

Summary
Background
Chondroitin sulfate (CS) is a glycosaminoglycan released by activated platelets.
Objective
Herewe test the hypothesis that CS released by activated platelets can trigger complement activation in the fluid phase.
Methods and results
Thrombin receptor-activating peptide (TRAP)-6 was used to activate platelets in platelet-rich plasma and blood, anticoagulated with the thrombin inhibitor lepirudin. TRAP activation induced fluid-phase complement activation, as reflected by the generation of C3a and sC5b-9, which could be attenuated by the C3 inhibitor compstatin. Chondroitinase ABC treatment of supernatants from activated platelets totally inhibited the activation, indicating that platelet-derived CS had initiated the complement activation. Furthermore, addition of purified CS to plasma strongly triggered complement activation. C1q was identified as the recognition molecule, as it bound directly to CS, and CS-triggered complement activation could be restored in C1q-depleted serum by adding purified C1q. TRAP activation of whole blood increased the expression of CD11b on leukocytes and generation of leukocyte–platelet complexes. It was demonstrated that these leukocyte functions were dependent on C3 activation and signaling via C5a, as this expression could be inhibited by compstatin and by a C5aR antagonist.
Conclusions
We conclude that platelets trigger complement activation in the fluid phase by releasing CS, which leads to inflammatory signals mediated by C5a.
Keywords: chondroitin sulfate, coagulation, complement, platelets, thrombin receptor-activating peptide (TRAP)
Introduction
Several studies have clearly shown that the levels of C3 and C4, and the C3/C4 ratio [1,2], are associated with increased risk of pathologic thrombotic processes such as myocardial infarction and stroke. Supporting direct involvement of complement in these conditions are generation of complement activation fragments in myocardial infarction and unstable angina [3-6]. The mechanism by which complement is activated in thrombotic and cardiovascular disease, and the function of this, are not very well understood.
Although the complement and coagulation systems have evolved independently from a common protease ancestor, cross-talk exists between the two systems [7]. An already well-established interaction involving the complement and coagulation systems is the regulation mediated by the C1 inhibitor C1-INH, which acts within both systems [8]. Early studies demonstrated that activated coagulation factors such as thrombin, plasmin and factor XIIa can cleave complement components [9,10]. Recently, it has been reported that C5 is activated by thrombin in C3-knockout mice, in which the terminal pathway cannot be activated by the C3b2Bb C5 convertase. Also, human C5 was shown to be cleaved by thrombin in this study, indicating that C5a may be generated in the absence of an intact complement system [11].
Despite these observations of direct contact between components of the two cascade systems, interactions between the two systems occur most frequently and vigorously through the mediating activity of cells such as polymorpho-nuclear neutrophils (PMNs), monocytes, and platelets [7]. In a series of papers, Sims and co-workers have shown that complement attack activates platelets and generates micro-particles via C5b-9 [12]. C5b-9 also triggers the expression of tissue factor (TF) in endothelial cells, and C5a is able to induce the expression of TF on PMNs and endothelial cells [13-15]. It is also well known that complement is activated in clotting blood, and significantly higher levels of complement activation products, for example C3a and sC5b-9, are found in human serum than in blood anticoagulated with EDTA, heparin, or lepirudin [7].
Kalowski and co-workers [16] have reported that thrombin and thromboplastin can induce complement activation when injected into rabbits. Induction of thrombocytopenia before treatment of the animals was able to attenuate this activation, suggesting that platelets were involved in the phenomenon. This activation has been further investigated in vitro in experiments in which platelets in platelet-rich plasma (PRP) or whole blood were activated by different stimuli and in which complement activation was reported to occur on activated platelets [17,18].
The presence of chondroitin sulfate (CS)-4 or CS-A in platelets as the main glycosaminoglycan (GAG) has been well established by both biochemical and histologic techniques [19,20]. Rapid release of CS from platelets has been demonstrated to occur in response to different agonists, including ADP, collagen, adrenalin, and thrombin, resulting in a rise in plasma CS by up to 2 μg mL−1 within 3 min after activation [21]. CS has also been shown to be exposed on the surface of platelets after activation [19]. The CS present in platelets, unlike that in blood plasma, is fully sulfated, and its molecular mass has been estimated to be approximately 28 kDa [20].
In the present study, we have investigated whether clotting induced by either contact activation or the TF pathway can trigger complement activation. Our analyses of the mechanism by which complement activation occurs in clotting blood have identified a novel fluid-phase mechanism in which activated platelets are able to trigger complement activation by releasing CS. CS has previously been shown to interact with C1q and act as a specific inhibitor of C1q [22,23]. We here postulate that this inhibition is a secondary phenomenon due to depletion of intact complement caused by activation of the classic pathway. Our studies also elucidate some of the physiologic consequences of platelet activation, which may be of importance for our understanding of the role of complement activation in thrombotic disease.
Materials and methods
Blood samples
Fresh non-anticoagulated human blood, obtained from healthy volunteers who had received no medication for at least 10 days, was collected in 50-mL tubes heparinized with the Corline surface (Corline Systems, Uppsala, Sweden). The blood was divided into aliquots for preparation of whole blood, PRP, and platelet-poor plasma (PPP). Blood was also drawn with the addition of the thrombin inhibitor lepirudin (70 μm; Refludan; Pharmion Ltd, Cambridge, UK). To obtain PRP, blood was centrifuged at 150 × g for 15 min at room temperature (RT). PPP was prepared by centrifuging twice at 2000 × g for 15 min at RT. The platelet content in PRP, counted using a Coulter AcT diff analyzer (Beckman Coulter, Miami, FL, USA) was increased by a factor of approximately 1.5 as compared to whole blood, whereas the platelet content in PPP was below the detection limit of the instrument (2 × 109 L−1).
