

Abstract
When investigators hypothesize that experimental interventions provide advantages other than improved effectiveness, they can use noninferiority (NI) trials to determine whether one can rule out the possibility that those interventions have unacceptably worse effectiveness than the standard “active control” regimen. To conduct valid NI trials, there must be evidence from historical studies that provides reliable, reproducible, and precise estimates of the effect of the active control, compared with that of placebo, on the specific outcomes investigators plan to use in the NI trial; this effect should be of substantial magnitude, and the estimates of the active control’s effect from historical studies must represent its effect in the planned NI trial had a placebo group been included. These conditions allow formulation of an NI margin such that, if the NI trial establishes that the effectiveness of the experimental intervention is not worse than the effectiveness of the active control by more than the NI margin, then one can conclude that the experimental regimen (1) preserves a substantial fraction of the effect of the active control and (2) will not result in a clinically meaningful loss of effectiveness. After general discussion of NI trial design issues, we consider the design of NI trials to evaluate antimicrobials in the treatment of community-acquired pneumonia. We present an extensive literature review, allowing estimation of the historical effect of active control regimens in community-acquired pneumonia primarily on the basis of evidence related to use of sulfonamides or penicillin. This review allows formulation of NI margins that are specific to age and bacteremia status of patients.
When effective interventions are available to treat or prevent a disease, there are instances in which investigators are interested in evaluating new experimental agents because they hypothesize that these agents provide advantages other than enhancing effectiveness. For instance, in the treatment of invasive aspergillosis, voriconazole may provide a better adverse-event profile than does amphotericin B deoxycholate. In community-acquired pneumonia (CAP), a new quinolone could allow improved convenience of administration, compared with that of penicillin. With regard to mother-to-child transmission of HIV, although intensive and expensive interventions can reduce mother-to-child transmission, these interventions are impractical or unaffordable in developing countries, where we need them most. If the experimental interventions, in fact, are not unacceptably worse in effectiveness relative to the standard therapy, then their improved safety, cost, or convenience would make them attractive alternative options for patients.
Randomized clinical trials comparing an experimental regimen and an active control regimen to determine whether the effectiveness of the experimental regimen is not unacceptably worse than that of the active control are called “noninferiority” (NI) trials. The present article provides insights into the criteria that investigators need to address when designing, conducting, and analyzing NI trials. We then put these criteria into practice by presenting the evidence needed to perform valid NI trials when evaluating antimicrobials for the treatment of CAP.
GENERAL PRINCIPLES IN DESIGNING NI TRIALS
NI trials inherently contain 2 questions regarding relative effectiveness: an explicit question relative to the active control and an implicit question relative to placebo [1]. For example, an NI trial of a new quinolone compared with an active control in CAP has the obvious appeal that it allows us to make a head-to-head comparison of the effectiveness of the new quinolone relative to that of an active control intervention (e.g., penicillin). However, we also want to learn from this study whether the new quinolone actually is effective at all relative to placebo. Because most NI trials do not include a group that receives a placebo, there is no “negative” control group. Therefore, one cannot directly differentiate whether similarity between the test and control interventions means that the two are similarly effective or similarly ineffective, because of the conditions of the experiment. We must gain insight regarding the effectiveness of the new intervention compared with no treatment indirectly, by comparing the new quinolone with penicillin in the NI trial and by examining evidence from similarly designed historical studies that provide reliable evidence about the effect of penicillin relative to placebo. Importantly, because these data on penicillin compared with placebo or no treatment come from trials conducted in the past, it is often unclear whether the historical estimates of effectiveness for penicillin apply under the conditions of the current study. Hence, NI trials share some of the inherent biases arising in historically controlled trials [2]. In addition to these design-related biases, the NI trial is particularly susceptible to operational bias due to irregularities in the quality of trial conduct, such as nonadherence to regimens, cross-ins, missing data, violations in entry criteria, or inconsistencies in assessing outcome measures. These irregularities tend to reduce sensitivity to true differences between study interventions, resulting in a bias toward demonstrating similarity of the 2 interventions [1]. Such bias is particularly problematic in NI trials, because it can lead to falsely declaring NI for an experimental regimen that truly is clinically meaningfully less effective or not effective at all. In summary, clinicians’ judgments that point estimates between an experimental intervention and an active control are “close enough” to each other do not provide de facto evidence that the experimental intervention is effective in comparison with placebo, let alone similar in its effectiveness to the active control intervention.
According to the International Conference of Harmonization guidelines [3, 4], the internationally accepted standards for clinical trials, for investigators to use any regimen, such as penicillin, as an active control in an NI trial in CAP, the estimate of its benefit should be (1) substantial in magnitude, compared with placebo or no treatment; (2) precisely estimated—ideally, previously reproduced in several randomized trials; and (3) relevant to the setting of the planned NI trial in which the active control is compared against the new agent, in this case a quinolone [4]. Thus, to justify the use of an active control in an NI trial, investigators must address the questions about the magnitude of effectiveness of that active control relative to placebo or no treatment, in what population and stage of disease studies have demonstrated this benefit, and how and when the outcomes were measured [2]. If the effectiveness of the active control is at all in question, or if one holds that historical data are not relevant to the setting of current trials, investigators either should use another control that satisfies these criteria or should choose a design other than NI, because NI trials are entirely based on having relevant historical evidence that establishes substantial effect of the control drug. These principles are reinforced by a recent US Food and Drug Administration guidance on the use of NI trials to support approvals of antibacterial drugs. That guidance pointed out the requirement in regulations for drug sponsors to supply the data that support a proposed NI margin [5].
The presumption that the historical estimates of effectiveness of the active comparator apply under the conditions of the NI trial is called the “constancy assumption.” The inability to find data to address the constancy of the effect of the control is one of the principal reasons it is not possible to conduct valid NI trials in many settings [1]. Why is the constancy assumption so critical? Suppose, for example, a new experimental intervention is compared against vancomycin, and the randomized NI trial appears to show similar results for the two agents. In comparison with vancomycin, is the new intervention “similarly effective” or “similarly ineffective”? It is tempting to conclude that the interventions are “similarly effective” because historical studies demonstrated the effectiveness of vancomycin when it was originally compared with no treatment. However, suppose investigators conduct the NI comparison in subjects with disease caused by vancomycin-resistant enterococci. In this setting, vancomycin might provide little, if any, benefit. If so, then the proper conclusion would be that the experimental agent and vancomycin are similarly ineffective.
The idea of maintaining the conditions of the experiment is familiar to laboratory investigators. If investigators change the conditions of laboratory experiments, in terms of the reagents used, the amount of those reagents, the incubation time for the experiment, or the way in which the results are measured, it is not clear whether the results of the experiment are valid or whether they are merely related to the conditions of the experiment. For this reason, laboratory investigators maintain the conditions of the experiment and use positive and negative controls whose behavior is known under the constant conditions of the experiment. If the controls do not perform as expected, then there is a problem with the experiment, and the results are not valid. In NI trials, a negative control is usually absent (e.g., a group receiving placebo or a less effective intervention), making it more challenging to assess whether the conditions of the experiment affect the results. Therefore, in designing NI trials, investigators must be rigorous in maintaining the conditions of the experiment by evaluating the details of the previous studies that demonstrated the effectiveness of the active control. In this regard, NI trials limit innovation, because investigators cannot make substantial changes to the study design by changing the definition of the disease, studying new populations, evaluating new end points or new timing of end points, or including new concomitant medications. In contrast, investigators can evaluate all of these factors in superiority trials.
