

Abstract
The complement system is one of the key players in the defence against infections. Its activation during the innate immune response leads to the generation of several proteins that contribute to the lysis and opsonization of microorganisms, regulate inflammatory reactions and bridge innate immunity with the subsequent adaptive immune response. Complement is also activated in overwhelming bacterial infections that lead to sepsis, and its protective functions play a role in this frequently lethal disorder. However, despite its role in protection, complement can also contribute to the development of severe complications that significantly worsen the prognosis of septic patients. Therefore, an understanding of the mechanisms involved in the activation of complement during sepsis is essential to our efforts to introduce rational therapies targeting complement to the treatment of patients suffering from this condition. This review presents a current view of the mechanisms involved in the activation of complement in sepsis, in the context of the multiple interactions between complement and other biological systems that are involved in the pathogenesis of this disorder.
Keywords: complement, sepsis, pathogenesis, coagulation
Introduction
Sepsis – terminology and basic facts
The pathways of complement activation
The role of the complement system in the pathogenesis of sepsis
Aetiology-dependent mechanisms of complement activation in sepsis
Sepsis-associated coagulopathy and complement activation
Concluding remarks
Introduction
Despite the laborious efforts of scientists and the pharmaceutical industry to develop new and more effective antimicrobial therapies, systemic infections remain a serious health issue even in well-developed and wealthy countries of the Western Hemisphere [1]. This situation is at least partially a result of mechanisms that enable microorganisms to quickly modify their genomes and acquire resistance to newly developed antibiotics [2]. Therefore, the race between drug discovery and the lethal effects of virulent pathogens on millions of patients worldwide seems to be endless.
The problems posed by infectious diseases have become even more serious as a result of the increasing number of patients who are receiving immunosuppressive treatment because of disseminated cancer or organ transplantation. In addition, the significant rise in average life expectancy in recent decades has contributed to an increase in the number of patients suffering from chronic diseases, which increase the vulnerability of these individuals to serious complications of infectious disease [3]. Under these circumstances, an ongoing discussion among scientists and physicians concerning infections and sepsis is both timely and necessary. In this review, we discuss the mechanisms that contribute to sepsis-associated activation of the complement system. Complement constitutes a crucial line of defence against microbial invasion [4], but it is also often identified as an inducer of excessive inflammatory host responses, which are thought to increase mortality from sepsis [5, 6].
Sepsis – terminology and basic facts
The currently accepted definition of sepsis describes it as a systemic inflammatory response syndrome (SIRS) induced by presumed or confirmed infection. SIRS is defined on the basis of clinical criteria, which include body temperature, heart and respiratory rate, blood pC02 and white blood cell count. Under unfavourable circumstances, sepsis can progress to severe sepsis and ultimately to septic shock. Severe sepsis is a syndrome associated with organ dysfunction, hypoperfusion or hypotension. Septic shockIs described as a combination of sepsis-induced hypotension that is unresponsive to adequate fluid resuscitation and the presence of perfusion abnormalities [7].
According to studies published in 2003, the prevalence of sepsis in the United States was estimated to be approximately 750,000 cases per year [8]. Considering the steady rise in the number of septic patients diagnosed each year, it has been predicted that this number will reach over 1 million cases by 2020. Despite the decrease in sepsis mortality rates over the past 20 years, the increasing number of sepsis cases has resulted in a tripling of the actual number of sepsis-associated deaths, to 215,000 deaths per year; remarkably, sepsis is responsible for 9.3% of all deaths in the United States [9]. These frightening numbers clearly indicate that the threat of infectious disease is one of the main problems facing twenty-first century medicine.
Contrary to the popular understanding of sepsis biology, which links this syndrome to infection with bacteria, the aetiology of sepsis is actually variable and includes fungi, parasites and viruses as well as bacteria [9]. Sepsis development is often preceded by localized respiratory or abdominal infections, although other organ systems, including the urogenital tract, can also be a source [1]. Currently, Gram-positive organisms outnumber Gram-negative bacteria as causative microflora, and an increasing number of sepsis cases are associated with fungal infections [8]. In a small but appreciable number of patients with a clinical presentation of sepsis, causative organisms cannot be identified [10]. However even in the absence of a clear aetiology, these individuals should be treated as septic patients.
The key events in sepsis pathogenesis involve complex multidimensional pathogen-host interactions, which are not only responsible for the clinical manifestations of sepsis but also strongly contribute to the clinical course, prognosis and complications, and potentially to the associated mortality [11]. Therefore, the overall picture of a septic patient is influenced by various factors that include aetiology, preexisting clinical conditions (‘comor-bidities’), the extent of the inflammatory and immune responses to pathogen invasion, and the disturbances of homeostasis that are induced by pathogen- and host-derived factors. The reciprocal interactions among all these variables contribute to the complexity of sepsis pathophysiology and further complicate our efforts to gain insight into the mechanisms regulating the host response during sepsis.
