

Abstract
Xanthine oxidoreductase (XOR) is a molybdenum-containing enzyme that under physiological conditions catalyzes the final two steps in purine catabolism, ultimately generating uric acid for excretion. Here we have investigated four naturally-occurring compounds that have been reported to be inhibitors of XOR in order to examine the nature of their inhibition utilizing in vitro steady-state kinetic studies. We find that luteolin, silibinin, and quercetin are mixed-type inhibitors of the enzyme in vitro and, unlike allopurinol, the inhibition is not time-dependent. These three natural products also decrease the production of superoxide by the enzyme. In contrast, and contrary to previous reports in the literature based on in vivo and other non-mechanistic studies, we find that curcumin did not inhibit the activity of purified XO, nor its superoxide production in vitro.
Xanthine oxidoreductase (XOR) is a 290 kDa molybdenum-containing enzyme that has been studied extensively from a biochemical perspective for more than 80 years. In human physiology, XOR catalyzes the final two steps of purine catabolism, transforming hypoxanthine to xanthine and then xanthine to uric acid by sequential oxidative hydroxylations at C-2 and C-8 of the purine ring, respectively (Figure 1).1 Xanthine oxidoreductase exists in two forms. The protein normally exists as a dehydrogenase (xanthine dehydrogenase, XDH), and utilizes NAD+ as its final electron acceptor in catalysis. Under certain conditions, most notably ischemia and/or hypoxia, XDH can be converted to an oxidase form (XO), which can no longer reduce NAD+ and instead utilizes O2 exclusively as the terminal electron acceptor in the course of turnover. This conversion may occur either by oxidation of sulfhydryl groups and/or by limited proteolysis.2, 3 Once in the oxidase form, the enzyme generates significant amounts of H2O2 and superoxide radicals, although XDH can also react with O2 and generate these reactive oxygen species (ROS).3 Under normal conditions, however, NAD+ effectively competes with O2 and limits the generation of ROS by the dehydrogenase.6,7 The specific conformational change responsible for the D to O conversion involves modification of the access channel to the flavin site (with its FAD cofactor) of XOR, eliminating NAD+ binding.3, 4 Once generated, these ROS can interfere with a multitude of cellular functions and processes. Two of the most extensively studied are the integrity of cellular membrane lipids and the interactions of superoxide with the vasodilator NO in circulation. In the latter case, superoxide from xanthine oxidase has been shown to degrade S-nitrothiols, which are considered to be a storage form of NO, as well as converting NO to the vaso-inactive compound peroxynitrite.8,9 Superoxide has also been shown to reduce H2O2 to form destructive hydroxyl radicals, and even carbonate radicals.10
Figure 1.

Catalytic mechanism of xanthine oxidoreductase conversion of xanthine to uric acid at the molybdenum-containing active site.
XOR is thus potentially a main player in many pathological conditions, and additionally is also thought to be involved in the pathogenesis of and secondary complications associated with other diseases.11,12,13 Hyperuricemia resulting from XOR activity is central to the pathogenesis of gout and gouty arthritis, as well as being causative in cases of high serum urate following (tumor) cellular necrosis in tumor lysis syndrome.14,15 XOR in its oxidase form is considered to be a main source of oxidative stress and destructive free radicals in ischemia-reperfusion injury associated with heart attacks and stroke, spinal cord injury, as well as being a destructive force in myocardial or renal hypoxia and infarctions.11,16,17
The mechanism of substrate hydroxylation by the molybdenum center of XOR is depicted in Figure 1, being initiated by abstraction of a proton from the Mo-OH group by an active site glutamate residue that is universally conserved in this family of molybdenum-containing enzymes.5 Subsequent to deprotonation, nucleophilic attack on substrate and concomitant hydride transfer to the metal center leads to an intermediate having product coordinated to the now reduced Mo center via the newly introduced hydroxy group. From the molybdenum center, electrons are passed sequentially via two [2Fe-2S] clusters to an FAD site, where they are finally passed to NAD+ or O2.
Inhibition of XOR is a primary objective in treating any case of hyperuricemia.11,18 Allopurinol was the first mechanism-based inhibitor of the enzyme to be developed, and is still the primary drug for combating hyperuricemia. Allopurinol is hydroxylated by the enzyme to alloxanthine (oxypurinol), which, once formed, coordinates tightly to the reduced form of the molybdenum center specifically (Figure 2) replacing the Mo-OH group of native enzyme.20 Although allopurinol has longstanding use in pharmacotherapy and is efficacious in both lowering urate levels in the body and retarding the metabolism of chemotherapeutic agents such as 6-mercaptopurine, some individuals exhibit hypersensitivity to the drug; in particular, side effects such as vasculitis (especially in those with already compromised renal function) may occur.11 The development of alternative XOR inhibitors thus remains desirable. One such inhibitor that is currently in clinical trials is febuxostat, whose crystal structure in complex with the enzyme has been determined (Figure 2).21,22,23 This structure shows the (hydroxylated) inhibitor bound to the molybdenum center and blocking access to the active site of the enzyme in a manner reminiscent of alloxanthine binding.19
Figure 2.