If historical studies show that the active control regimen—for example, penicillin—is reliably and reproducibly effective in comparison with placebo or no treatment, what are some of the factors that could explain why the effect of penicillin might be different in a current NI trial? First, the investigators may conduct the NI trial in subjects with disease that is less responsive to penicillin—for instance, subjects without the disease under study (e.g., viral pneumonia) or subjects whose disease is caused by bacterial pathogens resistant to penicillin. Second, subjects in the NI trial conducted in modern times may have access to enhanced levels of supportive care and different concomitant interventions that attenuate the effect of penicillin. Third, in the NI trial there may be lower adherence to penicillin. Fourth, the end points of the current NI trial could be different or could be assessed in a different manner than in the historical context. If the effect of the active control therapy in the setting of a current NI trial truly is smaller than the effect demonstrated in the historical context, then an ineffective experimental intervention may look similar to the active control therapy, and a false conclusion of benefit would result, because investigators falsely assumed the constancy assumption was fulfilled.
It would be useful to apply the concepts of appropriate NI trials to the design and conduct of NI trials for evaluating treatments for CAP. In a trial comparing the effect of the experimental intervention with that of the active control, one attempts to rule out that the effectiveness of the experimental regimen for the treatment of CAP is worse than that of the active control by more than a chosen margin. However, the devil is in the details. How does one choose a valid margin, which will depend not only on having evidence that the active control antimicrobial “works” in pneumonia but also on the magnitude of its effect?
For illustration, consider the setting of pneumococcal pneumonia and suppose the active control intervention is penicillin and the experimental intervention is a quinolone. Suppose the primary end point of the clinical trial is “failure.” In serious disease in older subjects, investigators might define failure as death within 14 days of enrollment in the study.
Suppose investigators randomized 300 subjects in a 1:1 manner between receiving the new quinolone and penicillin, and the failure rates were 25% and 20%, respectively. The point estimate and associated variability for the estimated difference in failure rates (new quinolone minus penicillin) are 5% ± 10% (figure 1).
Figure 1.
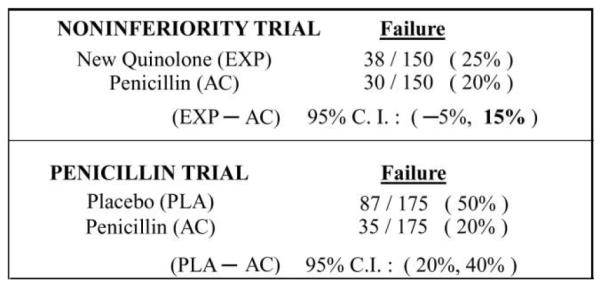
Illustration of a current community-acquired pneumonia noninferiority trial comparing failure rates for a new quinolone (experimental agent [EXP]) versus penicillin (active control [AC]) and a historical trial evaluating the AC, penicillin. PLA, placebo.
In figure 2, we plot the difference in the probability of failure for any regimen relative to that of the active control, penicillin. A regimen with a lower failure rate than penicillin would lie to the left of 0%, whereas one with a higher failure rate than penicillin would lie to the right of 0%. Given the data in figure 1, the new quinolone lies to the right of 0%, at +5%, with a 95% CI of −5% to 15%. The principal question in an NI trial relates to whether that upper limit of the increase in rate of failure of 15% is sufficiently low that it can be concluded that this new quinolone is more effective than placebo, had a placebo group been included in the trial, and that the quinolone preserves a clinically meaningful amount of the benefit of penicillin compared with placebo. In essence, investigators need to address 2 issues. First, how does one choose an “NI margin” before the trial that addresses effectiveness relative to active control and to placebo? Second, does 15% lie below that “NI margin”?
Figure 2.
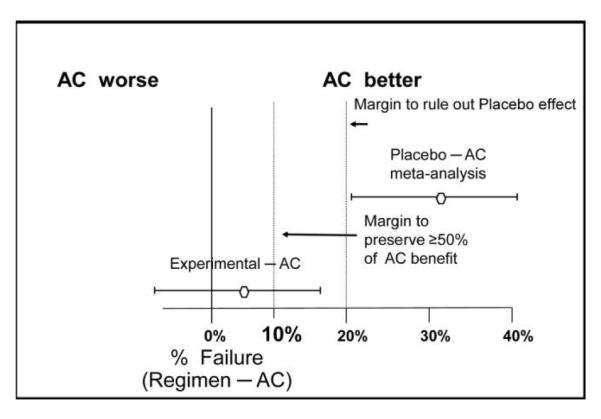
Illustration of the formulation of a noninferiority (NI) margin, using data from figure 1. A plot of the failure probability of any regimen compared with the active control (AC) is provided.
An initial step in determining the NI margin is obtaining evidence about the difference in failure rates with the placebo and active control interventions, essentially allowing estimation of where the placebo would be in figure 2. Optimally, this evidence regarding the effectiveness of the active control, penicillin, would come from previous randomized placebo-controlled trials, because such trials are less prone to random error and systematic biases than are other types of studies. Suppose historical trials involving 350 subjects show that the failure rates with placebo and penicillin are 50% and 20%, respectively (figure 1); then, we estimate that the placebo has a 30% higher failure rate than penicillin, with a 95% CI for the difference in failure rates (placebo minus penicillin) of 20%–40%. These data allow one to place the point estimate for the difference between active control and placebo at +30 on the graph in figure 2. However, this estimate has some associated random error. Also, one should take into account the potential uncertainty regarding the validity of the constancy assumption. For this reason, the “95-95” approach to defining the NI margin uses the lower limit of the (placebo minus penicillin) 95% CI, in this case 20%, to provide adequate confidence that the experimental regimen is more effective than placebo [1]. Because the upper limit of the 95% CI for the difference in failure rates in the NI trial between the new quinolone and penicillin lies below 20%, one has evidence that the experimental regimen is more effective than placebo.
However, why is it clinically acceptable for an experimental regimen to simply be better than placebo when an available active control regimen, penicillin, provides substantial beneficial effects in reducing the failure rate, especially when failure is mortality or a major morbidity outcome measure? The importance to patients of the effect of the active control regimen is the primary reason for performing an NI trial instead of a placebo-controlled trial. Given this basic consideration, it is logical to require that the effect of the experimental intervention should preserve a substantial fraction of the effect of active control therapy. If this fraction is, for example, one-half, then the NI margin in figure 2 would be 10%. Hence, in our example, the upper limit of the 95% CI for the difference in failure rates between the new quinolone and penicillin would need to lie below 10% for us to reasonably conclude that the new quinolone preserves at least half the effect of penicillin. When absolute benefits of the active control therapy are large, as for some antimicrobials in serious disease compared with no specific treatment, preserving a fraction larger than one-half of the benefit of the active control compared with placebo would be clinically important.
Choosing the NI margin to ensure preservation of a substantial fraction of the benefit of the experimental agent compared with placebo is an evidence-based step in margin formulation. This step is not based on judgment or a consensus of experts in the absence of data. The additional second step of justifying that the chosen NI margin is clinically acceptable does involve clinical judgment. Investigators should take into account the clinical relevance of potentially losing some of the benefit of the active control regimen. Investigators should base these considerations on the public health impact of the potential loss of effectiveness, rather than basing considerations only on sample size. For instance, if the experimental intervention provides safety advantages, clinicians and patients might be willing to accept a greater loss of effectiveness for that intervention in a self-resolving disease than in a serious and life-threatening disease in which the consequence of decreased effectiveness is death. The margin should be sufficiently small that one can rule out that the effect of the experimental regimen is clinically unacceptably worse than that of the active control. Hence, to justify 10% as an appropriate NI margin in figure 2, one should ensure that clinicians and patients would find a 10% increase in failure rate acceptable, given the reduction in toxicity, inconvenience, or cost provided by the experimental regimen. To justify the use of a larger NI margin and, hence, a smaller sample size, there is a strong temptation for investigators to pose a large loss of effectiveness as being clinically acceptable. However, to accept such an assumption, one must be able to conclude that a regimen that provides some increase in toxicity, cost, or inconvenience but achieves a 10% reduction in probability of death would not be viewed to be an important advance in clinical care. If an intervention that is 10% more effective is considered to be a clinical advance, it is difficult to justify that an intervention that may be 10% less effective than the active control is clinically acceptable.