The challenges that have limited our understanding of these complex processes are well illustrated by the recent evolution of thought concerning the role of inflammatory reactions in adverse outcomes in septic patients. It has long been accepted that death from sepsis-associated multi-organ failure is the result of an excessive inflammatory response to pathogens. Therefore, anti-inflammatory therapies have been seen as beneficial in decreasing the rate of sepsis-associated complications [12–14]. However, virtually all clinical trials of anti-inflammatory therapies have failed to yield improved outcomes for septic patients [11]. These disappointing results seem to be a consequence of inappropriately applying data obtained from animal studies or inadequate design of clinical trials. In some experiments, animals were infused with large doses of bacteria or bacterial products, which, as expected, induced a brisk inflammatory response. The factors contributing to this inflammation were found to be directly responsible for the death of experimental animals. However, the direct translation of results obtained from animal studies to the clinic led to the premature conclusion that anti-inflammatory treatment would be of benefit to all septic patients, regardless of the severity of their symptoms. This prediction did not take into account the fact that the character of sepsis-associated inflammatory reactions evolves with time and that the intense, dysregulated inflammation observed in severe sepsis is absent from patients with less pronounced symptoms. Furthermore, inhibiting inflammatory reactions in the early phases of sepsis can, in fact, reduce the host's capability to cope with the invading pathogens [15]. Obviously, the simple hypothesis that an excessive inflammatory reaction is the main factor responsible for an adverse outcome in sepsis must be reconsidered.
The pathways of complement activation
The complement system is an important contributor to the inflammatory reaction that occurs during sepsis. In mammals, the complement system has three well-characterized initial pathways of activation: the classical, lectin and alternative pathways. Although traditionally complement activation is attributed to the presence of invading pathogens, this process can also occur as a response to tissue damage. Therefore, complement can be currently viewed as an alarm system, which is capable of recognizing structures (danger-associated molecular patterns [DAMPs]) associated with a risk of the disturbance of homeostasis of either infectious or non-infectious origin. The classical pathway is activated by antibody-antigen complexes consisting of natural or immune response-elicited immunoglobulins bound to multivalent antigens on the surfaces of pathogens or altered host cells. The complement protein complex C1 (consisting of C1q with two molecules of C1 r and two molecules of C1s) binds to antibodies within these immune complexes, and this binding stimulates the C1s-mediated cleavage of the C4 complement component to C4a and C4b. In turn, C4b binds to pathogen or cell surfaces. The cleavage of C4 by C1s also exposes the binding site for C2, which, once bound, is also cleaved by C1s. These initial steps lead to the formation of the C3 convertase, C4b2a, on pathogen or host cell surfaces [16]. C1q is a pattern recognition receptor that is capable of distinguishing between self and non-self antigens through association with pathogen-associated molecular patterns (PAMPs) [17]. In addition, C1q serves as sensor of danger of non-infectious origin by binding to fragments of cellular or subcellular membranes (e.g. mito-chondrial membranes) and other modified host proteins and phospholipids [18–21]. Furthermore, C1q can activate the classical pathway through its interaction with pentraxin pattern recognition receptors, such as C-reactive protein (CRP) and serum amyloid protein (SAP) [22]. C3 convertase composed of C4b and C2a fragments is also generated as a result of complement activation through the lectin pathway. This pathway is triggered when pattern recognition receptors such as mannose-binding lectin (MBL) or ficolins bind to PAMPs or apoptotic host cells [23]. This binding activates MBL-associated serine proteases (MASPs), which are known to exist in three forms (MASP1, MASP2 and MASP3). Like C1s, MASP2 cleaves C4, leading to the formation of C3 convertase (MASP1 has also recently been shown to promote C4 cleavage through activation of MASP2 [24]). The convertases formed by the classical or lectin pathways cleave C3, the central component of the complement system, into the anaphylatoxin C3a and the opsonin C3b. The resulting C3b binds to the C4b2a complex, creating the C5 convertase (C4b2a3b), which then cleaves C5 into C5a (a potent anaphylatoxin) and C5b [25].