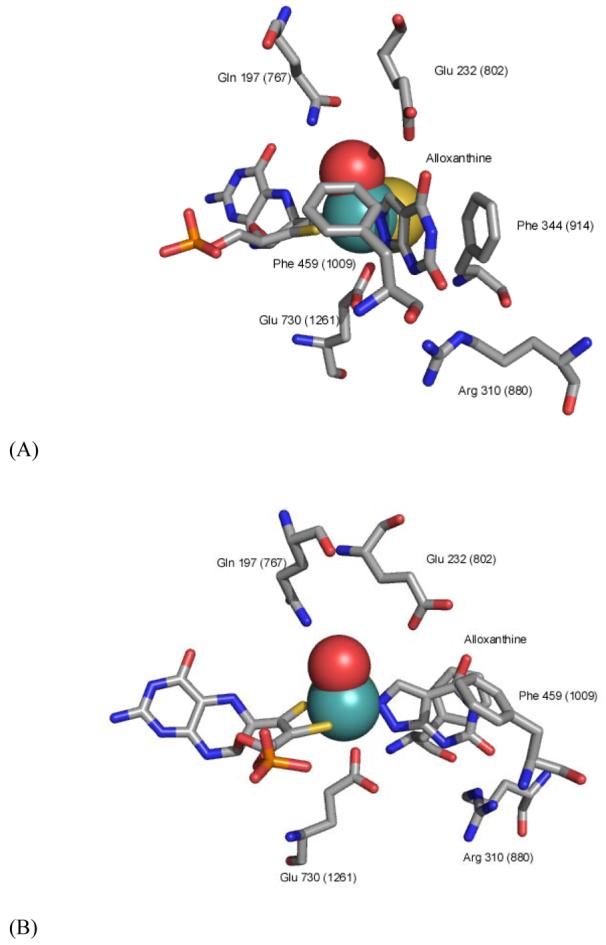
The active site of XDH from Rhodobacter capsulatus (PDB code 1JRP) showing the coordination of alloxanthine (reacted allopurinol) directly to the molybdenum center (modified from 37). Numbering in parentheses is for the bovine enzyme. Panel B is rotated 60 degrees to the right from panel A.
Investigations of the structure and function of xanthine oxidoreductase have most often utilized the enzyme isolated from bovine milk, although the enzyme from human, avian and bacterial sources have also been characterized, and have all been found to be homologous with, for example, the identical constituon of redox-active centers.32,33,34,35 In our own laboratory we have extensively investigated XO from bovine milk as well as XDH from the purple bacterium Rhodobacter capsulatus.36,37,38,39 Reviews of the current progress on such biochemical studies of the enzyme are available elsewhere.1,2
Although many previous reports have suggested the existence of naturally occurring XOR inhibitors ranging from flavonoids to a host of other natural plant products, and several of these studies have utilized the readily available enzyme from bovine milk, little work has focused on the detailed mechanism by which such inhibition may occur.24-31 Here we have examined the mechanism of inhibition exhibited four such compounds; silibinin, quercetin, curcumin, and luteolin. We find that luteolin, silibinin, and quercetin are inhibitors of the enzyme as manifested in their reduction in the initial rate of catalysis of xanthine to urate, and that luteolin and quercetin also proportionately reduce the rate of superoxide generation by XO with xanthine. In contrast, we find that curcumin does not inhibit the production of urate or superoxide by XO in vitro. Mechanistic details of the inhibition or for the lack of inhibition are discussed.
Results and Discussion
For each compound, absorbance spectra were taken each minute for 10 minutes to demonstrate stability of the stock solutions. Each compound was also monitored at 295 nm in the presence of 50 nM XO, and additionally at 550 nm with 75 μM cytochrome c as well as XO. No change in absorbance was observed at 295 nm for any of the compound tested, although a very slow increase in A550 in the cases of quercetin and luteolin was observed, indicating slow reduction of oxidized cytochrome c. None of the four compounds tested here caused any reduction of anaerobic enzyme, as monitored by the absorbance at 450 nm, indicating that none are hydroxylated by the enzyme (at least not on the timescale of the present assays).40
Allopurinol at concentrations of 10, 25, and 50 μM was used as a control for comparison to any inhibition exhibited by each of the natural products to be tested. Consistent with previous work, allopurinol inhibited XO in an uncompetitive manner reflecting binding of the hydroxylated product alloxanthine to the reduced (MoIV) form of the enzyme.20 Allopurinol was shown to reduce kcat/Km seen with xanthine, (reflecting the reaction of free enzyme and free xanthine at low [xanthine]) some 30-fold at a concentration of 50 μM, and >400-fold at 25 μM following a 10 min pre-incubation with the enzyme. Figure 5 compares the ΔA295 over 10 minutes in the presence of each compound at 50 μM with 100 μM xanthine and 5 nM XO, showing time-dependent inhibition of XO by allopurinol as noted previously by others42. Other kinetic data for the inhibition of XO by allopurinol are given in Table 1.
