequence alignments. In the complex, only residue K20me1 has well-defined electron density and poor and discontinued electron density is observed for the main chain of some flanking residues of the bound histone H4K20me1 peptide. Thus, among the 11 residues of the peptide, only mono-methylated lysine forms extensive interactions with 4MBT. As seen for other MBT–peptide complexes, K20me1 is accommodated in a semi-aromatic ‘cage’. Residues Phe570, Trp573, Tyr577, Leu552, form hydrophobic walls surrounding the methylated lysine side chain (Figure 2B). The aromatic rings of Phe570, Trp573, Tyr577 are approximately perpendicular to one another as seen in other methyl lysine-binding pockets (23–25,27). Phe570 forms the floor of the hydrophobic cage and Trp573, Tyr577 and Leu552 form the other three sides interacting with the mono-methylated lysine primarily via van der Waals’ and cation-π interactions. The fifth side of the cage harbors negatively charged Asp546, which interacts with the mono-methylammonium group of K20me1 via a salt bridge mediated by a hydrogen bond. Similar or identical residues are found in the binding pockets of other MBT family members with reported structures. L3MBTL1, Scm and SCML2 all use the same mechanism to selectively recognize lower methylation states of lysine histones. That is, tri-methylated lysine is too bulky to fit into the pocket and lacks the ability to form a hydrogen bond with the negatively charged aspartic acid; unmodified lysine would have much weaker van der Waals’ and cation-π interactions with the hydrophobic cage (13,23–25,27).
Although L3MBTL2 has a distorted rhombus architecture, the 4-MBT repeats of L3MBTL2 have similar 3D structures, and their globular β subunit cores can be well superimposed (Supplementary Figure 5). Comparison of the 4 MBT repeats (denoted MBT1–MBT4) also confirmed why the other 3 MBT repeats did not bind to methyl–lysine histones. In MBT1, a cage is formed by four residues Trp231, Tyr247, Phe250 and Ile286 from the globular beta core (Supplementary Figure 6), but this potential binding pocket is blocked by a loop spanning from Asn276 to Trp293 (colored in purple) (Supplementary Figure 6). Interestingly, a proline residue Pro283 from the loop occupies the cage, reminiscent of proline recognition by the L3MBTL1 pocket 1 (25). The MBT2 and MBT3 repeats lack corresponding aromatic residues identified in MBT4, which are essential for the methyl–lysine pocket formation. Therefore, although 4MBT contains 4 MBT repeats, only the fourth MBT repeat binds methyl–lysine histones.
Prediction of methyl–lysine histone-binding ability of other human MBT repeat proteins
The human genome encodes at least nine MBT repeat proteins and the binding specificities of three members have been reported, all of which selectively bind lower lysine methylation states of histones and do not exhibit sequence selectivity (23–25,27). On the basis of the sequence alignment of the MBT repeats of these nine MBT repeat proteins (Supplementary Figure 2), it is predicted that SCMH1, like SCML2, has a lower-methyl–lysine-binding pocket in its C-terminal MBT repeat. Interestingly, each of the first two MBT repeats of L3MBTL3 and L3MBTL4 contains the entire semi-aromatic pocket forming residues. Therefore, potentially each L3MBTL3 or L3MBTL4 molecule is able to recognize two mono- or di-methylated lysine marks from either the same nucleosome or neighboring nucleosomes in a chromatin context. In addition to L3MBTL2, there are three other four-MBT repeat proteins. MBTD1 is highly homologous to L3MBTL2 and is predicted to bind lower lysine methylation states of histones using the fourth MBT repeat. Surprisingly, none of the MBT repeats of SFMBT1 or SFMBT2 contains all the required residues to form an open semi-aromatic pocket to accommodate methyl–lysine peptides, although it is reported that SFMBT1 binds histone H3 and H4 through the four MBT repeat domain (20). Therefore, further structural and biochemical studies are warranted to delineate the binding specificities of the other MBT members.
Why does L3MBTL2 not have sequence selectivity?