It is important to understand that NI trials do not establish that the experimental agent is “not worse than” or “at least as effective as” the active control regimen unless the study demonstrates superiority of the experimental agent relative to the control. Indeed, an experimental intervention may be inferior to the active control and still achieve NI by ruling out the NI margin [1]. To illustrate, in figure 1, suppose that the failure rate for the new quinolone and penicillin remain 25% and 20%, respectively, but that the NI trial has a larger sample size, such that the 95% CI for the difference in failure rates is 2%–8%. Then, in figure 2, it would follow that the new quinolone is inferior to penicillin, because the 95% CI excludes 0%, and yet the results establish NI because the 95% CI also excludes 10%. This seemingly paradoxical phenomenon reflects issues of nomenclature in NI trials [2]. Despite the implications of the name “noninferiority,” achieving NI does not mean that the new quinolone is not inferior to penicillin; rather, it means it is “not unacceptably worse than” penicillin. This emphasizes why it is so important to ensure that the NI margin is sufficiently small to exclude any loss of effectiveness that patients and clinicians would consider clinically important.
In summary, to formulate a valid NI margin, (1) there must be data from historical studies that provide reliable, reproducible, and precise estimates of the effect of the active control regimen on the specific efficacy end point to be used in the NI trial; (2) this effect needs to be of substantial magnitude; and (3) these estimates of the effect of the active control regimen from historical studies must represent the effect of this control regimen in the planned NI trial (i.e., measured using similar patient populations, supportive care regimens, adherence to interventions, definitions of disease, and end points). Furthermore, the NI margin should be sufficiently small, such that establishing NI allows one to conclude that (1) the experimental regimen preserves a substantial fraction of the effect of the active control, and (2) the use of the experimental regimen rather than the active control regimen will not result in a clinically meaningful loss of effectiveness.
ESTIMATES OF EFFECTIVENESS FOR POTENTIAL ACTIVE CONTROL REGIMENS IN NI TRIALS IN CAP
Applying the principles outlined above, we examined the historical evidence of the effect of older antimicrobials, such as sulfonamides and penicillin, in the treatment of CAP. As noted, the goal of such an analysis was not only to determine whether active control antimicrobials are effective at all in CAP but to attempt to describe the magnitude of their effectiveness, the population in whom this effect is manifest, and the conditions of the previous studies that demonstrated this effect in terms of definitions and stage of disease, the populations, and the definitions and timing of outcomes.
Optimally, one would evaluate the historical evidence of effectiveness for a specific control drug, such as penicillin, and use that same drug as the control in future NI trials. However, current trials rarely use penicillin as the control intervention. Given the raising of US Food and Drug Administration—approved breakpoints for penicillin against Streptococcus pneumoniae in nonmeningeal disease, perhaps investigators should reassess the utility of penicillin as a control drug in certain situations, especially pneumococcal pneumonia. However, if one chooses to use a different drug, it must be justified that one can extrapolate the effects of the antimicrobials compared with nonspecific treatment in the early literature (sulfonamides and penicillin) to the active control drugs used in current NI trials. Several meta-analyses and systematic reviews are relevant to providing that justification [6–8]. To most closely approximate the effect of current active control drugs used in NI trials in CAP, such as β-lactams or quinolones, one should evaluate evidence from studies of medical interventions that have similar mechanisms of action—that is, interventions whose primary mode of action is inhibition of bacterial growth (e.g., sulfonamides and penicillins). The historical evidence from interventions with different mechanisms of action, such as the effects of serum therapy in the treatment of CAP, would be difficult to extrapolate to the effects of small-molecule antimicrobials. The mechanism of action of serum therapy is based on augmentation of the host immune response.
In evaluating the historical evidence, it is clear that the majority of patients had a microbiological diagnosis documented before initiation of treatment. Therefore, historical studies did not provide sensitivity analyses to evaluate the similarity of treatment effects in patients with and without a confirmed microbiological diagnosis. This is a point of difference between the historical data and recent NI trials, in which currently less than a third of subjects have documented microbiological diagnoses. The discussion below is predicated on the assumption that enrolled patients have a documented microbiological diagnosis. Failure to make an appropriate diagnosis may bias the study to make drugs appear more similar. In addition, the majority of the data from historical studies are in subjects infected with S. pneumoniae. One might reasonably extrapolate the data on pneumococcal pneumonia to other etiologies of pneumonia with similar pathogenesis, similar patient populations, and similar mortality among subjects who receive no specific therapy. However, these data cannot be extrapolated to pneumonias of different etiologies with different pathophysiologies and mortality, such as Chlamydophila, Mycoplasma, fungal, or viral pneumonias.
When evaluating treatment effects from historical data, it is important to take into account the quality of the previous evidence, because any bias in the historical evidence will affect the foundation on which future NI trials are built and potentially bias the results of the NI trials as well. Optimally, when seeking historical evidence regarding the effectiveness of the active control regimen, one should conduct a comprehensive systematic review of randomized, double-blinded, placebo-controlled superiority trials that have appropriate end points and analyses. However, in CAP, there are no randomized placebo-controlled trials of antimicrobials compared with supportive care alone. The process of randomization was not yet in wide-spread use at the time of the earliest studies of CAP. Randomization decreases selection bias, attempts to balance measured and unmeasured baseline characteristics of subjects, and also provides the statistical basis for hypothesis testing. Rather than randomized trials, there are 2 types of studies that provide evidence for evaluating the effectiveness of treatment for CAP. The first type is case series of patients receiving antimicrobials, compared with historical or concurrent groups of patients who received supportive care alone. The issues with historical controls are well known, in that there may be a lack of baseline comparability between subjects treated with the experimental intervention and those treated with supportive care [4]. Authors have noted that unfavorable supportive care provided in historical settings and selection biases of historical cases can lead to overestimating the treatment benefits of more recent active interventions when they are compared with historical controls [9]. International guidance notes that historical (external) controls can be useful in determining effectiveness when treatment effects are large and measured on objective end points, such as all-cause mortality [4]. US regulations also note that appropriately designed and conducted historically controlled trials are one of the types of adequate and well-controlled trials acceptable for regulatory review [10]. However, although historically controlled trials might provide adequate evidence about whether the experimental intervention is effective in comparison with no treatment in such settings, they may provide misleading information about the magnitude of effectiveness, given the biases with this type of trial design. Recent studies show that bias is more likely to affect results when the end points examined are subjective rather than objective [11]. Given the potential for selection bias in historically controlled studies as a result of the lack of randomization, one should exercise great care to ensure that comparisons are between patients with similar baseline risk factors for poor outcome. Randomization eliminates systematic imbalances for both measured and unmeasured baseline risk factors, but there is no protection from such bias in historically controlled studies.
The second type of data providing evidence for the magnitude of the benefit of active control interventions in CAP are from studies in which every other enrolled subject was administered antimicrobials or supportive care alone. Investigators even in the early 1900s were aware of the need to draw comparisons from subjects with similar baseline characteristics. The assignment of every other subject to various treatment groups, a process called “alternation,” was an early attempt to minimize selection bias. However, the lack of blinding of investigators to treatment assignment can still result in selection bias and, therefore, in groups that differ by important baseline risk factors. Recent studies show that a lack of blinding to treatment assignment (allocation concealment) can result in substantial bias in estimates of treatment effect [12]. We approached studies using alternation as containing the same potential selection biases as historically controlled studies.
Another issue when evaluating past evidence is the need for investigators to attempt to be as comprehensive as possible in acquiring data on the effects of the active control intervention. Authors have noted the effects of publication bias on assessment of the benefit of medical interventions, with a greater likelihood of publication for trials that demonstrate effectiveness of the test intervention [13]. Conversely, one should take care to avoid multiple counting of data that are published in more than one place or data that are used as the basis for historical controls repetitively in several trials. Multiple counting of data results in spurious precision of estimates of effects. We noted that in several instances, the same authors published the same data or updated data [12, 14–18] that included data from prior publications [12, 19–26]. We also noted instances in which authors repetitively used previously published evidence as comparison groups in their studies [27, 28]. In such cases, we used the publications that presented the most-detailed primary data, even if the numbers of subjects were larger in subsequent, less detailed publications. Increased sample size decreases random error but does not address systematic biases, such as selection bias, giving a more precise estimate of a potentially spurious value. Using primary data, we were able to evaluate the similarity of measured baseline characteristics in the studied subjects and attempt to control for selection bias.