In evolutionary terms, the alternative pathway is considered to be the oldest pathway of complement activation. Activation of complement through this pathway leads to the formation of a C3 convertase that is significantly different from those formed via the classical and lectin pathways. The internal thioester of C3 is spontaneously hydrolysed (referred to as the ‘tickover’ of C3) at a slow rate, leading to the formation of C3(H20), a conformationally altered form of C3. The binding of C3(H20) to the complement protein factor B changes the conformation of factor B, making it susceptible to cleavage by the constitutively active serum protease factor D into Ba and Bb fragments. These reactions result in the formation of the alternative pathway C3 convertase (C3(H20)Bb). Like the C4b2a convertase, this complex cleaves C3 into C3a and C3b, with small amounts of C3b binding to the hydroxyl or amino groups on susceptible surfaces; several studies show spontaneous C3b deposition on microorganisms and tumour cells (reviewed in ref. [26]). Surface-bound C3b can bind directly to factor B, and the factor B-C3b complexes can then be cleaved by factor D to form the alternative pathway C3 convertase (C3bBb). This ‘second’ C3 convertase represents an ‘amplification loop’ that results in further cleavage of C3, which may augment the process of complement activation induced by the classical or lectin pathways. Properdin (P) stabilizes the C3bBb convertase, and additional molecules of C3b resulting from C3 cleavage bind to this enzymatic complex, forming the alternative pathway C5 convertase, C3b3bBbR Like the C5 convertase produced by the classical or lectin pathways, the alternative pathway C5 convertase can cleave C5 to yield C5a and C5b fragments [27].
Cleavage of C5 by the various convertases and the binding of C5b to C6 begin the downstream terminal pathway of complement. C5b6 binds to C7, creating an amphiphilic complex that can be incorporated into the lipid bilayer of cell membranes. One C8 molecule is bound by one C5b-7 complex, which then binds one or more C9 molecules. The resulting C5b-9 complex, or membrane attack complex (MAC), creates a physical pore in the membrane that results in leakage and cell activation (at sublethal doses) or cell lysis [5]. Interestingly, these downstream events do not occur (i.e. no MAC is formed) when complement is activated by CRP. It is assumed that ligands bound by CRP, such as cell wall components of Streptococcus pneumonia[28], are opsonized by C3 fragments generated by CRP-induced complement activation and are targeted for phagocytosis [29].
Many recently published studies have demonstrated that complement activation can also occur through mechanisms that differ in several aspects from the traditionally recognized pathways of complement activation. For example, a new immunoglobulin-independent mechanism for activation of the classical pathway during S. pneumoniae infection has been described. Complement activation through this mechanism is a result of C1q binding to the C-type lectin SIGN-R1 [30]. This is the first known example of a cell-surface lectin directly initiating the classical pathway. SIGN-R1 is expressed at high levels by macrophages of the spleen marginal zone and lymph nodes [31, 32] and is the principal receptor for the S. pneumoniae capsular pneumococcal polysaccharide (CPS) [33, 34]. Mice lacking SIGN-R1 are more susceptible to pneumococcal septicemia, have deficits in C3 catabolism when challenged with S. pneumoniae or CPS, and lack proper localization of CPS to follicular dendritic cells (FDCs) in the follicles of the white pulp of the spleen [30, 33, 34]. It has also been shown that CPS does not bind to FDCs in C3-deficient mice, suggesting that both SIGN-R1 and C3 are necessary for CPS binding to FDCs. Thus, it appears that SIGN-R1 contributes to C3 fixation and, therefore, to the ability of the spleen to defend the host against certain encapsulated pathogens [30].
It has also recently been shown that properdin is able to directly activate the alternative pathway of complement. Individuals with properdin deficiency are more susceptible to infection by Neisseria and suffer mortality rates of 43–65% from resulting meningococcal disease. Properdin was shown to bind directly to C3b in vitro, with formation of the C3bBbP convertase after treatment with factor B and factor D [35]. It is therefore possible that properdin may initiate assembly of the alternative pathway convertase. Indeed, properdin-treated N. gonhorrheae bound C3b and formed the alternative pathway convertase in the presence of factor B and factor D [36]. This binding was likely through interaction with Neisseria lipo-oligosaccharide (LOS), a component of lipopolysaccharide (LPS) found on other bacteria such as E. coli. However, the LOS component is normally masked in LPS; thus, properdin does not bind to LPS-expressing bacteria. When E. coli strains that differed in their ability to bind properdin, due to their various mutant forms of LPS, were tested, it was seen that those with stronger properdin binding had faster complement activation (including C3b deposition and conversion to iC3b) through the alternative pathway [36]. Similarly, LPS from different types of bacteria had various abilities to bind to human properdin. In another study, it was observed that alternative pathway complement activation in sera from properdin-deficient mice was not induced by LPS from Salmonella typhosa, Salmonella minnesota (S) or E. coli[37]. Interestingly, both LOS and LPS were unable to induce complement activation in properdin-deficient sera. However, injection of LOS caused systemic complement activation in wild-type but not properdin-deficient mice, while LPS-induced systemic complement activation was still seen (though at reduced levels) in properdin-deficient animals. Thus, LOS appears to activate the alternative pathway in vivo, likely through interaction with properdin, whereas LPS activates complement through both alternative pathway-dependent and -independent mechanisms. Finally, recent work has demonstrated that neutrophil-derived properdin binds to early apoptotic T cells and initiates C3b deposition through alternative pathway activation. This deposition facilitates phagocytosis by CR3-bearing cells. Properdin-tagged T cells can also be taken up by phagocytes without prior complement activation, through the direct interaction of properdin with the phagocytic cells [38]. All of these studies point to an important role for properdin not only in supporting alternative pathway activation of complement by stabilizing the convertase, but also in directly activating this pathway in response to some pathogens.