Incubation of blood, PRP and PPP with thromboplastin
Blood (800 μL), PRP (400 μL) and PPP (400 μL) were activated by incubation in glass tubes. Incubation with 10 μL of rabbit brain thromboplastin-S (Biopool International, Ventura, CA, USA), diluted 1 : 100 in 0.9% NaCl, was performed in polypropylene tubes. The reaction was stopped by the addition of a stock solution giving final concentrations of 10 mm EDTA, 50 μg mL−1 soybean trypsin inhibitor, 1 mm theophylline, 20 mm benzamidine, and 1 μm polyethylene glycol (PGE)1 (all from Sigma Aldrich, St Louis, MO, USA), to block further activation of the platelet, contact, coagulation and complement systems. The tubes were then centrifuged at 3400 × g for 15 min at 4 °C, and the plasma (approximately 400 μL) was stored at − 70 °C until analysis.
Preparation of washed platelets
After centrifugation of lepirudin-anticoagulated PRP, the pellet containing the platelets was washed three times with Tyrode's buffer (pH 6.5), containing 3.5 mg mL−1 bovine serum albumin (BSA), 1 μm PGE1 (Sigma Aldrich), and 2 IU mL−1 heparin (Bioiberica, Barcelona, Spain). The platelets were resuspended in Tyrode's buffer (pH 7.45), without additions, and activated using 6.25 μg mL−1 thrombin receptor-activating peptide-6 (TRAP; Invitrogen, Molecular Probes, Carlsbad, CA, USA) dissolved in milliQ water, at 37 °C for 15 min. This concentration of TRAP was the lowest one able to induce maximal expression of P-selectin as determined by flow cytometry (not shown). After centrifugation, the super-natants were stored at − 70 °C.
Activation of platelets in blood and PRP by TRAP
Platelets were activated in lepirudin-anticoagulated PRP and whole blood by incubation with 6.25 μg mL−1 TRAP in polypropylene tubes. In some experiments, the blood was preincubated with 10 μm C3 inhibitor compstatin, Ac-ICV(1-MeW)QDWGAHRCT-NH2 [24], for 5 min at RT before activation. Complement activation was stopped by the addition of 10 mm EDTA (final concentration). PRP and blood were either directly used in further experiments or centrifuged to obtain plasma.
Activation of complement by CS
PPP was incubated with CS-A from bovine trachea, average molecular mass 28 kDa (product number C9819; Sigma Aldrich), dissolved in phosphate-buffered saline (PBS) or with supernatants from washed and TRAP-activated platelets for 15 min at 37 °C, and this was followed by addition of EDTA. Alternatively, the incubation was performed in the presence of 10 mm EDTA (where complement is totally inactivated) or 10 mm Mg2+-EGTA (where only the alternative pathway is operative). In some experiments, the platelet supernatants had been subjected to ultracentrifugation at 100 000 × g for 60 min, in order to remove platelet-derived microparticles, prior to the addition to PPP. Finally, in other sets of experiment, CS or supernatant from activated platelets was digested with 5 U of chondroitinase ABC (CSE; Sigma Aldrich) dissolved in PBS for 5 h at 37 °C before incubation with plasma.
Quantification of CS released from activated platelets
Platelets were activated in PRP with TRAP; this was followed by centrifugation, and the amount of CS released from the platelets was measured with toluidine blue. Plasma samples (25 μL) were incubated with 75 μL of a solution of toludine blue (25 μg mL−1; Fluka Chemie AG, Buchs, Swizerland) for 5 min at RT, and the color shift at 600 nm was compared to that generated by CS (Sigma Aldrich) serially diluted in plasma (standard curve).
Preparation of CS-coated microtiter plates
CS-coated surfaces were generated on a polyamine layer in microtiter plates using the cross-linking reagents 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide hydrochloride (EDC) and N-hydroxysuccinimide (NHS) (Pierce Biotechnology, Rockford, IL, USA). Microtiter plates (Maxisorb Immunoplates, Nunc, Copenhagen, Denmark) were coated with polyallylamine (Sigma Aldrich; 0.25 mg mL−1) dissolved in 25 mm borate buffer (pH 9.0) [25]. One hundred and fifty microliters of CS dissolved (2 mg mL−1) in 40% (v/v) ethanol containing 50 mm MES, pH 5.1 (Sigma Aldrich), was added to each well. One hundred and fifty microliters of 33 mm EDC and 6 mm NHS in 40% (v/v) ethanol containing 50 mm MES was mixed with the CS solution, and the plates were incubated on a shaker for 4 h. The surfaces were washed in 25 mm borate buffer (pH 9.0), and the remaining amine groups were acetylated using acetic acid anhydride (Merck, Darmstadt, Germany) diluted 1 : 1000 in 25 mm borate buffer (pH 10.5). The surface was rinsed repeatedly in milliQ water. The amount of bound CS was estimated to be 3 μg per well, using toluidine blue as described above.
Enzyme-linked immunosorbent assays (ELISAs)
PBS containing 0.05% Tween-20, 1% (w/v) BSA and 10 mm EDTA was used as dilution buffer, PBS containing 0.05% Tween-20 as washing buffer, and 1,2-phenylenediamine dihydrochloride in 0.1 m citrate, pH 5 as color substrate, in all ELISAs.