Figure 5.

Time course of the inhibition of XO by allopurinol, luteolin, silibinin, and quercetin. Reactions were monitored by A295 at 30 second intervals over 10 minutes. Each point represents an average of 3 trials.
Table 1.
Steady-state kinetic parameters for the inhibition of xanthine oxidase.
| kcat (s−1) | Km (μM) |
kcat/Km (μM−1·s−1) |
Ki (μM) | ||||
|---|---|---|---|---|---|---|---|
| Luteolin | 1.9 ± 0.7 | ||||||
| 10 μM | 6.2 ± 0.2 | 31 ± 5 | 0.20 | ||||
| 25 μM | 3.8 ± 0.2 | 127 ± 20 | 0.03 | ||||
| 50 μM | 2.6 ± 0.2 | 164 ± 30 | 0.02 | ||||
| Incubation | 5.8 ± 0.5 | 126 ± 30 | 0.05 | ||||
| Control | 12.1 ± 0.6 | 3.5 ± 0.8 | 3.5 | ||||
| Quercetin | 1.2 ± 0.7 | ||||||
| 10 μM | 5.9 ± 0.2 | 3.7 ± 0.3 | 1.6 | ||||
| 25 μM | 4.5 ± 0.4 | 43 ± 10 | 0.10 | ||||
| 50 μM | 1.9 ± 0.2 | 106 ± 30 | 0.02 | ||||
| Incubation | 4.3 ± 0.8 | 104 ± 50 | 0.04 | ||||
| Control | 10.5 ± 0.4 | 1.2 ± 0.3 | 8.8 | ||||
| Curcumin | |||||||
| 10 μM | 13.3 ± 0.6 | 2.8 ± 0.6 | 4.8 | ||||
| 25 μM | 11.8 ± 0.4 | 1.8 ± 0.3 | 6.5 | ||||
| 50 μM | 12.2 ± 0.5 | 2.4 ± 0.5 | 5.1 | ||||
| Incubation | 12.4 ± 0.8 | 3.2 ± 0.9 | 3.9 | ||||
| Control | 12.4 ± 0.4 | 2.1 ± 0.4 | 5.9 | ||||
| Silibinin | |||||||
| 10 μM | 5.0 ± 0.3 | 36 ± 8 | 0.14 | ||||
| 25 μM | 4.0 ± 0.4 | 30 ± 10 | 0.13 | ||||
| 50 μM | 4.3 ± 0.3 | 39 ± 10 | 0.11 | ||||
| Incubation | 3.8 ± 0.1 | 53 ± 7 | 0.07 | ||||
| Control | 10.9 ± 0.4 | 1.9 ± 0.3 | 5.6 | ||||
| Allopurinol | |||||||
| 10 μM | 8.1 ± 0.6 | 25 ± 7 | 0.32 | ||||
| 25 μM | 6.5 ± 0.6 | 43 ± 20 | 0.15 | ||||
| 50 μM | 6.9 ± 0.5 | 63 ± 20 | 0.11 | ||||
| Incubation | 1.3 ± 0.3 | 189 ± 90 | 0.007 | ||||
| Control | 10.1 ± 0.8 | 3.1 ± 1.0 | 3.3 | ||||
The reduction of cytochrome c by superoxide generated by XO in the course of turnover was significantly decreased at each concentration of allopurinol as shown in Table 2. The decrease was most pronounced following incubation of the enzyme with 25 μM allopurinol, as expected, and was proportionate to the decrease in xanthine consumption following incubation as monitored at 295 nm in the above experiments.
Table 2.
Initial rates of reduction for cytochrome c by xanthine oxidase + xanthine in the presence of natural product. Also shown are rates following 10 minute incubations of enzyme with 25 μM natural products and cytochrome c prior to the addition of xanthine. Final reaction mixtures were monitored over several minutes at 550 nm and contained 50 nM XO, 100 μM xanthine, natural product, and 75 μM cytochrome c. Error expressed as standard deviations from triplicate trials.