A growing body of research has also suggested that the complement cascade can be initiated by factors that contribute to hemostasis such as platelets and coagulation or fibrynolytic factors [39–41]. Thrombin has been shown to indirectly induce complement activation in rabbits via the classical pathway by activating platelets [42–44]. This mechanism may be important for the pathogenesis of sepsis, because platelets are activated during sepsis; furthermore, it is known that once platelets are activated, they release a serine/threonine Mn2+/Ca2+/Mg2+-dependent protein kinase (likely a casein kinase type I) that phosphorylates residues of the C3d region of C3 [45]. Thus, both C3 and several of its degradation fragments (C3b, iC3b and C3dg) are phosphorylated. The phosphorylation of C3b prevents its cleavage into iC3b by Factor I, resulting in prolonged complement activation. Therefore, parallel activation of complement and platelets during sepsis can lead to phosphorylation of activated complement components, a modification that may be important for regulating their activity [45].
Activation of complement can occur as a result of a direct cleavage of C3 or C5, without the involvement of convertases, through what has recently been termed the extrinsic protease pathway [5, 46]. Various in vitro studies have indicated that factors related to the kinin and coagulation cascades, as well as to fibrinolysis, are able to cleave complement proteins. Kallikrein isolated from rabbit plasma can cleave C5, resulting in the generation of active C5a, which induces neutrophil chemotaxis [47]. Plasmin cleaves iC3b to C3c and C3dg in vitro[48]. In addition, the Hageman factor fragment is capable of inducing complement activation through direct interaction with C1, which activates the C1r and C1s subunits [21]. Incubation of thrombin with C5 leads to C5a generation [49], and thrombin is also known to stimulate the cleavage of C5 into C5a in vivo[50]. During acute lung inflammation, C5a is produced in C3-deficient mice, which lack C3 cleavage products (i.e. C3b) [50]. It has also been shown that in the absence of C3, thrombin becomes the dominant enzyme during the inflammatory process that generates biologically active C5a. Thus, in addition to playing an indirect role in regulating the complement pathway through platelet activation, thrombin can directly cleave C5 to initiate the downstream terminal pathway of complement.
The various mechanisms of complement activation described above are summarized in Fig.1.
Figure 1.
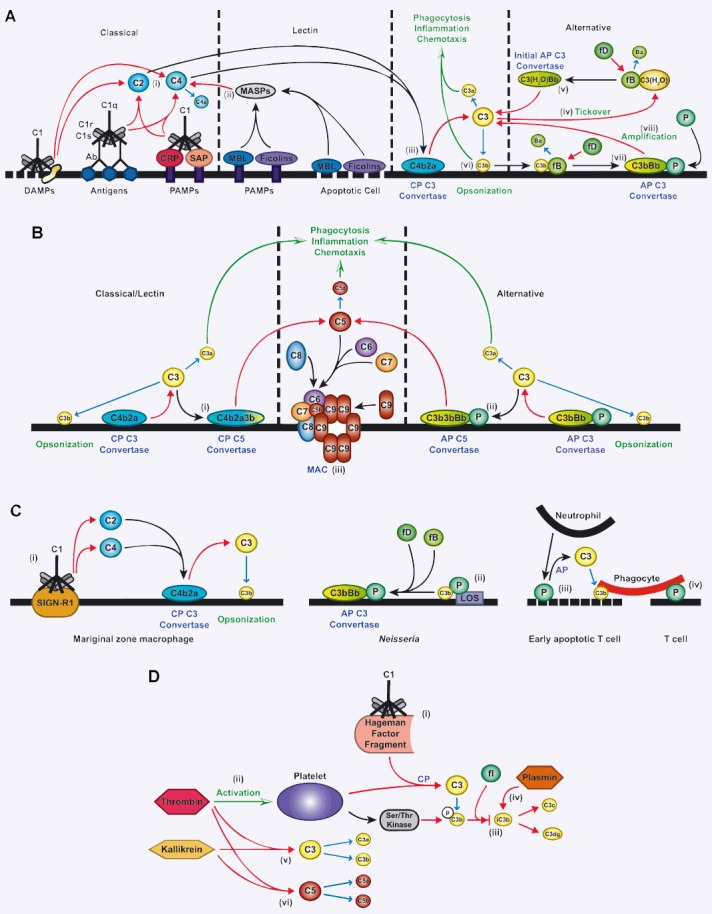
Activation of the complement system. (A) C1 can bind to various factors to initiate the classical pathway (CP). These include, most commonly antibody-antigen complexes, but also danger-associated molecular patterns (DAMPs) such as membrane fragments and proteins associated with tissue damage. C1 also binds to C-reactive protein (CRP) and serum amyloid protein (SAP), which recognize pathogen-associated molecular patterns (PAMPs) present on the surfaces of many pathogens. Binding by the C1 q subunit of C1 activates C1 r and C1 s, which results in the cleavage of C2 and C4 by C1s (i). Binding of mannose-binding lectin (MBL) or ficolins to PAMPs or apoptotic host cells activates MBL-associated serine proteases (MASPs), which cleave C4 (ii). The result of activation through either pathway is that C4a is released and C2a and C4b form the CP C3 convertase on the surface of the pathogen or apoptotic cell (iii), resulting in cleavage of C3 into C3a and C3b fragments. The alternative pathway (AP) can be initiated by spontaneous hydrolysis of C3 (‘tickover’) to form C3(H20) (iv). C3(H20) binds to factor B (fB), which is cleaved by factor D (fD) into Ba and Bb fragments, resulting in formation of the initial AP C3 convertase (v). Like the CP convertase, the AP C3 convertase cleaves C3 into C3a and C3b. The anaphylatoxin C3a induces chemotaxis and inflammation, while some C3b binds to the cell surface (opsonization) (vi), which promotes phagocytosis by CR3-bearing cells. Surface-bound C3b binds to factor B, and the resulting complex is cleaved by factor D to form the AP C3 convertase (vii). This convertase is stabilized by the binding of properdin (P). Through an amplification loop, the AP C3 convertase cleaves more C3 to augment complement activation induced by the classical or lectin pathways (viii). (B) After C3 is cleaved by the CP C3 convertase, C3b binds to the cell surface but also can bind to the C4b2a complex to form the CP C5 convertase (i). Similarly, C3b resulting from activity of the AP C3 convertase can bind to C3bBbP to form the AP C5 convertase (ii). These convertases cleave C5, leading to the generation of C5a, which acts similarly to C3a to promote inflammation and chemotaxis, and C5b. C5b is bound by C6 and C7, which can insert into the cell membrane and bind C8. One or multiple C9 molecules then bind, resulting in formation of the membrane attack complex (MAC) (iii). (C) C1 can bind to SIGN-R1 on marginal zone macrophages to result in formation of the CP C3 convertase, and subsequent C3 cleavage (i). P can bind directly to C3b through interaction with Neisseria lipo-oligosaccharide (LOS) and, in the presence of fB and fD, can form the AP C3 convertase (ii). P from neutrophils can bind to early apoptotic T cells to activate the AP and initiate C3b deposition, facilitating phagocytosis (iii). Phagocytosis can also be promoted through a direct interaction of P with phagocytes (without complement activation) when it binds to T cells (iv). (D) C1 can interact with the Hageman factor fragment, which results in complement activation through the CP (i). Thrombin induces C3 cleavage through the CP through the activation of platelets (ii). Activated platelets release a serine/threonine (ser/thr) kinase that can phosphorylate (p) C3b to block its cleavage into iC3b by Factor I (fl) (iii). Plasmin can directly cleave iC3b into C3c and C3dg (iv). Similarly, kallikrein and thrombin can directly cleave both C3 (v) and C5 (vi) to generate cleavage products.
The role of the complement system in the pathogenesis of sepsis
At first glance, the role of complement in sepsis pathogenesis might appear ambiguous or paradoxical: On the one hand, C3 deficiency, which eliminates most complement effector functions, clearly increases sepsis-associated mortality in animals [51–53]; these studies have emphasized the importance of complement as a defence mechanism against invading microbes. Conversely, other data have indicated that inhibition of C5a signalling improves the survival of experimental animals [54]. This apparent inconsistency between various studies may actually be an indication of the diversity of complement functions during the development and progression of sepsis. During the early stages of widespread bacterial infections, complement's pro-inflammatory and antimicrobial properties are critical for protecting the host from the detrimental effects of an uncontrolled spread of microbes, whereas in the later stages of sepsis development, C5a, in concert with cytokines, contributes to the development of multi-organ failure and circulatory insufficiency.
The complement system, which was originally viewed as an arm of humoural immunity, is now perceived as a central constituent of innate immunity, defending the host against infections, orchestrating inflammatory responses and connecting the innate and adaptive immune responses [5]. This broad spectrum of complement activities, together with the abundance of complement proteins in the plasma, enables this system to cope with local and systemic infections. In addition to plasma proteins that interact with various cells and mediators of the immune system, several membrane-bound regulators and receptors constitute an efficient regulatory module of the complement system that is designed to limit the activation of complement to pathogen surfaces or altered host cells [55].