C3a
C3a was detected using monoclonal antibody 4SD17.3 as the capture antibody and biotinylated polyclonal anti-C3a, followed by horseradish peroxidase (HRP)-conjugated streptavidin (Amersham, Little Chalfort, UK), for detection, using a standard of zymosan-activated serum calibrated against a solution of purified C3a. Data are given as ng mL−1.
sC5b-9
Soluble C5b-9 complexes were detected using monoclonal antibody aE11 [26] (Diatec, Oslo, Norway) as the capture antibody, and polyclonal rabbit anti-human C5 followed by HRP-conjugated polyclonal swine anti-rabbit Ig (both from Dako, Glostrup, Denmark) for detection. Zymosan-activated serum was used as a standard, and values are given as arbitrary units (AU) mL−1.
Thrombin–antithrombin (TAT)
TAT complexes were measured using antibodies from Enzyme Research Laboratories (South Bend, IN, USA). Anti-human thrombin was used as the capture antibody, and HRP-coupled anti-human antithrombin (AT) antibodies for detection. Standards were prepared by mixing purified thrombin (Sigma Aldrich) with an excess of AT in the presence of heparin (GE Healthcare, Uppsala, Sweden). The standard was diluted in normal citrate phosphate dextrose plasma and standardized against the Enzygnost TAT kit (Behring-Dade, Warburg, Germany). Values are expressed as ng mL−1.
Binding of C1q to CS
Purified C1q [27] or EDTA–plasma was incubated in wells of microtiter plates coated with CS at 37 °C for 60 min. Bound C1q was detected using sheep anti-human C1q (The Binding Site, Birmingham, UK) and rabbit anti-sheep Ig HRP (Dako). Anti-C3c (Dako) and anti-AT (American Diagnostica, Greenwich, CT, USA) were used as controls.
Binding of C3 fragments to CS
C1q-depleted serum [28] diluted 1 : 10 in the presence or absence of purified C1q (15 μg mL−1) and EDTA–plasma was incubated in wells of microtiter plates coated with CS at 37 °C for up to 60 min [29]. After washing, bound C3 fragments were detected using rabbit anti-human C3c–HRP.
Flow cytometry
Upregulation of CD11b on PMNs and monocytes, as well as conjugate formation between platelets and monocytes–granulocytes in whole TRAP-activated blood, were assessed by flow cytometry using anti-CD11b–allophycocyanin (APC), anti-CD14–fluorescein isothiocyanate, anti-CD16–peridin-chlorophyll-protein complex (PerCP), and anti-CD42a–phycoerythrin (PE) (BD Pharmingen, BD Biosciences, San José,CA, USA). After activation, 100 μL of blood was incubated with the antibodies for 1 h at 4 °C. The erythrocytes were then lysed by adding 2 mL of lysis buffer (Becton Dickinson, Franklin Lakes, NJ, USA), and the leukocytes were washed three times using PBS with 1% BSA and 0.1% NaN3 (Sigma Aldrich). In some experiments, the blood was preincubated with 50 μm compstatin and/or 5.5 μm C5aR antagonist (C5aRA) AcF[OpdC-haWR] [30] for 5 min at RT before activation. The samples were then analyzed using FACS Calibur (Becton Dickinson), and 15 000 cells (granulocytes and monocytes) were collected. Data were analyzed using Cellquest Professional (Becton Dickinson). Formation of complexes was measured as the proportion of anti-CD42a-positive granulocytes or monocytes. Data are presented as mean fluorescence intensity (MFI) given as percentage of the positive (TRAP-activated) control ([MFIexperiment – MFInegative control]/[MFIpositive control – MFInegative control]). The negative control represents the MFI of non-TRAP-activated platelets.
Statistics
Results are presented as mean ± SEM. Correlations were evaluated using Spearman's rank test. Differences between means were statistically evaluated using the Mann–Whitney non-parametric test. The flow cytometry data were analyzed using repeated measures anova followed by a Bonferroni multiple comparison test.
Results
Complement activation induced by clotting in PPP, PRP, and whole blood
To determine whether the clotting induced by either contact activation or the TF pathway can trigger complement activation, we incubated fresh non-anticoagulated PPP, PRP or blood in polypropylene tubes with thromboplastin (TF-activated clotting). Complement activation, as indicated by the generation of C3a (Fig. 1A) and sC5b-9 (not shown), occurred at only a low level in PPP, representing the background activation induced by the surface of the tubes and by the gas–fluid interphase. This activation was amplified in PRP, and particularly in whole blood.
Fig. 1.
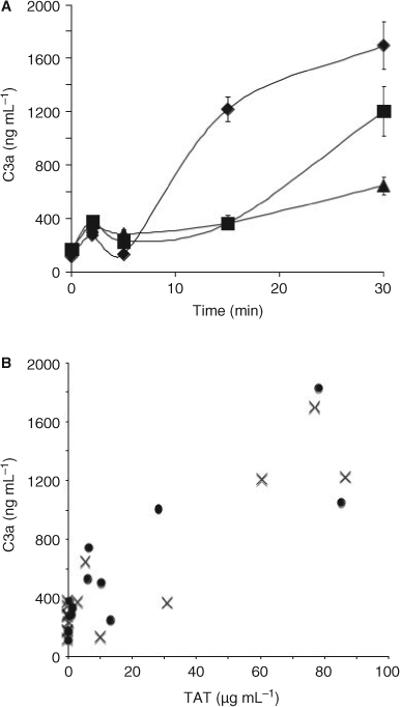
Clotting induced complement activation. (A) Generation of C3a in non-anticoagulated whole blood (diamonds), platelet-rich plasma (squares), and platelet-poor plasma (triangles), incubated in glass tubes. Data are mean ± SEM (n = 5). (B) Relationship between the generation of thrombin–antithrombin (TAT) (x-axis) and C3a (y-axis) in blood activated by incubation in glass tubes (crosses) or with thromboplastin (circles). Data points are means (n = 5).