All structures of MBT–peptide complexes to date adopt a ‘cavity insertion recognition mode’ (8), in which the methyl–lysine forms extensive interactions with a deep pocket in the protein, and the residues surrounding the methyl–lysine form few interactions with the protein (Figure 4A and B). In contrast, for proteins that use the surface groove recognition mode to bind histones, a well-defined and complementary binding groove is formed in the surface of the histone binders (Figure 4C and D). This complementary binding groove determines the binding sequence selectivity. This lack of a conserved peptide-binding surface is illustrated by a superimposition of the structures of all solved peptide-bound MBT repeats (Figure 5). While the structures of the MBT repeats are highly conserved and well superimposed, the orientation of the flanking peptide is highly variable. These MBT repeats do not form complementary binding grooves to constrain the surrounding binding sequence, and therefore the peptides bind in different orientations. While the currently published in vitro binding data of MBT domains with short histone peptides exhibit a broad range of binding, the limited reports from cell-based studies suggest that MBT protein function is dependent on modification of specific histone lysine sites in certain cases. For instance, the transcription repression activity of L3MBTL1 appears to be dependent on H4K20 mono-methylation during the cell cycle (18,19). Because MBT proteins often exist within multiprotein complexes, such as L3MBTL1 and Scm, in vivo sequence selectivity may potentially be conferred by other components in complexes such as HP1γ in L3MBTL1 complex (18), and Pc in the Scm-containing PRC1 complex.
Figure 4.

L3MBTL2 uses a cavity insertion recognition mode to recognize methyl–lysine histones. (A) Surface representation of the L3MBTL1 and H4K20me2 complex structure (PDB 2PQW). (B) Surface representation of the L3MBTL2 and H4K20me1 complex structure (PDB 3F70). (C) Surface representation of the HP1 and H3K9me3 complex structure (PDB 1KNE). (D) Surface representation of the Pc and H3K27me3 complex structure (PDB 1PFB). Peptides are shown in a stick model.
Figure 5.

Superposition of the structures of all solved peptide-bound MBT repeats viewed from two different directions (5A and 5B). L3MBTL1–H4K20me2 complex is colored in red (PDB 2PQW); L3MBTL1–H1.5 complex is colored in yellow (PDB 2RHI); Scm and RKmeS peptide complex is colored in blue (PDB 2R5M); and L3MBTL2–H4K20me1 complex is colored in green (this study). Lysine residues are shown in a stick model in (A).
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Structural Genomics Consortium, a registered charity (number 1097737) that receives funds from the Canadian Institutes for Health Research (CIHR); the Canadian Foundation for Innovation; Genome Canada through the Ontario Genomics Institute; GlaxoSmithKline; Karolinska Institutet; the Knut and Alice Wallenberg Foundation; the Ontario Innovation Trust; the Ontario Ministry for Research and Innovation; Merck & Co., Inc.; the Novartis Research Foundation; the Swedish Agency for Innovation Systems; the Swedish Foundation for Strategic Research and the Wellcome Trust. National Science foundation of China (30670429) [to C.Q. and J.R.M.], Wellcome Trust (050211) [to R.J.R.]; CIHR Canada Graduate Scholarship [to N.N.]; C.H.A. holds a Canada Research Chair in Structural Proteomics. Funding for open access charge: Canadian Institutes for Health Research (CIHR).
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
We would like to thank Alexey Bochkarev, Vladimir Lunin, Wolfram Tempel, Patrick Finerty Jr, Jitka Eryilmaz, Valerie Campagna-Slater, Ivona Kozieradzki, Farrell MacKenzie, Peter Loppnau and Lissete Crombet for advice and technical assistance.
REFERENCES
- 1.Grewal SI, Jia S. Heterochromatin revisited. Nat. Rev. 2007;8:35–46. doi: 10.1038/nrg2008. [DOI] [PubMed] [Google Scholar]
- 2.Cao R, Zhang Y. The functions of E(Z)/EZH2-mediated methylation of lysine 27 in histone H3. Curr. Opin. Gen. Dev. 2004;14:155–164. doi: 10.1016/j.gde.2004.02.001. [DOI] [PubMed] [Google Scholar]