The anti-microbial properties of complement can be divided into three distinct categories: (i) opsonization and subsequent killing of microorganism by phagocytes, (ii) lysis of pathogens (Neisseria species) and (iii') coordination of inflammatory events associated with the response to infection [4]. Opsonization involves the coating of bacterial surfaces with complement proteins such as C3b and iC3b. These cleavage products of C3 are covalently bound to the pathogen surfaces and act as ligands for receptors expressed by phagocytes. Engagement of these receptors with their ligands significantly facilitates the phagocytosis and killing of bacteria by neutrophils and macrophages [25]. Various defects in opsonization are known to contribute to an increased susceptibility to infections caused by pyogenic bacteria such as
Haemophilus influenzae and S. pneumoniae[25]. Lysis of pathogens occurs as a result of sequential activation of complement proteins on pathogen surfaces, forming the MAC, which then creates pores in the bacterial cell wall and ultimately leads to bacterial lysis [5]. Although direct lysis of pathogens by MAC is a rare form of defence against invading pathogens, it has been shown that inherited deficiencies in components of the MAC are associated with an increased susceptibility to Neisserial diseases, particularly Neisseria meningitides[56]. Complement-mediated lysis is a major mechanism for neutralizing Neisseria species, which are capable of intracellular survival.
The complement system plays an invaluable role in promoting and coordinating the inflammatory process that is triggered in response to pathogens. The anaphylatoxins C3a and C5a are actively involved in the regulation of various critical events during an inflammatory response, such as changes in vascular flow and blood vessel calibre, increased vascular permeability, and leukocyte extravasation and chemotaxis [5]. These processes are essential for recruiting and activating the cells that are involved in the innate immune response, including neutrophils, monocytes and macrophages [57]. Anaphylatoxins not only directly coordinate cellular inflammatory responses by binding to the reciprocal receptors expressed by peripheral blood leukocytes or macrophages but also provide coordination in an indirect manner by controlling cytokine production and secretion [5]. These cytokines, in turn, are capable of potentiating inflammation.
The essential goal of inflammatory reactions is to eliminate hazardous factors, which in the case of infection are microbes [57]. The contribution of the complement system is necessary to achieving this goal. When invading pathogens are successfully eliminated through innate and/or adaptive immune responses, acute inflammation subsides. Regulatory complement proteins can immediately shut down the activation process in the absence of threatening infection. However, under unfavourable circumstances, pathogens can escape surveillance by the immune system. In these situations, the ‘frustrated’ inflammatory response continues its efforts to neutralize the danger, leading to the destruction of host tissue and further exacerbation of inflammation [58]. This large-scale inflammatory response, currently referred to as SIRS, is a hallmark of sepsis. When this response occurs, a steady activation of complement and the release of other inflammatory mediators, combined with an inability to destroy pathogens, creates a vicious circle leading ultimately to multiple organ dysfunction and immunosuppression [59]. Disturbances in various organ and circulatory system functions accompany severe sepsis. At this stage, the decline in the overall condition of the patient and a decrease in the patient's ability to cope with the existing infection together facilitate the spread of microbes, contributing to a worsening of the individual's clinical status. With severe sepsis, the prognosis is poor, and the risk of septic shock becomes higher.
A crucial requirement for successful sepsis therapy is the capacity to break this vicious cycle of progressive infection and exacerbating inflammation. The most effective way to do so is through elimination of the infectious agent. In many cases, therapy based on the empiric selection of antibiotics is sufficient to prevent severe sepsis complications. However, early surgical intervention is also critical in controlling and eliminating the focus of infection if sepsis is related to perforation or obstruction of the gastrointestinal, biliary or urinary tract, or if the abscess, which is the source of pathogens, requires drainage. Extensive clinical experience has confirmed that early intervention is essential for successful therapy [9]. In order to have the best chance of curing septic patients, appropriate treatment should be introduced before severe deregulation of the inflammatory response develops.
Currently, therapeutic interventions targeting the complex network of inflammatory and immune responses appear to be a risky approach, given that inhibition of these responses may significantly impair the ability of the host to naturally cope with infections [15]. However, the combination of appropriate antibiotic therapy introduced early during sepsis combined with inhibition of inflammation in the late stages of the disease process appears to be a promising mode of treatment of septic patients in the near future.
Aetiology-dependent mechanisms of complement activation in sepsis
Although various factors influence the activation of complement during sepsis, the specific aetiology of the syndrome in a particular individual has a decisive impact on the initiation of the complement cascade. In the last three decades, Gram-positive microorganisms have become the leading etiological factors in sepsis, with Staphylococcus aureus and S. pneumoniae being the most commonly isolated pathogens. In the group of Gram-negative bacteria, E. coli, Klebsiella spp and Pseudomonas aeruginosa are unquestionably the leaders [8].
Recently published data from human studies have indicated that complement activation is initiated differently by Gram-positive and Gram-negative bacteria that cause bacterial septicemia [60]. In patients with confirmed Gram-positive bacteria-induced sepsis, there is a significant consumption of C1q, but not MBL, in the acute phase of the disease; the opposite pattern is seen in sepsis caused by Gram-negative bacteria. These data suggest that the classical pathway of complement activation plays an essential role in the sepsis induced by Gram-positive pathogens. C1q can bind directly to bacterial surfaces or to immune complexes formed by bacterial antigens and antibodies. In the case of Gram-negative pathogens, the activation of complement is induced in sepsis through the binding of MBL (the lectin pathway) predominantly to LPS, which is a component of the outer cell wall of Gram-negative bacteria [60].