| Rate of cyt c reduction (nM · s−1) | ||
|---|---|---|
| Luteolin | ||
| Control | 160 ± 0.6 | |
| 10 μM | 100 ± 3 | |
| 25 μM | 82 ± 9 | |
| 50 μM | 71 ± 3 | |
| Incubation | 110 ± 10 | |
| (Control) | (170 ± 20) | |
| Quercetin | ||
| Control | 170 ± 1 | |
| 10 μM | 140 ± 20 | |
| 25 μM | 100 ± 7 | |
| 50 μM | 80 ± 6 | |
| Incubation | 110 ± 7 | |
| (Control) | (170 ± 20) | |
| Curcumin | ||
| Control | 160 ± 20 | |
| 10 μM | 160 ± 7 | |
| 25 μM | 150 ± 6 | |
| 50 μM | 150 ± 6 | |
| Incubation | 150 ± 10 | |
| (Control) | (170 ± 20) | |
| Silibinin | ||
| Control | 160 ± 0.6 | |
| 10 μM | 160 ± 7 | |
| 25 μM | 150 ± 17 | |
| 50 μM | 140 ± 13 | |
| Incubation | 120 ± 4 | |
| (Control) | (170 ± 20) | |
| Allopurinol | ||
| Control | 170 ± 10 | |
| 10 μM | 140 ± 10 | |
| 25 μM | 140 ± 6 | |
| 50 μM | 120 ± 9 | |
| Incubation | 28 ± 1 | |
| (Control) | (170 ± 20) | |
Inhibition of xanthine oxidase by luteolin
As shown in Table 1, among the natural products examined here luteolin had the greatest effect on kcat/Km, with a Ki of 1.9 ± 0.7 μM. At 10 μM, the inhibitory effect of luteolin when presented to XO with xanthine resulted in a nearly 20-fold reduction in turnover of the free enzyme as assessed by kcat/Km. There was further inhibition of turnover with the free enzyme at luteolin concentrations of 25 and 50 μM culminating in a 170-fold reduction in kcat/Km; observed rate constants were reduced up to sixfold compared to control at these three concentrations. The double-reciprocal plot of [XO]/Vobs vs. 1/[xanthine] showed a series of linear relationships at each [luteolin], that intersected near the origin at x = 0 as shown in Figure 4. The secondary plot of the slopes vs. [luteolin] resulted in a linear relationship and the value for Ki above. Any additional effect following incubation of the enzyme with luteolin prior to the introduction of xanthine was negligible compared to that seen when the enzyme was exposed to xanthine and luteolin simultaneously.
Figure 4.

Double-reciprocal plots of [XO]/Vobs versus 1/[xanthine] for each concentration of luteolin (panel A) and quercetin (panel B). Also shown is the resulting plot of slopes versus 1/[inhibitor] used to determine the Ki for each respective compound. The intersection of the linear fits at x = 0 for these two inhibitors indicates that the mode of inhibition is competitive. Panel C depicts the analogous double-reciprocal plot for silibinin, with linear fits that are not consistent with competitive inhibition of the enzyme.
At 100 μM xanthine, 5 nM XO, and 50 μM luteolin, significant inhibition was observed with a linear increase in A295 over 10 min (Figure 5). Pre-incubation of the enzyme with luteolin did not result in any additional effect of the inhibition, a very similar case to that with quercetin discussed below. Also, the extent of inhibition of superoxide generation was proportionate to inhibition of xanthine turnover (Table 2).
Inhibition of xanthine oxidase by quercetin
Inhibition of XO by quercetin was next examined. The rate for overall turnover is reduced by approximately 60% at a concentration of 10 μM quercetin, and this value decreases further to less than 20% of the uninhibited kcat at 50 μM. The effects on Km are also pronounced, with a 100-fold increase in the presence of 50 μM quercetin and a >400-fold reduction in kcat/Km from a value of 8.78 to 0.04. The Ki for competitive inhibition by quercetin was determined to be 1.2 ± 0.7 μM. As shown in Figure 4, the double-reciprocal plot for quercetin again resulted in linear fits that intersected near the origin at x = 0, and the secondary plot of these slopes vs. [quercetin] crossed at a value for y = 0 to give the value above for Ki. Pre-incubation of XO with 25 μM quercetin did not result in any additional decrease in turnover rate, although the Km for xanthine was slightly more than twice that seen without pre-incubation. The production of superoxide by XO with xanthine as substrate was reduced at 25 μM and 50 μM quercetin, with resulting decreases analogous to those with luteolin and most significant at 50 μM quercetin. As for the case of luteolin, the inhibition of superoxide production showed the same inhibitor concentration dependence as for the turnover of xanthine.
Inhibition of xanthine oxidase by silibinin
Inhibition of XO by silibinin in the steady-state appeared to be independent of the silibinin concentration. At silibinin concentrations of 10, 25, and 50 μM, the kcat for xanthine turnover is reduced by approximately 50 % in each case, and the value of kcat/Km is reduced 50-fold. Pre-incubation of the enzyme with 25 μM silibinin resulted in no additional decrease in kcat, but rather a significantly increased Km, and the value for kcat/Km was further decreased from 5.6 to 0.07, reflecting an 80-fold reduction when compared to the control. No value for Ki could be obtained for inhibition by silibinin, as the linear fits of double-reciprocal plots were nearly identical for the three silibinin concentrations, and thus a secondary plot of the slopes vs. [silibinin] did not cross the x-axis at a meaningful point.