- 3.Fischle W, Wang Y, Jacobs SA, Kim Y, Allis CD, Khorasanizadeh S. Molecular basis for the discrimination of repressive methyl-lysine marks in histone H3 by Polycomb and HP1 chromodomains. Genes Dev. 2003;17:1870–1881. doi: 10.1101/gad.1110503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jacobs SA, Khorasanizadeh S. Structure of HP1 chromodomain bound to a lysine 9-methylated histone H3 tail. Science. 2002;295:2080–2083. doi: 10.1126/science.1069473. [DOI] [PubMed] [Google Scholar]
- 5.Min J, Zhang Y, Xu RM. Structural basis for specific binding of Polycomb chromodomain to histone H3 methylated at Lys 27. Genes Dev. 2003;17:1823–1828. doi: 10.1101/gad.269603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nielsen PR, Nietlispach D, Mott HR, Callaghan J, Bannister A, Kouzarides T, Murzin AG, Murzina NV, Laue ED. Structure of the HP1 chromodomain bound to histone H3 methylated at lysine 9. Nature. 2002;416:103–107. doi: 10.1038/nature722. [DOI] [PubMed] [Google Scholar]
- 7.Flanagan JF, Mi LZ, Chruszcz M, Cymborowski M, Clines KL, Kim Y, Minor W, Rastinejad F, Khorasanizadeh S. Double chromodomains cooperate to recognize the methylated histone H3 tail. Nature. 2005;438:1181–1185. doi: 10.1038/nature04290. [DOI] [PubMed] [Google Scholar]
- 8.Taverna SD, Li H, Ruthenburg AJ, Allis CD, Patel DJ. How chromatin-binding modules interpret histone modifications: lessons from professional pocket pickers. Nat. Struct. Mol. Biol. 2007;14:1025–1040. doi: 10.1038/nsmb1338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Joshi AA, Struhl K. Eaf3 chromodomain interaction with methylated H3-K36 links histone deacetylation to Pol II elongation. Mol. Cell. 2005;20:971–978. doi: 10.1016/j.molcel.2005.11.021. [DOI] [PubMed] [Google Scholar]
- 10.Kim J, Daniel J, Espejo A, Lake A, Krishna M, Xia L, Zhang Y, Bedford MT. Tudor, MBT and chromo domains gauge the degree of lysine methylation. EMBO Rep. 2006;7:397–403. doi: 10.1038/sj.embor.7400625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Huang Y, Fang J, Bedford MT, Zhang Y, Xu RM. Recognition of histone H3 lysine-4 methylation by the double tudor domain of JMJD2A. Science. 2006;312:748–751. doi: 10.1126/science.1125162. [DOI] [PubMed] [Google Scholar]
- 12.Lee J, Thompson JR, Botuyan MV, Mer G. Distinct binding modes specify the recognition of methylated histones H3K4 and H4K20 by JMJD2A-tudor. Nat. Struct. Mol. Biol. 2008;15:109–111. doi: 10.1038/nsmb1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Botuyan MV, Lee J, Ward IM, Kim JE, Thompson JR, Chen J, Mer G. Structural basis for the methylation state-specific recognition of histone H4-K20 by 53BP1 and Crb2 in DNA repair. Cell. 2006;127:1361–1373. doi: 10.1016/j.cell.2006.10.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Collins RE, Northrop JP, Horton JR, Lee DY, Zhang X, Stallcup MR, Cheng X. The ankyrin repeats of G9a and GLP histone methyltransferases are mono- and dimethyllysine binding modules. Nat. Struct. Mol. Biol. 2008;15:245–250. doi: 10.1038/nsmb.1384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wismar J. Molecular characterization of h-l(3)mbt-like: a new member of the human mbt family. FEBS Lett. 2001;507:119–121. doi: 10.1016/s0014-5793(01)02959-3. [DOI] [PubMed] [Google Scholar]
- 16.Wismar J, Loffler T, Habtemichael N, Vef O, Geissen M, Zirwes R, Altmeyer W, Sass H, Gateff E. The Drosophila melanogaster tumor suppressor gene lethal(3)malignant brain tumor encodes a proline-rich protein with a novel zinc finger. Mech. Dev. 1995;53:141–154. doi: 10.1016/0925-4773(95)00431-9. [DOI] [PubMed] [Google Scholar]
- 17.Maurer-Stroh S, Dickens NJ, Hughes-Davies L, Kouzarides T, Eisenhaber F, Ponting CP. The Tudor domain ‘Royal Family': Tudor, plant Agenet, Chromo, PWWP and MBT domains. Trends Biochem. Sci. 2003;28:69–74. doi: 10.1016/S0968-0004(03)00004-5. [DOI] [PubMed] [Google Scholar]
- 18.Trojer P, Li G, Sims RJ, 3rd, Vaquero A, Kalakonda N, Boccuni P, Lee D, Erdjument-Bromage H, Tempst P, Nimer SD, et al. L3MBTL1, a histone-methylation-dependent chromatin lock. Cell. 2007;129:915–928. doi: 10.1016/j.cell.2007.03.048. [DOI] [PubMed] [Google Scholar]