This relatively clear picture becomes obscured when we consider other reports relating to the mechanisms of complement activation by particular species of bacteria. Gram-positive S. aureus, which has developed numerous mechanisms to evade complement attack [4], has been reported to activate the complement cascade through the lectin pathway, with no involvement of alternative pathway amplification [61]. In line with these findings, it has also been reported that L-ficolin, which initiates the lectin pathway, binds to lipoteichoic acid (LTA), a cell wall component found in a majority of Gram-positive bacteria [62]. Other studies, however, have suggested that the lectin pathway preferentially facilitates complement-mediated opsonophagocytosis of the fungus Candida albicans, but not bacteria [63].
Both Gram-positive bacterial strains (e.g. S. aureus and S. pneumoniae) and Gram-negative E. coli have been shown to activate the C1q-dependent classical pathway [63]. In accordance with these findings, the classical pathway of complement activation has been shown to be essential for innate immune responses to S. pneumoniae. Interestingly, studies using various complement-deficient mouse strains and mice lacking secretory IgM have demonstrated that the proportion of a population of S. pneumoniae bound by C3 depends on the classical pathway, whereas the intensity of this binding is alternative pathway-dependent.
The classical pathway of complement activation is initiated by the binding of natural IgM to S. pneumoniae surfaces. The lack of IgM in deficient mice results in rapidly progressing sepsis and alterations in macrophage function [64]. However, it appears that an antibody-independent mechanism of classical pathway activation by S. pneumoniae is equally important. As described earlier, it has recently been reported that the direct binding of C1q to SIGN-R1, a C-type lectin that is an uptake receptor expressed at high levels by spleen marginal zone and lymph node macrophages, contributes significantly to complement activation and opsonization of S. pneumoniae by C3 cleavage products [30].
A detailed discussion of the large number of other studies investigating the mechanisms of complement activation by various pathogens goes far beyond the scope of this review. However, the overall picture that emerges from these investigations indicates the enormous diversity in the mechanisms of complement activation that contribute to the response to infection. It appears that the simple assignment of a single pathway to particular pathogen is an inappropriate approach that can lead to an oversimplification of this process. In addition, activation of complement can also be enhanced in a pathogen-independent manner by acute phase proteins, whose levels increase during sepsis. For example, as mentioned earlier, the acute phase protein CRP can bind to C1q, and this interaction triggers the classical pathway [29].
Sepsis-associated coagulopathy and complement activation
Coagulation is generally activated during the course of sepsis [65]. Sepsis-associated coagulopathy can occur in various forms, including disseminated intravascular coagulation (DIC), a particularly severe complication that significantly and adversely affects the prognosis in sepsis [66]. DIC is a condition in which an increased tendency toward coagulation leads to the formation of multiple thrombi in the microcirculation. This massive formation of thrombi is responsible for the consumption of clotting factors, which in turn leads to haemorrhagic diathesis [67]. Clinical studies have suggested that DIC contributes to the development of multi-organ dysfunction, ultimately increasing mortality in patients with sepsis. In addition, treatment regimens that attenuate coagulation have been postulated to improve overall survival [68].
Increased thrombogenicity in sepsis is a result of the upregulation of tissue factor (TF) expression on circulating leukocytes and endothelial cells in the blood. Under normal physiological conditions, contact between TF and blood clotting factors is largely prevented by anatomical barriers, and leukocytes and endothelial cells do not express TF. However, when these cells are stimulated by inflammatory mediators, including complement effectors, they become key contributors to the increased tendency toward clotting in various clinical conditions that are associated with inflammation [39]. Several studies have demonstrated that complement activation can be triggered by the activation of the coagulation or contact systems [40], as described in the previous section of this review. Thus, cross-talk between the coagulation and complement systems represents another way of amplifying complement activation in sepsis.
Concluding remarks
The existence of rare human complement deficiencies and the results of various animal studies have established an essential role for the complement system in the defence against infections, including those that may eventually lead to sepsis. Complement creates several barriers that prevent the spread of microorganisms and contribute to their clearance. At virtually every step in the development of the infectious process, including the activation of adaptive immunity, complement proteins are engaged in the battle against pathogens. These multiple tasks require prompt and efficient activation of the complement cascade in coordination with other immune mechanisms, resulting in the generation of a number of effector molecules that participate in the response to infection. This review has highlighted how such activation is achieved by multiple pathways acting in concert to ensure both the development of a response that is proportional to the scale of danger and a timely delivery of complement effectors to the sites of infection. It also appears that in addition to the previously well-studied pathways of complement activation, various newly discovered mechanisms can either initiate or amplify the activation of the complement cascade in sepsis.