In Fig. 1B, the thrombin generation per se, regardless of activating agent, reflected as TAT complexes are shown to correlate with the complement activation monitored as C3a levels in experiments where clotting was induced by either glass or thromboplastin (r = 0.72, P < 0.0001).
Complement activation induced by platelets
The amplification of complement activation was inhibited by the thrombin inhibitor lepirudin, further supporting the idea that complement activation was elicited by the clotting process. In order to study the effect of protease-activated receptor (PAR)-1 and PAR-4 stimulation of platelets in the absence of clotting, we developed an in vitro model where PPP, PRP and whole blood were anticoagulated using lepirudin. The platelets were then stimulated by incubation with TRAP for up to 60 min, and complement activation was monitored (Fig. 2A,B). A background level of complement activation, as indicated by the generation of C3a and sC5b-9, was seen in PPP incubated with (not shown) or without TRAP. The same level of activation was found in non-TRAP-activated PRP. This level of complement activation is elicited by the polystyrene surfaces of the plastic tubes [29]. Addition of TRAP to PRP significantly increased the formation of both C3a and sC5b-9, indicating that complement was activated by the platelets.
Fig. 2.

Complement activation induced by TRAP-activated platelets. Lepirudin-anticoagulated platelet-poor plasma (PPP) (squares), platelet-rich plasma (PRP) (circles) or PRP activated by incubation with 6.25 μg mL−1 thrombin receptor-activating peptide (TRAP) (triangles) at 37 °C for up to 60 min. The reaction was stopped by addition of 10 mm EDTA, and the generation of C3a (A) and sC5b-9 (B) was assessed by enzyme-linked immunosorbent assay. In addition, the generation of C3a (C) and sC5b-9 (D) measured in PRP and TRAP-activated PRP in the absence (gray bars) or presence (white bars) of 10 μm complement inibitor compstatin (white bars). Data are mean ± SEM (n = 5; *P < 0.05). AU, arbitrary units.
Addition of the complement inhibitor compstatin to the PPP and non-activated PRP lowered the surface-induced generation of C3a and sC5b-9 to background levels (P < 0.05) (Fig. 2C,D). Compstatin also inhibited complement activation in the TRAP-activated PRP to almost the same level (P < 0.05). TRAP treatment of PRP from an individual deficient in mannan-binding lectin (MBL) (< 40 μg mL−1) did not affect the platelet-dependent complement activation, excluding the possibility that activation occurred via the lectin pathway.
Activation of complement by CS released from activated platelets
In order to study the release of CS from platelets, we activated PRP with TRAP and measured the release of CS into the supernatant. The estimated increase in the concentration of CS was 4 μg mL−1 in PRP from a basal level of 2 μg mL−1, as determined using a colorimetric assay. Next, the ability of CS to activate complement was demonstrated by incubating CS in lepirudin-anticoagulated PPP (Fig. 3A,B), resulting in generation of C3a and sC5b-9 in a dose-dependent fashion. Inhibition of complement by addition of EDTA, Mg2+-EGTA, or compstatin was able to inhibit this activation, and digestion of CS with chondroitinase ABC prior to the addition to PPP also abrogated the reaction. C3a and sC5b-9 were likewise generated when supernatants from activated platelets were incubated with lepirudin-treated PPP for 30 min at 37 °C (Fig. 3C,D). Less than 40% of the complement-activating activity was lost by ultracentrifugation of the supernatants at 100 000 × g, indicating that part of the activity was dependent on the presence of microparticles.
Fig. 3.

Complement activation by CS in the fluid phase. Platelet-poor plasma (PPP) was incubated with increasing concentrations of chondroitin sulfate (CS) for 30 min at 37 °C, and the generation of C3a (A) and sC5b-9 (B) in PPP (diamonds), without additives and in the presence of 10 mm EDTA (triangles) or 10 mm Mg2+-EGTA (squares), was measured by enzyme-linked immunosorbent assay (ELISA). CS (25 μg mL−1) and supernatants from isolated and thrombin receptor-activating peptide-activated platelets (PS) were treated with 5 U of chondroitinase ABC (CSE) for 5 h at 37 °C, and then incubated with PPP for 30 min at 37 °C (white bars). In the same experiments, CS and PS were incubated in PPP in the absence (black bars) or presence (gray bars) of 10 mm Mg2+-EGTA. The generation of C3a (C) and sC5b-9 (D) was analyzed by ELISA. Data are mean ± SEM (n = 3). AU, arbitrary units.
Triggering of complement activation by C1q binding to CS
The binding of purified C1q to CS immobilized to microtiter plates increased in a dose-dependent fashion when serial dilutions (0−300 μg mL−1) were incubated for 60 min (not shown). Similar binding of C1q was observed when undiluted EDTA–plasma was incubated in the wells for up to 60 min (Fig. 4A). Measurements of C3 or AT binding were both negative. As a functional verification of this binding reaction, we measured the level of complement activation that occurred in C1q-depleted serum, with or without the addition of purified C1q. The serum preparations were incubated in CS-coated microtiter wells for up to 60 min, and the binding of C3 fragments, as an indicator of complement activation, was quantified (Fig. 4B). There was no binding of C3 in the absence of C1q, but when purified C1q was added, the complement activity was restored as monitored by a time-dependent increase in bonding. No binding of C3 fragments was observed in EDTA–plasma (where complement is inactivated; Fig. 4B).
Fig. 4.