With these points in mind, we evaluated the medical literature to attempt to determine the effect of antimicrobials in the treatment of CAP, in comparison with no specific treatment. We identified data through internet searches, books on CAP, and early uses of antimicrobials [16, 29, 30] and searched the references of the articles found by this method for other relevant articles. The focus was on adults, so data on patients aged <12 years were excluded. The antimicrobials that investigators evaluated in these trials were primarily sulfonamide derivatives and penicillin, with some data on early tetracyclines and other drugs. In the most serious cases, investigators combined antimicrobials with serum therapy. Given the goal of an overall estimate of treatment benefit for antimicrobials in general rather than for a specific drug, we pooled the data on treatment effects for sulfonamides, penicillin, tetracyclines, and other drugs. We included patients who received serum therapy in addition to drugs, to include the more seriously ill patients. In addition, studies done during the 1940s did not show a clear additive benefit of serum therapy combined with antimicrobials [31].
The primary outcome measure used in early CAP trials was all-cause mortality. This is because CAP is a potentially lethal disease, and, therefore, all-cause mortality is the most important measure. No studies attempted to evaluate cause-specific mortality. Investigators acknowledged that pneumonia can result in worsening of other conditions that cannot be separated from pneumonia [24, 32]. Evaluation of other, less important end points without taking into account all-cause mortality may result in biased estimates of effect, because more patients will be alive to experience end points such as complications of illness in the group receiving an intervention that results in lower mortality [33]. In addition, although some studies noted faster resolution of illness and a general sense of well-being in subjects who received active treatment, these studies did not provide reliable descriptions of which variables investigators actually measured (content validity) or of how investigators obtained these measures or when the measurements occurred (criterion validity). This makes it impossible to determine a reliable and reproducible effect size for antimicrobials for end points other than all-cause mortality. As one investigator noted [15, p. 15], “It is difficult to assess impartially the results of the treatment in pneumonia other than by consideration of the case-mortality rate. The dramatic improvement which may occur at the crisis tends to focus undue attention on the treatment being employed at the time.” This statement points out that clinical impressions are not valid measurements of outcomes in a disease like pneumonia, which can resolve quickly even in the absence of specific treatment in those who do recover, especially in the setting of nonblinded studies. Investigators attempted to use defervescence as a surrogate for symptomatic and general improvement in CAP, but “normalization” of body temperature is not a direct or sufficient measure of patient benefit in CAP. Body temperature is a biomarker that does not capture the important aspects of how patients function or survive. Body temperature lacks one of the primary characteristics of a valid surrogate end point—namely, that it is on the causal pathway of the disease [34, 35]. Fever is a result, not a cause, of pneumonia. In addition, patients with low body temperatures have higher mortality in pneumonia [24]. If body temperature were the only important measure in pneumonia, then antipyretics would be a sufficient treatment for the disease. Conversely, serum therapy frequently caused increases in body temperature yet decreased overall mortality, and antimicrobials themselves may result in drug fever in patients cured of pneumonia [19–21]. Investigators also noted a frequent “secondary pyrexia” in subjects whose body temperatures initially normalized, which, when taken into account, decreased the differences between treated and untreated groups. For instance, Agranat et al. [36, p. 310] note, “The temperature became normal in three days in about half of treated cases compared with about a quarter of controls…but a secondary pyrexia was fairly common and was considered quite usual…. The average duration of pyrexia in hospital was little affected by the drug, the figures being 4.8 and 5.6 days for treated and control groups.” Others noted a lack of correlation between decreasing body temperature and the clinical condition of the patient. For instance, Flippin [37, p. 8] noted, “Most of the clinical reports have stressed the frequency in which the initiation of drug treatment is followed within 24 to 36 hours or less by a critical drop in temperature…. Resolution of pneumonia then follows within a variable period of days, although we cannot say if this is hastened or retarded by the fall in temperature.” Defervescence also would not be a useful end point in current NI trials, because the majority of subjects enrolled are not febrile at baseline.
A recent analysis [38, p. 1] of outcomes in CAP trials concluded that “Studies of patients with community-acquired pneumonia have established certain expected rates of outcomes, including mortality, clinical complications, and time to resolution of symptoms…. However, there are no well-controlled studies that provide definitive estimates of the magnitude of the impact of antimicrobial therapy on these outcomes for patients with community-acquired pneumonia.” It also seems incongruous to speak of antimicrobials as “life-saving drugs,” which indeed they are in CAP, yet not evaluate the life-saving potential of these drugs as an end point. Some have asserted that “rescue therapy” in current trials—that is, administering an effective drug to patients who are “clinically failing”—makes the historical assessments of mortality less relevant today. However, a review of the literature showed there was “rescue therapy” in historical studies. Patients who were experiencing treatment failure while receiving sulfonamides or penicillin received serum, and vice-versa. Current studies of treated patients show rates of mortality (∼30%) among those with serious illness that are similar to rates in the past [39]. In addition, the overall mortality rate for pneumonia has not changed during the past 60 years. Historical studies also showed that most deaths occurred early in the course of the disease, so rescue therapy might have less impact than anticipated [27]. Data showing a potential benefit of earlier administration of antimicrobials would also mitigate any effects of “rescue therapy.” Although end points such as complications of disease (e.g., empyema, meningitis, and endocarditis) and resolution of symptoms are clinically meaningful end points for patients, there is a lack of evidence on which to base a quantifiable, reliable, and reproducible NI assessment for these outcomes. Investigators can assess such outcomes in current clinical trials as secondary end points testing superiority hypotheses. For these reasons, we have concentrated on all-cause mortality in evaluating the historical evidence in CAP.
Some of the studies excluded from their analyses subjects who died within the first few days of treatment or included subjects in the no-specific-treatment groups in whom the condition was first diagnosed at autopsy. Both of these inclusions/exclusions would bias toward showing a greater difference between no specific treatment and treatment with antimicrobials. It is interesting to note that current NI trials also inappropriately exclude subjects who do not receive ≥3 days of drug treatment, which may bias results [40]. We attempted to include all subjects who died and to exclude subjects whose condition was first diagnosed at autopsy, when possible (i.e., when the authors provided the numbers of such subjects in the study).
As noted above, none of the early studies in CAP was truly randomized with allocation concealment. Many investigators made conscious decisions about treatment assignment, which increases the potential for imbalances between groups in patient characteristics that may predict a poor outcome independent of treatment, a condition called “confounding.” Indeed, several investigators note that they made no attempts at either randomization or alternation but instead relied on post hoc comparisons of subjects according to baseline risk factors [41].
An example using actual data from one of the studies comparing no specific treatment with sulfanilamide treatment illustrates the potential issues associated with confounding. In this study, one group of 18 subjects had a mortality rate of 50% (9/18). A second group of 18 subjects within the same study had a mortality rate of 6% (1/18), resulting in a significant treatment difference of 44% (2-sided P = .003). The surprising finding is that the first group received sulfanilamide, and the second group received no specific therapy. The author of this study concluded that sulfanilamide was not very useful when used as a sole agent [20]. However, an evaluation of the baseline characteristics of the 2 groups shows that the subset of patients in the no-specific-treatment group were all aged <30 years and were without concomitant bacteremia. In the sulfanilamide group, 13 of 18 subjects were aged >30 years, 10 were bacteremic, and 11 had multilobar involvement. The patient groups were dissimilar in important baseline characteristics, and this imbalance raises concerns about direct unadjusted comparisons between the groups.