In the presence of 50 μM silibinin, the production of superoxide by XO was decreased by approximately 20 % relative to the control at 100 μM xanthine. The reaction of silibinin alone with XO under anaerobic conditions did not yield any apparent enzyme reduction, indicating that silibinin does not reduce the enzyme. The reduction of cytochrome c by XO was also not appreciably affected by silibinin.
The effect of curcumin on xanthine oxidase
Curcumin did not demonstrate any appreciable inhibition of purified bovine xanthine oxidase at 10 μM, 25 μM, 50 μM, or 100 μM in our steady-state assays. Neither did pre-incubation of XO with 25 μM curcumin for 10 minutes result in a significant change in either kcat or Km. Superoxide production was not significantly altered by any concentration of curcumin, either initially or following pre-incubation. These results contrast with other reports, principally involving in vivo studies, which suggest that curcurmin inhibits xanthine oxidase in this concentration range.
The present in vitro study was undertaken given the conflicting literature reports concerning the inhibition of XO by the compounds investigated. The issue is particularly important given the desirability of developing novel clinical inhibitors of XO due to the adverse events sometimes associated with acute and chronic administration of allopurinol. We find that xanthine oxidase is inhibited by luteolin, quercetin, silibinin, but not curcumin. The mode of inhibition by luteolin and quercetin was competitive, while that by silibinin was a mixed type of inhibition. Inhibition by luteolin and quercetin is not surprising given the resemblance to the known substrate lumazine, a pteridine (Figure 3) known to be hydroxylated to violapterin.43 On the other hand, it is perhaps surprising that the bulkier silibinin is indeed an inhibitor. This natural compound does possess a benzopyran ring, however, which resembles the analogous moieties of quercetin and luteolin.
Figure 3.

Natural compounds in the current study, previously reported to be inhibitors of xanthine oxidase. Also shown are allopurinol, xanthine, and lumazine.
With regard to superoxide production by XO, three of the four compounds tested appear to decrease the initial production of superoxide by the reaction of xanthine oxidase with xanthine, as followed by a ΔA550 associated with the reduction of cytochrome c. A modest decline in superoxide is only apparent at the highest concentration of silibinin, and the significant declines in the cases of luteolin and quercetin are nearly proportionate to the inhibition of urate production at each concentration (see Tables 1 and 2). This observation, that the decreases in superoxide production are proportional to the decreased turnover of xanthine, strongly suggests that each inhibitor here acts at the molybdenum-containing active site of XOR to competitively inhibit the reduction of the enzyme by xanthine, rather than acting directly at the FAD-containing flavin site that interacts with O2. This results in a slower timecourse of superoxide production at a given xanthine concentration. Thus any decrease in the reduction of cytochrome c via the scavenging of superoxide radicals by the inhibitor itself (acting as an antioxidant), or by direct interaction (i.e. interposition) at the flavin site of XO to inhibit final electron transfer to O2, is unlikely here.
Reports from various in vivo studies have suggested that luteolin, quercetin, or silibinin may approach the effectivenesss of allopurinol as inhibitors of xanthine oxidase.24-31 The present results demonstrate that these three compounds are each inhibitors in vitro, although there is no time-dependent increase in inhibition analogous to that with allopurinol and the effective Ki's determined here do not approach those for allopurinol. Given the unknown pharmacokinetics of these natural products, the effective dose could therefore be considerably higher than for either allopurinol or the more recently approved febuxostat.
In the case of the non-flavonoid curcumin, the lack of inhibition is at variance with previous reports that did not utilize purified enzyme.31 We note that curcumin is not planar like xanthine, lumazine, or other known inhibitors of XO, and does not otherwise structurally resemble any of these compounds. Given that the solvent access channel to the deeply buried active site of XO narrows to <10 Å approximately half-way in, the central sp3 hybridized carbon of curcumin (Fig. 3) it is likely that curcumin is unable to access the substrate binding site. Similarly, the physical shape of curcumin, with a bend of approximately 110°, makes it unlikely that it simply occludes the solvent access channel as seen with febuxostat. Finally, we note that curcumin lacks a reactive carbon center that would constitute a possible site of hydroxylation by XO. Thus with the lack of a plausible mechanistic or structural basis for inhibition, our results showing a lack of XO inhibition by curcumin are not surprising. Previous reports demonstrating inhibition of xanthine oxidoreductase by curcumin using other methods may indicate that suppression of superoxide production or the production of uric acid in these experiments is an indirect consequence of curcumin's presence. Our results clearly indicate that curcumin does not directly inhibit the activity of bovine xanthine oxidase, and thus further investigations are warranted.