- 19.Kalakonda N, Fischle W, Boccuni P, Gurvich N, Hoya-Arias R, Zhao X, Miyata Y, Macgrogan D, Zhang J, Sims JK, et al. Histone H4 lysine 20 monomethylation promotes transcriptional repression by L3MBTL1. Oncogene. 2008;27:4293–4304. doi: 10.1038/onc.2008.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wu S, Trievel RC, Rice JC. Human SFMBT is a transcriptional repressor protein that selectively binds the N-terminal tail of histone H3. FEBS Lett. 2007;581:3289–3296. doi: 10.1016/j.febslet.2007.06.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bornemann D, Miller E, Simon J. Expression and properties of wild-type and mutant forms of the Drosophila sex comb on midleg (SCM) repressor protein. Genetics. 1998;150:675–686. doi: 10.1093/genetics/150.2.675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Breen TR, Duncan IM. Maternal expression of genes that regulate the bithorax complex of Drosophila melanogaster. Dev. Biol. 1986;118:442–456. doi: 10.1016/0012-1606(86)90015-1. [DOI] [PubMed] [Google Scholar]
- 23.Grimm C, de Ayala Alonso AG, Rybin V, Steuerwald U, Ly-Hartig N, Fischle W, Muller J, Muller CW. Structural and functional analyses of methyl-lysine binding by the malignant brain tumour repeat protein Sex comb on midleg. EMBO Rep. 2007;8:1031–1037. doi: 10.1038/sj.embor.7401085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Min J, Allali-Hassani A, Nady N, Qi C, Ouyang H, Liu Y, MacKenzie F, Vedadi M, Arrowsmith CH. L3MBTL1 recognition of mono- and dimethylated histones. Nat. Struct. Mol. Biol. 2007;14:1229–1230. doi: 10.1038/nsmb1340. [DOI] [PubMed] [Google Scholar]
- 25.Li H, Fischle W, Wang W, Duncan EM, Liang L, Murakami-Ishibe S, Allis CD, Patel DJ. Structural basis for lower lysine methylation state-specific readout by MBT repeats of L3MBTL1 and an engineered PHD finger. Mol. Cell. 2007;28:677–691. doi: 10.1016/j.molcel.2007.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang WK, Tereshko V, Boccuni P, MacGrogan D, Nimer SD, Patel DJ. Malignant brain tumor repeats: a three-leaved propeller architecture with ligand/peptide binding pockets. Structure. 2003;11:775–789. doi: 10.1016/s0969-2126(03)00127-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Santiveri CM, Lechtenberg BC, Allen MD, Sathyamurthy A, Jaulent AM, Freund SM, Bycroft M. The malignant brain tumor repeats of human SCML2 bind to peptides containing monomethylated lysine. J. Mol. Biol. 2008;382:1107–1112. doi: 10.1016/j.jmb.2008.07.081. [DOI] [PubMed] [Google Scholar]
- 28.Sathyamurthy A, Allen MD, Murzin AG, Bycroft M. Crystal structure of the malignant brain tumor (MBT) repeats in Sex Comb on Midleg-like 2 (SCML2) J. Mol. Biol. 2003;278:46968–46973. doi: 10.1074/jbc.M306469200. [DOI] [PubMed] [Google Scholar]
- 29.Nady N, Min J, Kareta MS, Chedin F, Arrowsmith CH. A SPOT on the chromatin landscape? Histone peptide arrays as a tool for epigenetic research. Trends Biochem. Sci. 2008;33:305–313. doi: 10.1016/j.tibs.2008.04.014. [DOI] [PubMed] [Google Scholar]
- 30.Klymenko T, Papp B, Fischle W, Kocher T, Schelder M, Fritsch C, Wild B, Wilm M, Muller J. A Polycomb group protein complex with sequence-specific DNA-binding and selective methyl-lysine-binding activities. Genes Dev. 2006;20:1110–1122. doi: 10.1101/gad.377406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schuetz A, Allali-Hassani A, Martin F, Loppnau P, Vedadi M, Bochkarev A, Plotnikov AN, Arrowsmith CH, Min J. Structural basis for molecular recognition and presentation of histone H3 by WDR5. EMBO J. 2006;25:4245–4252. doi: 10.1038/sj.emboj.7601316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J. Appl. Crystallography. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Perrakis A, Harkiolaki M, Wilson KS, Lamzin VS. ARP/wARP and molecular replacement. Acta Crystallographica. 2001;57:1445–1450. doi: 10.1107/s0907444901014007. [DOI] [PubMed] [Google Scholar]
- 34.Terwilliger TC, Grosse-Kunstleve RW, Afonine PV, Moriarty NW, Adams PD, Read RJ, Zwart PH, Hung LW. Iterative-build OMIT maps: map improvement by iterative model building and refinement without model bias. Acta Crystallographica. 2008;64:515–524. doi: 10.1107/S0907444908004319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cowtan K. dm: an automated procedure for phase improvement by density modification. Joint CCP4 and ESF-EACBM Newsletter on Protein Crystallography. 1994;31:5. [Google Scholar]
- 36.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallographica. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.