Given the issue of confounding, we evaluated the evidence from the early studies of CAP to attempt to identify baseline characteristics that may influence outcome independent of treatment. We found at least 7 such characteristics outlined in the historical data associated with higher mortality: greater age of the patient, presence of bacteremia, the type of pneumococcus causing the infection (with type III associated with the highest mortality), the presence of multilobar disease, a chest radiography finding of bronchopneumonia (compared with lobar pneumonia), the presence of comorbid illness, and the timing of initiation of treatment.
Ideally, it would be best to take all of these factors into account to obtain adjusted estimates of the effect of treatment on mortality. Unfortunately, the articles providing mortality data on patients receiving either no specific treatment or antimicrobial treatment are inconsistent in their presentation of the distribution of patient characteristics at baseline. Age and bacteremia status are most consistently presented. Therefore, we followed an approach similar to that of Finland [14] by categorizing patients by 2 of the most important factors: presence of bacteremia (yes vs. no) and age (12–29 vs. 30–49 vs. ≥50 years). More-recent studies show that the effects of age and bacteremia on outcome are still important today [42]. In our extensive review of the literature, several articles did not provide data by age or bacteremia status [36, 43, 44], several provided data by age alone [18, 25, 27, 28, 45–47], and several provided data by bacteremia status alone [17, 18, 45–48]. Fortunately, 10 articles provided data by age and bacteremia status simultaneously [19–22, 24, 26, 49–52]. Table 1 provides the mortality reported in these 10 articles, by intervention group (i.e., no specific treatment vs. treatment with sulfonamides or penicillin) and by age and bacteremia status. Data are presented simply as “mortality,” because the articles were not specific regarding the duration of follow-up. Our analyses of effects on mortality, using data in table 1, rely on an assumption that those receiving no specific treatment had the same duration of follow-up as those receiving antibiotics.
Table 1.
Mortality, by age and by bacteremia status
| Age 12–29 years |
Age 30–49 years |
Age ≥50 years |
||||
|---|---|---|---|---|---|---|
| Reference, intervention | Bacteremic | Not bacteremic | Bacteremic | Not bacteremic | Bacteremic | Not bacteremic |
| Tilghman and Finland [24], no antibiotics | 30/49 | 29/222 | 137/178 | 64/323 | 239/255 | 168/289 |
| Finland and Brown [20] | ||||||
| No antibiotics | 0/3 | 1/18 | 15/22 | 2/18 | 13/15 | 11/20 |
| Antibiotics | 0/1 | 0/2 | 1/5 | 3/4 | 3/4 | 2/2 |
| Brown and Finland [19] | ||||||
| No antibiotics | 1/1 | 1/13 | 5/7 | 3/23 | 12/13 | 1/7 |
| Antibiotics | 0/0 | 0/0 | 0/0 | 0/1 | 2/5 | 1/2 |
| Heintzelman et al. [49] | ||||||
| No antibiotics | 0/0 | 0/0 | 1/1 | 2/4 | 1/1 | 4/4 |
| Antibiotics | 0/0 | 0/0 | 0/0 | 0/6 | 1/1 | 1/2 |
| Finland et al. [22] | ||||||
| No antibiotics | 1/2 | 5/115 | 6/7 | 15/140 | 20/21 | 88/187 |
| Antibiotics | 4/14 | 4/50 | 18/54 | 6/75 | 25/57 | 14/104 |
| Ruegsegger et al. [50], antibiotics | 0/1 | 0/9 | 0/4 | 3/26 | 1/2 | 2/7 |
| Finland and Brown [21], antibiotics | 0/0 | 0/2 | 2/3 | 0/0 | 1/1 | 0/0 |
| Bullowa and Wilcox [26], no antibiotics | 49/71 | 54/668 | 155/213 | 132/714 | 83/90 | 81/208 |
| Winters et al. [52], antibiotics | 1/4 | 0/29 | 2/14 | 2/42 | 7/14 | 2/20 |
| Meakins and Hanson [51], antibiotics | 0/1 | 0/5 | 0/1 | 0/17 | 0/0 | 1/6 |
NOTE. Data are no. of deaths/no. of patients.
Results from an informal meta-analysis of the data in table 1 are presented in table 2. Several important insights are evident. First, when one looks only at the data from patients receiving no specific therapy, it is apparent that both age and bacteremia status are independently strongly predictive of mortality rate. This confirms that analyses of effects of sulfonamides or penicillin on mortality, when nonrandomized comparator groups are used, could be biased (i.e., confounded) if these characteristics are imbalanced across treatment groups. Second, antimicrobials substantially reduce mortality risk in bacteremic patients, independently of age, and in older nonbacteremic patients. However, although still effective in younger nonbacteremic patients, antimicrobials achieve a smaller absolute difference in mortality in this population. This suggests that age and bacteremia are not only baseline predictors of outcome but also effect modifiers; that is, the magnitude of the absolute differences in mortality between treatment and no treatment differ, depending on the chosen baseline characteristics. For instance, the magnitude of treatment effects on an absolute scale is larger in CAP in bacteremic subjects of any age than in younger nonbacteremic subjects. The role of baseline characteristics as predictors is critical in addressing bias from confounding in nonrandomized trials, and their role as effect modifiers is critical in addressing the validity of the constancy assumption—that is, what types of patients need to be enrolled in current NI trials so that such trials are capable of differentiating effective from ineffective drugs. An NI margin developed on the basis of historical data from bacteremic patients would not be applicable to a current trial enrolling predominantly young nonbacteremic subjects, even if the age of subjects in the historical studies and current NI trials is similar. Also of note, the current Patient Outcome Research Team (PORT) scoring system includes data on age and comorbid illness but does not include data on bacteremia status [39]. Therefore, using PORT scoring alone is insufficient in selecting patients for a current NI trial. A recent study showed that bacteremic and nonbacteremic subjects had similar PORT scores, but they still differed by bacteremia status in their risks of death [42]. This study also confirmed, with more-recent data, the greater mortality among bacteremic patients compared with nonbacteremic patients. Because it is apparent that age and bacteremia status are both predictors (confounding factors) and effect modifiers, we did not include data from articles that did not provide information on both characteristics.
Table 2.
Formulation of the noninferiority margin, as a function of age and bacteremia status, for patients receiving penicillin or a sulfonamide as an active control regimen
| Bacteremic subjects |
Nonbacteremic subjects |
|||
|---|---|---|---|---|
| Age group, variable | No specific treatment |
Antibiotic treatment |
No specific treatment |
Antibiotic treatment |
| 12–29 years | ||||
| No. of deaths/no. of patients (% mortality) | 81/126 (64.29) | 5/21 (23.81) | 90/1036 (8.69) | 4/97 (4.12) |
| % Difference (95% CI) | 40 (20–61) | 5 (0–9) | ||
| Proposed margin | 10 | None | ||
| 30–49 years | ||||
| No. of deaths/no. of patients (% mortality) | 319/428 (74.53) | 23/81 (28.40) | 218/1222 (17.84) | 14/171 (8.19) |
| % Difference (95% CI) | 46 (35–57) | 10 (5–14) | ||
| Proposed margin, % | 10 | 2.5 | ||
| ≥50 years | ||||
| No. of deaths/no. of patients (% mortality) | 368/395 (93.16) | 40/84 (47.62) | 353/715 (49.37) | 23/143 (16.08) |
| % Difference (95% CI) | 46 (35–57) | 33 (26–40) | ||
| Proposed margin, % | 10 | 10 | ||
NOTE. On the basis of the clinical relevance of the increase in mortality, the maximum margin is assumed to be 10%.
Using methods described above in “General Principles in Designing NI Trials,” we computed an NI margin for each of the 6 age-by-bacteremia settings. None is larger than 10%, even though this may represent preserving more than half the effect of the comparator drug, because of the recognition that it would be clinically unacceptable to use an experimental drug with an absolute mortality rate 10% higher than that of a comparator antimicrobial. These results suggest that investigators could perform an NI trial with a 10% margin (based on an absolute difference) in bacteremic patients or in older nonbacteremic patients, assuming that patients in the NI trial are similar to these historical patients in other characteristics (e.g., microbiologically defined disease and comorbid illness) that would predict a 15% mortality rate among subjects receiving the comparator antimicrobials. This would correlate with subjects of any PORT score with concomitant bacteremia or nonbacteremic subjects with class IV and V PORT scores.