Our work here demonstrates that luteolin, silibinin, and quercetin inhibit xanthine oxidase, and suggests that these compounds, or derivatives of them, may be useful leads in the development of clinically useful inhibitors of XO.24-30 The nature of this inhibition, particularly the somewhat stronger effect of quercetin and luteolin relative to that of silibinin, is interesting and merits further characterization. It seems likely that luteolin, silibinin, and quercetin position in the active site of XO with the dihydroxybenzene functionality of their benzopyran moiety directed toward the molybdenum cofactor, their benzopyran intercalated between Phe914 and Phe1009, their C1 carbonyl groups directed toward Arg880, and their additional moiety projecting backward into the solvent channel. Given our recent success at obtaining crystal structures of bovine XO with substrate species bound in the enzyme's active site, it should be possible to obtain structures of XO with luteolin, quercetin, and possibly silibinin.44 This would allow direct observation of each compound's interaction(s) with active site residues, and thus allow further insight not only into the role that these residues may be playing in normal catalysis, but also into possibly new means of inhibiting the enzyme.
Experimental Section
Xenobiotics and enzyme
Silibinin, quercetin, luteolin, curcumin, and xanthine were purchased from Aldrich (Sigma-Aldrich, St. Louis, MO). All were at >95% purity and were used without further purification. Stock solutions of luteolin, quercetin, silibinin, and curcumin were prepared at a concentration of 5 mM in 0.1-0.2 M aqueous KOH, as were 33.3 mM and 1 mM xanthine stock solutions. All other reagents were purchased either from Aldrich or Fisher (Thermo Fisher Scientific, Waltham, MA) and were of the highest purity commercially available.
Xanthine oxidase was isolated and purified from bovine milk according to the previously reported method of Massey and coworkers.40,41 The enzyme as isolated was routinely approximately 70% active based on the activity-to-flavin ratio, consistent with previous literature, the nonfunctional enzyme lacking a catalytically essential Mo=S ligand in the active site.40 The enzyme was stored in buffer containing 1 mM salicylate at −80°C, and passed down a Sephadex G-25 column to remove the salicylate prior to use.
Steady-state kinetics and absorption spectra
Inhibition of XO was determined as follows. Each assay contained 50 nM XO, with xanthine concentrations ranging from 1 μM to 800 μM in the presence or absence of a known concentration of each natural compound in question, or in the presence of allopurinol as a standard reference. Each of the four compounds in question was assayed at 10 μM, 25 μM, 50 μM, and 100 μM over this concentration range of xanthine, monitoring the ΔA295 associated with the generation of uric acid (ε295 = 9600 M−1cm−1).41 The kinetic constants kcat and Km were determined from plots of the initial reaction velocity Vobs versus [xanthine] for each concentration of inhibitor, and values for Ki determined from secondary plots of the slope of the primary double reciprocal plot versus [inhibitor] using regression data generated from double-reciprocal plots of [enzyme]/Vobs versus 1/[xanthine] at 10, 25, and 50 μM concentrations of inhibitor. All data analysis was performed on SigmaPlot 10 (Systat Inc., San Jose, CA).
In addition to the above, assays were conducted in which the enzyme was first pre-incubated for 10 min with 25 μM of each potential inhibitor prior to the addition of xanthine to initiate the assay. Controls were run in the absence of any inhibitor in the reaction mix, both before and after testing each respective compound in the assays above.
Superoxide production was monitored indirectly by monitoring the non-enzymatic reduction of cytochrome c by O2•−. The capacity for each compound to influence superoxide production was determined by including 75 μM oxidized cytochrome c to the reaction mix, monitoring the reaction at 550 nM (Δε550 = 19.6 mM−1cm−1).45 Stock solutions of cytochrome c were prepared by incubating concentrated solutions for 10 minutes with 2-3 mM ferricyanide, followed by passage through a Sephadex G-25 column; final concentrations were determined from serial dilutions of the stock, using ε410 nm = 106 mM−1cm−1.46 Assays were conducted in the presence of 10 μM, 25 μM, and 50 μM of each compound tested with 50 nM XO and 100 μM xanthine.
To ascertain whether each compound could be hydroxylated by enzyme, 2 μM XO was titrated under anaerobic conditions with 25 μM of each compound. Reduction of the enzyme, as reflected in a bleaching throughout the visible region, was monitored over 10 min and quantified at 450 nm as described previously using Δε450 = 26.6 mM−1cm−1.40
Each reaction was run at 25°C in 0.1 M MOPS, 0.2 mM EDTA, with 0.1 M KCl (for ionic strength) at a pH of 7.4, with a final reaction volume of 1 mL (3 ml in the case of anaerobic titrations). All assays were conducted using a Hewlett-Packard 8452A diode array spectrophotometer interfaced with the Hewlett-Packard Chemstation (Palo Alto, CA).
Acknowledgements
The authors would like to thank Dr. M. Dunn for the use of his laboratory facilities at UCR. This work was supported by NIH grant GM 075036.