Complement activation by surface-bound CS. (A) Binding of C1q (squares), C3 (triangles) and antithrombin (circles) in EDTA–plasma to chondroitin sulfate (CS) conjugated to microtiter plates. (B) Binding of C3 fragments from C1q-depleted serum to CS conjugated to microtiter plates in the presence (squares) or absence (diamonds) of purified C1q. EDTA–plasma was used as control (circles). Data represent mean ± SEM (n = 3). AU, arbitrary units.
Activation of monocytes and granulocytes
The physiologic relevance of platelet-triggered complement activation was investigated by monitoring the expression of CD11b on monocytes and granulocytes, as well as the formation of conjugates between platelets and monocytes–granulocytes. Addition of TRAP in the presence or absence of non-functional linear compstatin peptide to lepirudin-treated whole blood led to identical upregulation of CD11b on monocytes and granulocytes (Fig. 5A) and formation of granulocyte–platelet and monocyte–platelet complexes (Fig. 5B). Addition of either compstatin or C5aRA inhibited this expression completely on monocytes (P < 0.05) and by ∼ 60% on granulocytes (P < 0.05).
Fig. 5.
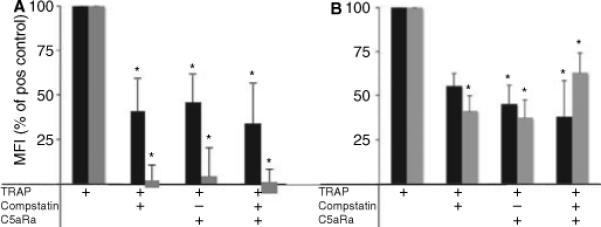
Activation of granulocytes and monocytes in TRAP-activated whole blood. (A) CD11b expression on granulocytes (black bars) and monocytes (gray bars). (B) Formation of granulocyte–platelet (black bars) and monocyte–platelet (gray bars) conjugates in whole blood activated with 6.25 μg mL−1 thrombin receptor-activating peptide (TRAP) as assessed by flow cytometry. The activation was inhibited using 50 μm compstatin and/or 5.5 μm C5aRA, as indicated in the table below the figure. Data are expressed as percentage mean fluorescence intensity (MFI) of the positive control (mean ± SEM; n = 6) as described in Materials and methods (*P < 0.05).
In addition, the granulocyte–platelet and monocyte–platelet complex formation was also significantly inhibited by addition of either compstatin or C5aRA (P < 0.05), indicating that the upregulation was triggered by complement activation induced by TRAP-activated platelets. This observation confirmed the interpretation that the cellular activation was complement-mediated.
Discussion
In the present work, we have demonstrated that clotting in whole blood, elicited by either the contact system or the TF pathway, leads to activation of the complement system. In clotting PRP and whole blood, complement activation was observed at levels well above the background level of activation induced by contact with the surface of the polystyrene plastic tube. These findings therefore indicate that the complement activation was mediated by the blood cells in the sample, and the results are in agreement with an earlier study in which we detected lower amounts of TAT in PRP than in whole blood [31]. As both PRP and whole blood exhibited this property, and the complement activation was inhibited by the specific thrombin inhibitor lepirudin, we focused our investigation on platelets. In order to study platelet-induced complement activation, we used a system in which platelets were activated by TRAP in PRP or whole blood that had been anticoagulated with lepirudin, allowing us to activate platelets specifically without causing a clotting reaction.
Significantly increased production of both C3a and sC5b-9, indicating complement activation, was found in the fluid phase of TRAP-activated PRP and whole blood, and this complement activation was confirmed by the inhibition seen in the presence of the C3 inhibitor compstatin. Incubation of PPP with supernatants from activated platelets resulted in a similar generation of C3a and sC5b-9, demonstrating that complement activation takes place in the fluid phase of TRAP-activated PRP and whole blood. Less than 40% of this activity was lost if the supernatants were ultracentrifuged, indicating that some of the activity was associated with microparticles. CS is a GAG that is released by activated platelets [19]. In the present study, we have demonstrated that CS is a strong activator of complement, making this substance a likely trigger for platelet-induced complement activation. This potential role was confirmed by incubating CS and supernatants from activated platelets with chondroitinase ABC, which abrogated the complement activation.
Specific chelation of Ca2+ with EGTA totally inhibited complement activation [32], indicating that the activation was triggered by the C1 complex in the classic pathway, which requires Ca2+ for its function, and ruling out complement activation by the alternative pathway [17]. The involvement of the lectin pathway is less likely, as PRP from an individual deficient in MBL still activated complement after TRAP stimulation to the same extent.
Interactions between C1q and polyanions such as heparin and DNA are well established [29], and heparin at low concentrations tends to activate complement [29,33]. Kirschfink and collaborators have previously demonstrated interaction between C1q and GAGs secreted by human B-cell lines [34], but in contrast to platelets, which mainly contain CS-A [19,20], leukocytes and leukocyte cell lines release high amounts of heparan sulfate (25−30%) in addition to CS-A and CS-C [35]. In the present study, we have shown that C1q, both in purified form and in whole EDTA–plasma, can bind specifically to surface-conjugated CS.
The surface-bound CS did not activate complement in C1q-depleted serum, but its ability to activate complement was restored when the C1q-depleted serum was reconstituted with purified C1q. Binding of C1q to CS is also consistent with the previously described interaction between these two molecules [22,23]; in these two studies, CS was shown to inhibit C1qspecific hemolytic activity. In light of our results, it is obvious that this inhibition was accomplished by classic pathway activation, thereby depleting the plasma of intact complement components, which subsequently leads to inhibition of the hemolytic activity of C1q.