A controversial issue from table 2 relates to whether younger nonbacteremic patients derive benefit from antimicrobials that is sufficient to justify an NI trial having a very small margin. On the basis of the absolute risk scale traditionally implemented in the design of NI trials with anti-infectives, along with data from table 2 to justify a margin, an NI trial would not be possible in that setting. However, suppose that (1) treatment effects are instead assessed on an OR scale, (2) a strong assumption is made that age and bacteremia status are not effect modifiers on the OR scale, and (3) one extrapolates the use of the 1.67 OR margin that corresponds to using a 10% margin in the absolute risk scale when there is ∼20%–25% mortality with the comparator antimicrobial regimen. This would correspond to using a 1% margin in a trial with 1.5% mortality among subjects receiving the control antimicrobial and a 2% margin in a trial with 3% mortality among subjects receiving the control antimicrobial.
The use of a 1.67 OR margin to allow inclusion of younger nonbacteremic subjects in an NI mortality trial is controversial, given the lack of evidence about the validity of assumption 2 above from the data in table 2. However, if mortality data for patients receiving penicillin or a sulfonamide are added from those articles [31, 33, 34, 50–52] that provide data by age alone (rather than by age and bacteremia status), with care to avoid double counting from articles that have overlapping samples, then one can provide more-precise estimates of the magnitude of treatment effect in nonbacteremic patients in the <30-year and 30–49-year age categories. Use of this added information likely underestimates the effect of antimicrobials in younger nonbacteremic patients, given that some of the patients in these 6 additional articles who received antimicrobials were bacteremic. This approach yields estimates of mortality among subjects receiving antimicrobials of 25 (2.39%) of 1044 nonbacteremic patients aged 12–29 years and 125 (7.65%) of 1634 nonbacteremic patients aged 30–49 years. We can use these estimates of mortality for nonbacteremic patients receiving antimicrobials, along with data from table 2, for mortality among nonbacteremic patients in these age groups who received no specific treatment, to obtain margins using methods described above in “General Principles in Designing NI Trials.” On the basis of these margins, and factoring in issues of monotonicity and variability, one can support the use of the 1.67 OR NI margin in a trial enrolling adults from any age group or bacteremia status.
CONCLUSIONS
Superiority trials, including add-on trials in which all subjects receive a standard-of-care regimen and then are randomized to receive an additional experimental intervention or a placebo (e.g., combination therapy trials), provide a direct and interpretable approach for evaluating the benefit-to-risk profile of the new therapeutic regimen. When a standard “active control” regimen provides clinically meaningful benefit with regard to mortality or measures of major morbidity and the most clinically relevant question is whether investigators can use the experimental regimen in place of the active control without meaningful loss in effectiveness, then an NI trial provides a potential design option to address that scientific question. Unfortunately, in many if not most clinical settings, it is not possible to design a valid NI trial. It is insufficient to know only that an active intervention “works” in a given setting. To conduct a valid NI trial, historical studies must provide reliable, reproducible, and precise estimates of the effect of the active control regimen on the specific effectiveness end point that investigators plan to use in the NI trial; this effect needs to be of substantial magnitude, and the estimates from historical studies of the active control’s effect must represent its effect in the planned NI trial had a placebo group been included in that trial. These conditions allow formulation of an NI margin such that, if the NI trial establishes that effectiveness of the experimental intervention is not worse than that of the active control by more than the NI margin, then one can conclude that the experimental regimen (1) preserves a substantial fraction of the effect of the active control and (2) will not result in a clinically meaningful loss of effectiveness.
In considering the use of an NI trial design, investigators should also address the issues recently discussed by authors of a Lancet article who questioned the ethics of NI trials in some settings [53]. Suppose an active control regimen provides proven benefit with regard to mortality or irreversible morbidity. Is it is ethical to ask participants to be randomized between that proven effective regimen and an experimental intervention that one hopes is as good as—but could be meaningfully worse than—the active control? Do potential research subjects understand that this is what is asked of them in NI trials? If participants are going to be asked to take such a risk, the experimental agent should be expected to provide other important benefits, such as meaningful improvements in safety, convenience, or cost. In the area of anti-infectives, some have proposed that it is reasonable to allow a loss of effectiveness in current trials in the hope that new drugs might be superior in effectiveness in disease caused by resistant pathogens, either now or in the future. However, clinicians should provide to current patients, in clinical practice or trials, interventions that are not inferior to available treatment. When the focus is on subjects infected with resistant pathogens, better diagnostics can help in identifying and enrolling such patients in trials intended to determine whether new interventions have superior effectiveness. Because the relationship between in vitro “resistance” and clinical outcomes is not clear in some cases [38], better diagnostics would allow one to evaluate that relationship as well.
To use any intervention as an active control in an NI trial, investigators must reliably determine the proper clinical end point that captures the essence of the intended clinical benefit, the magnitude of the treatment effect on that end point, the population in whom this effect is manifest, and the conditions of the previous studies that demonstrated that effect. In the setting of CAP, the historical evidence supports the conclusions that serum therapy, sulfonamides, penicillin, and certain other drugs provide clear benefit to patients, compared with no specific treatment, in terms of decreased all-cause mortality among patients with microbiologically confirmed disease. Although investigators noted a general impression of benefit with regard to time to resolution of illness, they did not use any standardized measurement of such benefit, and the magnitude of effect is unclear. US regulations require that outcome measures be “well-defined and reliable” [10]. The end point in current CAP NI trials, based on clinician judgment that a research subject does not need additional antimicrobials, is neither well-defined nor reliable [40]. Furthermore, there is a lack of historical data on which to formulate an NI margin for such a clinical-judgment end point. With such end points in NI trials, investigators’ knowledge that all subjects are receiving active therapy may also bias outcome evaluations toward assessing the treatment of most patients as being a success, even in double-blinded trials, reducing sensitivity to detection of true differences between treatments [54]. Although clinicians use signs and symptoms related to resolution of illness to guide decisions about clinical care, these measures are not sufficiently validated for use as primary end points in registrational clinical trials. Furthermore, these surrogate end points are not needed in trials of short-term, acute diseases when one can measure the more relevant clinical end points in a short time period. More research in superiority trials using properly developed and evaluated patient-reported outcome instruments may help us to define useful end points for future trials in addition to, but not in place of, all-cause mortality [55].
Given that many antimicrobials do provide important beneficial effects on mortality risk, it is important to preserve those benefits by studying new interventions in CAP in adequately designed, conducted, and analyzed trials. Clinicians see a subset of antimicrobials that reach clinical practice after demonstration of safety and effectiveness, but this does not mean that all experimental antimicrobials are de facto safe and effective. Since 1964, antimicrobials have the highest rates of approval by regulatory agencies of any therapeutic class, but this rate is 28%, not 100%, among agents with investigational new drug applications, pointing out that antimicrobials do fail to demonstrate safety and effectiveness in clinical trials [56]. Antimicrobials shown to be ineffective in clinical trials also had promising results from in vitro and animal studies to support a hypothesis for further testing in humans. One should separate the issues of developing an appropriate hypothesis from providing confirmatory evidence in clinical trials. Demonstration of effectiveness under specified conditions of use is needed to justify the conclusion that the benefit-to-risk ratio is favorable, because adverse events are inherent with all drugs. This principle is embodied in US law regarding the approval of all drugs, including antimicrobials [57].