References and Notes
- 1.Hille R. Eur. J. Inorg. Chem. 2006;2006:1913–1926. [Google Scholar]
- 2.Hille R, Nishino T. FASEB J. 1995;9:995–1003. [PubMed] [Google Scholar]
- 3.Nishino T, Okamoto K, Kawaguchi Y, Hori H, Matsumura T, Eger BT, Pai EF, Nishino T. J. Biol. Chem. 2005;280:24888–24894. doi: 10.1074/jbc.M501830200. [DOI] [PubMed] [Google Scholar]
- 4.Enroth C, Eger BT, Okamoto K, Nishino T, Nishino T, Pai EF. Proc. Natl. Acad. Sci. U. S. A. 2000;97:10723–10728. doi: 10.1073/pnas.97.20.10723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Huber R, Hof P, Duarte RO, Moura JJG, Moura I, LeGall J, Hille R, Archer M, Roma∼o M. Proc. Natl. Acad. Sci. U. S. A. 1996;93:8846–8851. doi: 10.1073/pnas.93.17.8846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Harris CM, Massey V. J. Biol. Chem. 1997;272:8270–8379. [Google Scholar]
- 7.McCord JM. N. Engl. J. Med. 1985;312:159–163. doi: 10.1056/NEJM198501173120305. [DOI] [PubMed] [Google Scholar]
- 8.Millar TM. FEBS Letters. 2004;562:129–133. doi: 10.1016/S0014-5793(04)00218-2. [DOI] [PubMed] [Google Scholar]
- 9.Li H, Cui H, Liu X, Zweier JL. J. Biol. Chem. 2005;280:16594–16600. doi: 10.1074/jbc.M411905200. [DOI] [PubMed] [Google Scholar]
- 10.Bonini MG, Miyamoto S, Mascio PD, Augusto O. J. Biol. Chem. 2004;279:51836–51843. doi: 10.1074/jbc.M406929200. [DOI] [PubMed] [Google Scholar]
- 11.Pacher P, Nivorozhkin A, Szabo C. Pharmacol. Rev. 2006;58:87–114. doi: 10.1124/pr.58.1.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Berry CE, Hare JM. J. Physiol. 2004;555.3:589–606. doi: 10.1113/jphysiol.2003.055913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Martin HM, Hancock JT, Salisbury V, Harrison R. Infect. Immun. 2004;72:4933–4939. doi: 10.1128/IAI.72.9.4933-4939.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Falasca GF. Clinics in Dermatology. 2006;24:498–508. doi: 10.1016/j.clindermatol.2006.07.015. [DOI] [PubMed] [Google Scholar]
- 15.Smalley RV, Guaspari A, Haase-Statz S, Anderson SA, Cederberg D, Hohneker JA. J. Clin. Oncol. 2000;18:1758–1763. doi: 10.1200/JCO.2000.18.8.1758. [DOI] [PubMed] [Google Scholar]
- 16.Yellon DM, Hausenloy DJ. N. Engl. J. Med. 2007;357:1121–35. doi: 10.1056/NEJMra071667. [DOI] [PubMed] [Google Scholar]
- 17.Li C, Jackson RM. Am. J. Physiol. Cell. Physiol. 2002;282:227–241. doi: 10.1152/ajpcell.00112.2001. [DOI] [PubMed] [Google Scholar]
- 18.Terkeltaub RA. N. Engl. J. Med. 2003;349:1647–1655. doi: 10.1056/NEJMcp030733. [DOI] [PubMed] [Google Scholar]
- 19.Okamoto K, Eger BT, Nishino T, Kondo S, Pai EF, Nishino T. J. Biol. Chem. 2003;278:1848–1855. doi: 10.1074/jbc.M208307200. [DOI] [PubMed] [Google Scholar]
- 20.Massey V, Komai H, Palmer G, Elion GB. J. Biol. Chem. 1970;245:2837–2844. [PubMed] [Google Scholar]
- 21.Khosravan R, Grabowski BA, Wu JT, Joseph-Ridge N, Vernillet L. Clin. Pharmacokinet. 2006;45:821–841. doi: 10.2165/00003088-200645080-00005. [DOI] [PubMed] [Google Scholar]
- 22.Becker MA, Schumacher HR, Jr., Wortmann RL, MacDonald PA, Eustace D, Palo WA, Streit J, Joseph-Ridge N. N. Engl. J. Med. 2005;353:2450–2461. doi: 10.1056/NEJMoa050373. [DOI] [PubMed] [Google Scholar]
- 23.Schumacher HR., Jr Expert. Opin. Investig. Drugs. 2005;14:893–903. doi: 10.1517/13543784.14.7.893. [DOI] [PubMed] [Google Scholar]
- 24.Mo SF, Zhou F, Lv YZ, Hu QH, Zhang DM, Kong LD. Biol. Pharm. Bull. 2007;30:1551–1556. doi: 10.1248/bpb.30.1551. [DOI] [PubMed] [Google Scholar]