In platelets, the CS-A chains are attached to a serine- and glycine-rich protein core forming the serglycin macromolecule. Serglycins of different compositions have been implicated as carriers of proteases [36]. Considering the fact that we do not see any complement activation in C1q-depleted serum, and that the activity is restored upon addition of purified C1q (which is not proteolytically activated), it is not likely that other non-complement proteases are responsible for the activation.
Taken together, these findings show that the binding of C1q to CS in the fluid phase triggers complement activation in TRAP-activated PRP. This reaction adds an additional mechanism to the previously reported mechanisms of complement activation, which have been confined to the platelet surface. Del Conde and co-workers [17] have proposed that P-selectin, by binding C3b, triggers the activation of complement, principally through the alternative pathway. In contrast, Peerschke et al. [18] have reported that complement is activated on the surface of activated platelets. In their assay using platelets fixed to microtiter plates with polylysine and glutaraldehyde, they demonstrated that the activation involved classic pathway components. The mechanism for complement activation on the platelet surface is yet to be established.
In the present study, stimulation of whole blood with TRAP led to upregulation of CD11b on monocytes and granulocytes. The release of CS from TRAP-activated platelets in PRP was shown to add approximately 4 μg mL−1 CS to the plasma. The effect of this amount of CS would be multiplied in the vicinity of the platelets, leading to enhanced complement activation around the platelets. The physiologic relevance of this complement activation was demonstrated by the finding that TRAP-mediated platelet activation leads to CD11b upregulation on leukocytes and generation of leukocyte–platelet complexes (Fig. 5). This link was confirmed by our observation that compstatin inhibited these functions. The dependence on the C5a receptor was demonstrated by attenuation of CD11b expression and inhibition of leukocyte–platelet complex formation in the presence of a C5aRA. The upregulation of CD11b and generation of leukocyte–platelet complexes may have implications for a wide variety of processes, from clotting to tissue regeneration. The high affinity between CD11b and fibrin [37] and the chemotactic properties of the complement-generated anaphylatoxins and fibrin degradation products [38] suggest that complement activation may be important for leukocytic infiltration of blood clots and leukocyte-mediated fibrinolysis [39,40], previously reported to be dependent on C3 [41].
In conclusion, we have demonstrated that complement is activated by CS released from thrombin receptor-activated platelets, as indicated by C3a and C5b-9 generation in the fluid phase of PRP and blood. This activation leads to the generation of C5a, which upregulates CD11b on both PMNs and monocytes (Fig. 6). These findings may have important implications for a wide range of clinical conditions that involve platelet activation. Release of CS will contribute to the complement activation that occurs in thrombotic conditions such as myocardial and stroke [4-6,42]. Also, platelet activation is a major contributor to the increased risk for thrombosis in systemic lupus erythematosis [43]. This activation will add to the complement activation that typically accompanies exacerbations of the disease. Therefore, CS-mediated activation of complement provides a new potential target for reducing the pathologic consequences of platelet activation, and adds to mechanisms that link platelets to inflammation and the immune system.
Fig. 6.

Model for how chondroitin sulfate (CS) released from thrombin receptor-activated platelets induces complement activation in the fluid phase of platelet-rich plasma and blood. This activation leads to the generation of C5a, which subsequently upregulates CD11b on polymorphonuclear neutrophils (PMNs) and monocytes (Mo) and to formation of leukocyte–platelet complexes. PAR, protease-activated receptor.
Acknowledgements
This work was supported by National Institute of Health Grants AI068730, EB003968, GM-62134, and GM069736, Swedish Research Council Grants 2006-5595 and 15244, and grants from the Natural Sciences Faculty, University of Kalmar.
Footnotes
Disclosure of Conflict of Interests
The authors state that they have no conflict of interest.
References
- 1.Palikhe A, Sinisalo J, Seppanen M, Haario H, Meri S, Valtonen V, Nieminen MS, Lokki ML. Serum complement C3/C4 ratio, a novel marker for recurrent cardiovascular events. Am J Cardiol. 2007;99:890–5. doi: 10.1016/j.amjcard.2006.11.034. [DOI] [PubMed] [Google Scholar]
- 2.Muscari A, Bozzoli C, Puddu GM, Sangiorgi Z, Dormi A, Rovinetti C, Descovich GC, Puddu P. Association of serum C3 levels with the risk of myocardial infarction. Am J Med. 1995;98:357–64. doi: 10.1016/S0002-9343(99)80314-3. [DOI] [PubMed] [Google Scholar]