Our review of the evidence showed the challenges in precisely estimating the magnitude of the effects of antimicrobials in CAP; in part, these challenges exist because there are no trials in which patients were randomized between experimental antimicrobial and “no specific treatment” regimens. Evidence provided in table 2 regarding the absolute difference in mortality confirms that patient characteristics such as age and bacteremia status are effect modifiers as well as significant confounding factors in analyses estimating the magnitude of treatment effect. Hence, the magnitude of the absolute difference in mortality that patients receive from active control regimens used in current NI trials in CAP differs across patient populations. Analyses evaluating treatment effects by age alone do not take into account other important confounding factors and effect modifiers, such as bacteremia; thus, they still contain residual confounding. It is likely that even our analysis by age and bacteremia status will have some residual confounding by other factors, such as the presence of multilobar disease and comorbid illness, but the primary data from the historical studies often were not presented in sufficient detail to adjust for the effects of these other factors. Fortunately, these factors appear to be correlated with age and presence of bacteremia, so they may be partially addressed in our analysis. It is apparent that antimicrobials in CAP induce a larger absolute difference in mortality in bacteremic and/or older patients, who would have >15% mortality risk even when receiving standard antimicrobial therapy. This would correlate with subjects of any PORT score with concomitant bacteremia or nonbacteremic subjects with class IV and V PORT scores. Hence, if investigators are to study a new antimicrobial regimen in CAP by use of an NI trial, they should use all-cause mortality as the preferred end point and, if a margin of 10% for the absolute difference in mortality is used, they should study high-risk (bacteremic and/or older) patients as a preferred patient population. Use of an NI margin based on an OR of 1.67 rather than the absolute difference in mortality as traditionally used in antimicrobial development would allow expansion of the patient population to anyone with CAP. However, with this OR approach to formulation of the NI margin, including subjects with less serious disease would substantially increase the sample size of the trial. There is an incentive to study more seriously ill patients, in part to reduce trial size but also to provide robust evidence for safety and effectiveness in the group of patients in whom the drugs can make the most difference in decreasing mortality.
Acknowledgments
We are very appreciative of comments on earlier drafts of this article by Katherine Odem-Davis.
Financial support. National Institutes of Health (NIH)/National Institute of Allergy and Infectious Diseases (NIAID) (R37 AI 29168, “Statistical Issues in AIDS Research,” to T.R.F.). For J.H.P., this project has been funded in whole or in part with federal funds from the National Cancer Institute, NIH, under contract N01-CO-12400. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the US government.
Footnotes
The content of this publication does not necessarily reflect the views or policies of the US Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the US government.
Supplement sponsorship. This article was published as part of a supplement entitled “Workshop on Issues in the Design and Conduct of Clinical Trials of Antibacterial Drugs for the Treatment of Community-Acquired Pneumonia,” sponsored by the US Food and Drug Administration and the Infectious Diseases Society of America.
Potential conflicts of interest. T.R.F. has received consulting fees from Boehringer-Ingelheim, Bristol-Myers Squibb, Eli Lilly, GlaxoSmithKline, Novartis, Pfizer, Roche, Schering-Plough, Theravance, and Wyeth; interactions with these companies were not on projects related to communityacquired pneumonia. J.H.P. has received consulting fees from Acureon, Astellas, Astra-Zeneca, Basilea, Centegen, Cerexa, Concert, Cubist, Destiny, Forest, Johnson & Johnson, Merck, Methylgene, MPEX, Octoplus, Takeda, Theravance, and Wyeth.
References
- 1.Fleming TR. Current issues in non-inferiority trials. Stat Med. 2008;27:317–32. doi: 10.1002/sim.2855. [DOI] [PubMed] [Google Scholar]
- 2.Powers JH. Non-inferiority and equivalence trials: deciphering the similarity of medical interventions. Stat Med. 2008;27:343–52. doi: 10.1002/sim.3138. [DOI] [PubMed] [Google Scholar]
- 3.Statistical principles for clinical trials (ICH E-9) [Accessed 11 September 2008];International Conference on Harmonisation. Available at: http://www.ich.org/MediaServer.jser?@_ID=485&@_MODE=GLB.
- 4.Choice of control group and related issues in clinical trials (ICH E-10) [Accessed 11 September 2008];International Conference on Harmonisation. Available at: http://www.ich.org/MediaServer.jser?@_ID=486&@_MODE=GLB. [PubMed]
- 5.US Food and Drug Administration [Accessed 11 September 2008];Guidance for industry antibacterial drug products: use of non-inferiority studies to support approval. Available at: http://www.fda.gov/cder/guidance/7884dft.htm.
- 6.Loeb M. Community acquired pneumonia. Clin Evid. 2006;15:2015–24. [PubMed] [Google Scholar]
- 7.Mills GD, Oehley MR, Arrol B. Effectiveness of beta lactam antibiotics compared with antibiotics active against atypical pathogens in non-severe community acquired pneumonia: meta-analysis. BMJ. 2005;330:456. doi: 10.1136/bmj.38334.591586.82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shefet D, Robenshtok E, Paul M, Leibovici L. Empirical atypical coverage for inpatients with community-acquired pneumonia: systematic review of randomized controlled trials. Arch Intern Med. 2005;165:1992–2000. doi: 10.1001/archinte.165.17.1992. [DOI] [PubMed] [Google Scholar]
- 9.Sacks H, Chalmers TC, Smith H., Jr. Randomized versus historical controls for clinical trials. Am J Med. 1982;72:233–40. doi: 10.1016/0002-9343(82)90815-4. [DOI] [PubMed] [Google Scholar]
- 10.Food and Drug Administration, Department of Health and Human Services [Accessed 11 September 2008];Applications for FDA approval to market a new drug [21 USC 314.126] Available at: http://a257.g.akamaitech.net/7/257/2422/10apr20061500/edocket.access.gpo.gov/cfr_2006/aprqtr/21cfr314.126.htm.
- 11.Wood L, Egger M, Gluud LL, et al. Empirical evidence of bias in treatment effect estimates in controlled trials with different interventions and outcomes: meta-epidemiological study. BMJ. 2008;336:601–5. doi: 10.1136/bmj.39465.451748.AD. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schulz KF, Chalmers I, Hayes RJ, Altman DG. Empirical evidence of bias: dimensions of methodological quality associated with estimates of treatment effects in controlled trials. JAMA. 1995;273:408–12. doi: 10.1001/jama.273.5.408. [DOI] [PubMed] [Google Scholar]
- 13.Turner EH, Matthews AM, Linardatos E, Tell RA, Rosenthal R. Selective publication of antidepressant trials and its influence on apparent efficacy. N Engl J Med. 2008;358:252–60. doi: 10.1056/NEJMsa065779. [DOI] [PubMed] [Google Scholar]
- 14.Finland M. Chemotherapy in bacteremias. Conn State Med J. 1943;7:92–100. [Google Scholar]
- 15.Evans GM, Gaisford WF. Treatment of pneumonia with 2-(p-aminobenzenesulphonamido) pyridine. Lancet. 1938;232:14–9. [Google Scholar]