- 25.Wu JW, Tsai TH. Drug Metabolism and Disposition. 2007;35:1603–1610. doi: 10.1124/dmd.107.014894. [DOI] [PubMed] [Google Scholar]
- 26.Yu Z, Fong WP, Cheng CHK. J. Pharm. Exp. Therap. 2006;316:169–175. doi: 10.1124/jpet.105.092684. [DOI] [PubMed] [Google Scholar]
- 27.Cos P, Ying L, Calomme M, Hu JP, Cimanga K, Poel BV, Pieters L, Vlietinck AJ, Berghe DV. J. Nat. Prod. 1998;61:71–76. doi: 10.1021/np970237h. [DOI] [PubMed] [Google Scholar]
- 28.Knekt P, Kumpulainen J, Järvinen R, Rissanen H, Heliövaara M, Reunanen A, Hakulinen T, Aromaa A. Am. J. Clin. Nutr. 2002;76:560–568. doi: 10.1093/ajcn/76.3.560. [DOI] [PubMed] [Google Scholar]
- 29.Nijveldt RJ, van Nood E, van Hoorn DEC, Boelens PG, van Norren K, van Leeuwen PAM. Am. J. Clin. Nutr. 2001;74:418–425. doi: 10.1093/ajcn/74.4.418. [DOI] [PubMed] [Google Scholar]
- 30.Chun OK, Chung SJ, Song WO. J. Nutrition. 2007;137:1244–1252. doi: 10.1093/jn/137.5.1244. [DOI] [PubMed] [Google Scholar]
- 31.Lin JK, Shih CA. Carcinogenesis. 1994;15:1717–1721. doi: 10.1093/carcin/15.8.1717. [DOI] [PubMed] [Google Scholar]
- 32.Benboubetra M, Baghiani A, Atmani D, Harrison R. J. Dairy Sci. 2004;87:1580–1584. doi: 10.3168/jds.S0022-0302(04)73311-1. [DOI] [PubMed] [Google Scholar]
- 33.Wright RM, Vaitaitis GM, Wilson CM, Repine TB, Terada LS, Repine JE. Proc. Natl. Acad. Sci. 1993;90:10690–10694. doi: 10.1073/pnas.90.22.10690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yamaguchi Y, Matsumura T, Ichida K, Okamoto K, Nishino T. J. Biochem. 2007;141:513–524. doi: 10.1093/jb/mvm053. [DOI] [PubMed] [Google Scholar]
- 35.Xiang Q, Edmondson DE. Biochemistry. 1996;35:5441–5450. doi: 10.1021/bi952880d. [DOI] [PubMed] [Google Scholar]
- 36.Leimkühler S, Kern M, Solomon PS, McEwan AG, Schwarz G, Mendel RR, Klipp W. Molecular Microbiology. 1998;27:853–869. doi: 10.1046/j.1365-2958.1998.00733.x. [DOI] [PubMed] [Google Scholar]
- 37.Truglio JJ, Theis K, Leimkühler S, Rappa R, Rajagopalan KV, Kisker C. Structure. 2002;10:115–125. doi: 10.1016/s0969-2126(01)00697-9. [DOI] [PubMed] [Google Scholar]
- 38.Pauff JM, Hemann CF, Jünemann N, Leimkühler S, Hille R. J. Biol. Chem. 2007;282:12785–12790. doi: 10.1074/jbc.M700364200. [DOI] [PubMed] [Google Scholar]
- 39.Ryan MG, Ratnam K, Hille R. J. Biol. Chem. 1995;270:19209–19212. doi: 10.1074/jbc.270.33.19209. [DOI] [PubMed] [Google Scholar]
- 40.Kim JH, Hille R. J. Biol. Chem. 1993;268:44–51. [PubMed] [Google Scholar]
- 41.Massey V, Brumby PE, Komai H, Palmer G. J. Biol. Chem. 1969;244:1682–1691. [PubMed] [Google Scholar]
- 42.Williams JW, Bray RC. Biochem. J. 1981;195:753–760. doi: 10.1042/bj1950753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hemann C, Ilich P, Stockert AL, Choi EY, Hille R. J. Phys. Chem. B. 2005;109:3023–3031. doi: 10.1021/jp046636k. [DOI] [PubMed] [Google Scholar]
- 44.Pauff JM, Zhang J, Bell CE, Hille R. J. Biol. Chem. 2008;283:4818–4824. doi: 10.1074/jbc.M707918200. [DOI] [PubMed] [Google Scholar]
- 45.Bonagura CA, Bhaskar B, Sundaramoorthy M, Poulos TL. J. Biol. Chem. 1999;274:37827–37833. doi: 10.1074/jbc.274.53.37827. [DOI] [PubMed] [Google Scholar]
- 46.Guo M, Bhaskar B, Li H, Barrows TP, Poulos TL. Proc. Natl. Acad. Sci. U.S.A. 2004;101:5940–5945. doi: 10.1073/pnas.0306708101. [DOI] [PMC free article] [PubMed] [Google Scholar]