- 3.Iltumur K, Karabulut A, Toprak G, Toprak N. Complement activation in acute coronary syndromes. APMIS. 2005;113:167–74. doi: 10.1111/j.1600-0463.2005.apm1130303.x. [DOI] [PubMed] [Google Scholar]
- 4.Langlois PF, Gawryl MS. Detection of the terminal complement complex in patient plasma following acute myocardial infarction. Atherosclerosis. 1988;70:95–105. doi: 10.1016/0021-9150(88)90103-7. [DOI] [PubMed] [Google Scholar]
- 5.Yasuda M, Kawarabayashi T, Akioka K, Teragaki M, Oku H, Kanayama Y, Takeuchi K, Takeda T, Kawase Y, Ikuno Y. The complement system in the acute phase of myocardial infarction. Jpn Circ J. 1989;53:1017–22. doi: 10.1253/jcj.53.1017. [DOI] [PubMed] [Google Scholar]
- 6.Yasuda M, Takeuchi K, Hiruma M, Iida H, Tahara A, Itagane H, Toda I, Akioka K, Teragaki M, Oku H. The complement system in ischemic heart disease. Circulation. 1990;81:156–63. doi: 10.1161/01.cir.81.1.156. [DOI] [PubMed] [Google Scholar]
- 7.Markiewski MM, Nilsson B, Nilsson Ekdahl K, Mollnes TE, Lambris JD. Complement and coagulation: strangers or partners in crime? Trends Immunol. 2007;28:184–92. doi: 10.1016/j.it.2007.02.006. [DOI] [PubMed] [Google Scholar]
- 8.Cicardi M, Zingale L, Zanichelli A, Pappalardo E, Cicardi B. C1 inhibitor: molecular and clinical aspects. Springer Semin Immunopathol. 2005;27:286–98. doi: 10.1007/s00281-005-0001-4. [DOI] [PubMed] [Google Scholar]
- 9.Ghebrehiwet B, Silverberg M, Kaplan AP. Activation of the classical pathway of complement by Hageman factor fragment. J Exp Med. 1981;153:665–76. doi: 10.1084/jem.153.3.665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Spath P, Gabl F. Critical role of the conversion of the third complement component C3 (beta 1C/beta 1A) for its immunochemical quantitation. Clin Chim Acta. 1976;73:171–5. doi: 10.1016/0009-8981(76)90319-3. [DOI] [PubMed] [Google Scholar]
- 11.Huber-Lang M, Sarma JV, Zetoune FS, Rittirsch D, Neff TA, McGuire SR, Lambris JD, Warner RL, Flierl MA, Hoesel LM, Gebhard F, Younger JG, Drouin SM, Wetsel RA, Ward PA. Generation of C5a in the absence of C3: a new complement activation pathway. Nat Med. 2006;12:682–7. doi: 10.1038/nm1419. [DOI] [PubMed] [Google Scholar]
- 12.Sims PJ, Wiedmer T. The response of human platelets to activated components of the complement system. Immunol Today. 1991;12:338–42. doi: 10.1016/0167-5699(91)90012-I. [DOI] [PubMed] [Google Scholar]
- 13.Ikeda K, Nagasawa K, Horiuchi T, Tsuru T, Nishizaka H, Niho Y. C5a induces tissue factor activity on endothelial cells. Thromb Haemost. 1997;77:394–8. [PubMed] [Google Scholar]
- 14.Ritis K, Doumas M, Mastellos D, Micheli A, Giaglis S, Magotti P, Rafail S, Kartalis G, Sideras P, Lambris JD. A novel C5a receptor–tissue factor cross-talk in neutrophils links innate immunity to coagulation pathways. J Immunol. 2006;177:4794–802. doi: 10.4049/jimmunol.177.7.4794. [DOI] [PubMed] [Google Scholar]
- 15.Tedesco F, Pausa M, Nardon E, Introna M, Mantovani A, Dobrina A. The cytolytically inactive terminal complement complex activates endothelial cells to express adhesion molecules and tissue factor procoagulant activity. J Exp Med. 1997;185:1619–27. doi: 10.1084/jem.185.9.1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kalowski S, Howes EL, Jr, Margaretten W, McKay DG. Effects of intravascular clotting on the activation of the complement system: the role of the platelet. Am J Pathol. 1975;78:525–36. [PMC free article] [PubMed] [Google Scholar]
- 17.Del Conde I, Cruz MA, Zhang H, Lopez JA, Afshar-Kharghan V. Platelet activation leads to activation and propagation of the complement system. J Exp Med. 2005;201:871–9. doi: 10.1084/jem.20041497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Peerschke EI, Yin W, Grigg SE, Ghebrehiwet B. Blood platelets activate the classical pathway of human complement. J Thromb Haemost. 2006;4:2035–42. doi: 10.1111/j.1538-7836.2006.02065.x. [DOI] [PubMed] [Google Scholar]
- 19.Ward JV, Packham MA. Characterization of the sulfated glycosaminoglycan on the surface and in the storage granules of rabbit platelets. Biochim Biophys Acta. 1979;583:196–207. doi: 10.1016/0304-4165(79)90427-6. [DOI] [PubMed] [Google Scholar]
- 20.Okayama M, Oguri K, Fujiwara Y, Nakanishi H, Yonekura H, Kondo T, Ui N. Purification and characterization of human platelet proteoglycan. Biochem J. 1986;233:73–81. doi: 10.1042/bj2330073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Donato JL, Nogueira MD, Marcondes S, Antunes E, Nader HB, Dietrich CP, de Nucci G. The kinetics of chondroitin 4-sulfate release from stimulated platelets and its relation to thromboxane A2 formation and granule secretion. Braz J Med Biol Res. 1994;27:2163–7. [PubMed] [Google Scholar]
- 22.Silvestri L, Baker JR, Roden L, Stroud RM. The C1q inhibitor in serum is a chondroitin 4-sulfate proteoglycan. J Biol Chem. 1981;256:7383–7. [PubMed] [Google Scholar]
- 23.Ghebrehiwet B, Galanakis DK. C1q inhibitor (chondroitin-4-sulfate proteoglycan): structure and function. Behring Inst Mitt. 1993;93:214–23. [PubMed] [Google Scholar]