- 16.Bullowa JGW. The course, symptoms and physical findings. In: Bullowa JGW, editor. The management of pneumonias. Oxford University Press; New York: 1937. [Google Scholar]
- 17.Flippin HF, Lockwood JS, Pepper DS, Schwartz L. The treatment of pneumococcic pneumonia with sulfapyridine. JAMA. 1939;112:529–34. [Google Scholar]
- 18.Pepper DS, Flippin HF, Schwartz L, Lockwood JS. The results of sulfapyridine therapy in 400 cases of typed pneumococcic pneumonia. Am J Med Sci. 1939;198:22–35. [Google Scholar]
- 19.Brown JW, Finland M. Specific treatment of pneumococcus type II pneumonia. Am J Med Sci. 1939;197:369–81. [Google Scholar]
- 20.Finland M, Brown JW. Specific treatment of pneumococcus type I pneumonia. Am J Med Sci. 1939;197:151–68. [Google Scholar]
- 21.Finland M, Brown JW. Specific treatment of pneumococcus type V and type VII pneumonias. Am J Med Sci. 1939;197:381–93. [Google Scholar]
- 22.Finland M, Spring WC, Jr, Lowell FC. Specific treatment of the pneumococcic pneumonias; an analysis of the results of serum therapy and chemotherapy at the Boston City Hospital from July 1938 through June 1939. Ann Intern Med. 1940;13:1567–93. [Google Scholar]
- 23.Finland M, Lowell FC, Spring WC., Jr. Clinical and laboratory studies on the use of serum and sulfapyridine in the treatment of the pneumococcal pneumonias. N Engl J Med. 1940;222:739–47. [Google Scholar]
- 24.Tilghman RC, Finland M. Clinical significance of bacteremia in pneumococcal pneumonia. Arch Intern Med. 1937;59:602–19. [Google Scholar]
- 25.Gaisford WF. Results of the treatment of 400 cases of lobar pneumonia with M&B 693. Proc Royal Soc Med. 1939;32:1070–6. doi: 10.1177/003591573903200923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bullowa JGW, Wilcox C. Incidence of bacteremia in the pneumonias and its relation to mortality. Arch Intern Med. 1935;55:558–73. [Google Scholar]
- 27.Austrian R, Gold J. Pneumococcal bacteremia with especial reference to bacteremic pneumococcal pneumonia. Ann Intern Med. 1964;60:759–76. doi: 10.7326/0003-4819-60-5-759. [DOI] [PubMed] [Google Scholar]
- 28.Dowling HF, Lepper MH. The effect of antibiotics (penicillin, aureomycin, and terramycin) on the fatality rate and incidence of complications in pneumococcic pneumonia: a comparison with other methods of therapy. Am J Med Sci. 1951;222:396–403. doi: 10.1097/00000441-195110000-00005. [DOI] [PubMed] [Google Scholar]
- 29.Lesch JE. The first miracle drugs; how the sulfa drugs transformed medicine. 1st ed Oxford University Press; Oxford: 2007. [Google Scholar]
- 30.Podolsky SH. Pneumonia before antibiotics; therapeutic evolution and evaluation in twentieth-century America. Johns Hopkins University Press; Baltimore, Maryland: 2006. [Google Scholar]
- 31.Plummer N, Liebmann J, Solomon S, Kammerer WK, Kalkstein M, Ensworth HK. Chemotherapy versus combined chemotherapy and serum. JAMA. 1941;116:2366–71. [Google Scholar]
- 32.Wyckoff J, DuBois EF, Woodruff IO. The therapeutic value of digitalis in pneumonia. JAMA. 1930;95:1243–9. [Google Scholar]
- 33.Lubsen J, Kirwan BA. Combined endpoints: can we use them? Stat Med. 2002;21:2959–70. doi: 10.1002/sim.1300. [DOI] [PubMed] [Google Scholar]
- 34.Fleming TR, DeMets DL. Surrogate end points in clinical trials: are we being misled? Ann Intern Med. 1996;125:605–13. doi: 10.7326/0003-4819-125-7-199610010-00011. [DOI] [PubMed] [Google Scholar]
- 35.Fleming TR. Surrogate endpoints and FDA’s accelerated approval process. Health Aff (Millwood ) 2005;24:67–78. doi: 10.1377/hlthaff.24.1.67. [DOI] [PubMed] [Google Scholar]
- 36.Agranat AL, Dreosti AO, Ordman D. Treatment of pneumonia with 2-(p-aminobenzenesulphonoamido) pyridine (M&B 693) Lancet. 1939;233:309–17. [Google Scholar]
- 37.Flippin HF. Sulfapyridine in the treatment of pneumococci pneumonia based upon treatment of 350 cases. Chest. 1939;5:8–9. [Google Scholar]
- 38.Metlay JP, Singer DE. Outcomes in lower respiratory tract infections and the impact of antimicrobial drug resistance. Clin Microbiol Infect. 2002;8(Suppl 2):1–11. doi: 10.1046/j.1469-0691.8.s.2.4.x. [DOI] [PubMed] [Google Scholar]
- 39.Fine MJ, Auble TE, Yealy DM, et al. A prediction rule to identify low-risk patients with community-acquired pneumonia. N Engl J Med. 1997;336:243–50. doi: 10.1056/NEJM199701233360402. [DOI] [PubMed] [Google Scholar]
- 40.Powers JH. Reassessing the design, conduct and analysis of clinical trials in therapy for community-acquired pneumonia. Clin Infect Dis. 2008;46:1152–6. doi: 10.1086/533442. [DOI] [PubMed] [Google Scholar]
- 41.Finland M. Controlling clinical therapeutic experiments with specific serums. N Engl J Med. 1941;225:495–506. [Google Scholar]
- 42.Musher DM, Alexandraki I, Graviss EA, et al. Bacteremic and nonbacteremic pneumococcal pneumonia: a prospective study. Medicine (Baltimore) 2000;79:210–21. doi: 10.1097/00005792-200007000-00002. [DOI] [PubMed] [Google Scholar]
- 43.Davies DT, Hodgson HG, Whitby LEH. A study of pneumococcal pneumonia. Lancet. 1935;1:791–6. [Google Scholar]
- 44.Ormiston G, Woodman D, Lewis FJW. Pneumonia in infants, children, and adults. Quarterly J Med. 1942;48:155–80. [Google Scholar]
- 45.Anderson T, Cairns JG. Treatment of pneumonia with sulphapyridine and serum. Lancet. 2008;i:449–51. [Google Scholar]
- 46.Graham D, Warner WP, Dauphinee JA, Dickson RC. The treatment of pneumococcal pneumonia with Dagenan (M&B 693) Can Med Assoc J. 1939;40:325–32. [PMC free article] [PubMed] [Google Scholar]
- 47.Plummer N, Ensworth HK. Sulfapyridine in the treatment of pneumonia. JAMA. 1939;113:1847–54. [Google Scholar]
- 48.Don CSD, Luxton RW, Donald HR, et al. Treatment of pneumonia with M.&B. 693. Lancet. 1940;ii:311–4. [Google Scholar]
- 49.Heintzelman JHL, Hadley PB, Mellon RR. The use of p-aminobenezenesulphonamide in type 3 pneumococcus pneumonia. Am J Med Sci. 1937;193:759–63. [Google Scholar]
- 50.Ruegsegger JM, Hamburger M, Cockrell SL. The comparative use of sulfapyridine and specific serum in pneumococcal pneumonia. Ohio State Med J. 1940;36:257–61. [Google Scholar]
- 51.Meakins JC, Hanson FR. The treatment of pneumococcic pneumonia with sulfapyridine. Can Med Assoc J. 1939;40:333–6. [PMC free article] [PubMed] [Google Scholar]
- 52.Winters WL, Rhoads PS, Fox WW, Rosi R. The treatment of pneumococcic pneumonia with sulfapyridine. Ann Intern Med. 1941;14:1827–37. [Google Scholar]
- 53.Garattini S, Bertele’ V. Non-inferiority trials are unethical because they disregard patients’ interests. Lancet. 2007;370:1875–7. doi: 10.1016/S0140-6736(07)61604-3. [DOI] [PubMed] [Google Scholar]
- 54.Snapinn SM. Noninferiority trials. Curr Control Trials Cardiovasc Med. 2000;1:19–21. doi: 10.1186/cvm-1-1-019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lamping DL, Schroter S, Marquis P, Marrel A, Duprat-Lomon I, Sagnier PP. The community-acquired pneumonia symptom questionnaire: a new, patient-based outcome measure to evaluate symptoms in patients with community-acquired pneumonia. Chest. 2002;122:920–9. doi: 10.1378/chest.122.3.920. [DOI] [PubMed] [Google Scholar]
- 56.DiMasi JA. Success rates for new drugs entering clinical testing in the United States. Clin Pharmacol Ther. 1995;58:1–14. doi: 10.1016/0009-9236(95)90066-7. [DOI] [PubMed] [Google Scholar]
- 57.Powers JH. Increasing the efficiency of clinical trials of antimicrobials: the scientific basis of substantial evidence of effectiveness of drugs. Clin Infect Dis. 2007;45(Suppl 2):S153–62. doi: 10.1086/519253. [DOI] [PubMed] [Google Scholar]