- 24.Katragadda M, Magotti P, Sfyroera G, Lambris JD. Hydrophobic effect and hydrogen bonds account for the improved activity of a complement inhibitor, compstatin. J Med Chem. 2006;49:4616–22. doi: 10.1021/jm0603419. [DOI] [PubMed] [Google Scholar]
- 25.Christensen K, Larsson R, Emanuelsson H, Elgue G, Larsson A. Coagulation and complement activation. Biomaterials. 2001;22:349–55. doi: 10.1016/s0142-9612(00)00190-3. [DOI] [PubMed] [Google Scholar]
- 26.Mollnes TE, Lea T, Froland SS, Harboe M. Quantification of the terminal complement complex in human plasma by an enzyme-linked immunosorbent assay based on monoclonal antibodies against a neoantigen of the complex. Scand J Immunol. 1985;22:197–202. doi: 10.1111/j.1365-3083.1985.tb01871.x. [DOI] [PubMed] [Google Scholar]
- 27.Tenner AJ, Lesavre PH, Cooper NR. Purification and radiolabeling of human C1q. J Immunol. 1981;127:648–53. [PubMed] [Google Scholar]
- 28.Sjoholm AG, Selander B, Ostenson S, Holmstrom E, Soderstrom C. Normal human serum depleted of C1q, factor D and properdin: its use in studies of complement activation. APMIS. 1991;99:1120–8. doi: 10.1111/j.1699-0463.1991.tb01309.x. [DOI] [PubMed] [Google Scholar]
- 29.Nilsson UR, Larm O, Nilsson B, Storm KE, Elwing H, Nilsson Ekdahl K. Modification of the complement binding properties of polystyrene: effects of end-point heparin attachment. Scand J Immunol. 1993;37:349–54. doi: 10.1111/j.1365-3083.1993.tb02564.x. [DOI] [PubMed] [Google Scholar]
- 30.Strey CW, Markiewski M, Mastellos D, Tudoran R, Spruce LA, Greenbaum LE, Lambris JD. The proinflammatory mediators C3a and C5a are essential for liver regeneration. J Exp Med. 2003;198:913–23. doi: 10.1084/jem.20030374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hong J, Larsson A, Ekdahl KN, Elgue G, Larsson R, Nilsson B. Contact between a polymer and whole blood: sequence of events leading to thrombin generation. J Lab Clin Med. 2001;138:139–45. doi: 10.1067/mlc.2001.116486. [DOI] [PubMed] [Google Scholar]
- 32.Nilsson B, Larsson R, Hong J, Elgue G, Ekdahl KN, Sahu A, Lambris JD. Compstatin inhibits complement and cellular activation in whole blood in two models of extracorporeal circulation. Blood. 1998;92:1661–7. [PubMed] [Google Scholar]
- 33.Mollnes TE, Brekke OL, Fung M, Fure H, Christiansen D, Bergseth G, Videm V, Lappegard KT, Kohl J, Lambris JD. Essential role of the C5a receptor in E. coli-induced oxidative burst and phagocytosis revealed by a novel lepirudin-based human whole blood model of inflammation. Blood. 2002;100:1869–77. [PubMed] [Google Scholar]
- 34.Kirschfink M, Blase L, Engelmann S, Schwartz-Albiez R. Secreted chondroitin sulfate proteoglycan of human B cell lines binds to the complement protein C1q and inhibits complex formation of C1. J Immunol. 1997;158:1324–31. [PubMed] [Google Scholar]
- 35.Makatsori E, Karamanos NK, Papadogiannakis N, Hjerpe A, Anastassiou ED, Tsegenidis T. Synthesis and distribution of glycosaminoglycans in human leukemic B- and T-cells and monocytes studied using specific enzymic treatments and high-performance liquid chromatography. Biomed Chromatogr. 2001;15:413–17. doi: 10.1002/bmc.91. [DOI] [PubMed] [Google Scholar]
- 36.Kolset SO, Tveit H. Serglycin – structure and biology. Cell Mol Life Sci. 2008;65:1073–85. doi: 10.1007/s00018-007-7455-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Goel MS, Diamond SL. Neutrophil enhancement of fibrin deposition under flow through platelet-dependent and -independent mechanisms. Arterioscler Thromb Vasc Biol. 2001;21:2093–8. doi: 10.1161/hq1201.100255. [DOI] [PubMed] [Google Scholar]
- 38.Gross TJ, Leavell KJ, Peterson MW. CD11b/CD18 mediates the neutrophil chemotactic activity of fibrin degradation product D domain. Thromb Haemost. 1997;77:894–900. [PubMed] [Google Scholar]
- 39.Kolev K, Komorowicz E, Owen WG, Machovich R. Quantitative comparison of fibrin degradation with plasmin, miniplasmin, neutrophil leukocyte elastase and cathepsin G. Thromb Haemost. 1996;75:140–6. [PubMed] [Google Scholar]
- 40.Simon DI, Ezratty AM, Francis SA, Rennke H, Loscalzo J. Fibrin(ogen) is internalized and degraded by activated human monocytoid cells via Mac-1 (CD11b/CD18): a nonplasmin fibrinolytic pathway. Blood. 1993;82:2414–22. [PubMed] [Google Scholar]
- 41.Taylor FB, Jr, Muller-Eberhard HJ. Qualitative description of factors involved in the retraction and lysis of dilute whole blood clots and in the aggregation and retraction of platelets. J Clin Invest. 1970;49:2068–85. doi: 10.1172/JCI106425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mocco J, Wilson DA, Komotar RJ, Sughrue ME, Coates K, Sacco RL, Elkind MS, Connolly ES., Jr Alterations in plasma complement levels after human ischemic stroke. Neurosurgery. 2006;59:28–33. doi: 10.1227/01.NEU.0000219221.14280.65. [DOI] [PubMed] [Google Scholar]
- 43.Ekdahl KN, Bengtsson AA, Andersson J, Elgue G, Ronnblom L, Sturfelt G, Nilsson B. Thrombotic disease in systemic lupus erythematosus is associated with a maintained systemic platelet activation. Br J Haematol. 2004;125:74–8. doi: 10.1111/j.1365-2141.2004.04858.x. [DOI] [PubMed] [Google Scholar]