Acknowledgments
We thank Dr. D. McClellan for her excellent editorial assistance. This work was supported by the National Institutes of Health grants AI30040: AI068730and AI071028.
References
- 1.Angus DC, Linde-Zwirble WT, Lidicker J, Clermont G, Carcillo J, Pinsky MR. Epidemiology of severe sepsis in the United States: analysis of incidence, outcome, and associated costs of care. Crlt Care Med. 2001;29:1303–10. doi: 10.1097/00003246-200107000-00002. [DOI] [PubMed] [Google Scholar]
- 2.Rehm SJ. Staphylococcus aureus: the new adventures of a legendary pathogen. Cleve Clin J Med. 2008;75:177–92. doi: 10.3949/ccjm.75.3.177. [DOI] [PubMed] [Google Scholar]
- 3.Wheeler AP, Bernard GR. Treating patients with severe sepsis. N Engl J Med. 1999;340:207–14. doi: 10.1056/NEJM199901213400307. [DOI] [PubMed] [Google Scholar]
- 4.Lambris JD, Ricklin D, Geisbrecht BV. Complement evasion by human pathogens. Nat Rev Microbiol. 2008;6:132–42. doi: 10.1038/nrmicro1824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Markiewski MM, Lambris JD. The role of complement in inflammatory diseases from behind the scenes into the spotlight. Ain J Pathol. 2007;171:715–27. doi: 10.2353/ajpath.2007.070166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Guo RF, Ward PA. Role of C5a in inflammatory responses. Annu Rev Immunol. 2005;23:821–52. doi: 10.1146/annurev.immunol.23.021704.115835. [DOI] [PubMed] [Google Scholar]
- 7.Bone RC, Balk RA, Cerra FB, Delllnger RP, Fein AM, Knaus WA, Scheln RM, Slbbald WJ. Definitions for sepsis and organ failure and guidelines for the use of innovative therapies in sepsis. The ACCP/SCCM Consensus Conference Committee. American College of Chest Physicians/Society of Critical Care Medicine. Chest. 1992;101:1644–55. doi: 10.1378/chest.101.6.1644. [DOI] [PubMed] [Google Scholar]
- 8.Martin GS, Mannlno DM, Eaton S, Moss M. The epidemiology of sepsis in the United States from 1979 through 2000. N Engl J Med. 2003;348:1546–54. doi: 10.1056/NEJMoa022139. [DOI] [PubMed] [Google Scholar]
- 9.O'Brien JM, Jr, All NA, Aberegg SK, Abraham E. Sepsis. Am J Med. 2007;120:1012–22. doi: 10.1016/j.amjmed.2007.01.035. [DOI] [PubMed] [Google Scholar]
- 10.Alberti C, Brun-Bulsson C, Burchardl H, Martin C, Goodman S, Artlgas A, Slclgnano A, Palazzo M, Moreno R, Boulme R, Lepage E, Le GR. Epidemiology of sepsis and infection in ICU patients from an international multicentre cohort study. Intensive Care Med. 2002;28:108–21. doi: 10.1007/s00134-001-1143-z. [DOI] [PubMed] [Google Scholar]
- 11.van der PT, Opal SM. Host-pathogen interactions in sepsis. Lancet Infect DIs. 2008;8:32–43. doi: 10.1016/S1473-3099(07)70265-7. [DOI] [PubMed] [Google Scholar]
- 12.Beutler B, Mllsark IW, Ceraml AC. Passive immunization against cachectin/tumor necrosis factor protects mice from lethal effect of endotoxin. Science. 1985;229:869–71. doi: 10.1126/science.3895437. [DOI] [PubMed] [Google Scholar]
- 13.Tracey KJ, Fong Y, Hesse DG, Manogue KR, Lee AT, Kuo GC, Lowry SF, Ceraml A. Anti-cachectin/TNF monoclonal antibodies prevent septic shock during lethal bacteraemia. Nature. 1987;330:662–4. doi: 10.1038/330662a0. [DOI] [PubMed] [Google Scholar]
- 14.Ohlsson K, Bjork P, Bergenfeldt M, Hageman R, Thompson RC. Intelleukin-1 receptor antagonist reduces mortality from endotoxin shock. Nature. 1990;348:550–2. doi: 10.1038/348550a0. [DOI] [PubMed] [Google Scholar]
- 15.Elchacker PQ, Parent C, Kalll A, Esposlto C, Cul X, Banks SM, Gerstenberger EP, Fltz Y, Danner RL, Natanson C. Risk and the efficacy of antiinflammatory agents: retrospective and confirmatory studies of sepsis. Am J Respir Crit Care Med. 2002;166:1197–205. doi: 10.1164/rccm.200204-302OC. [DOI] [PubMed] [Google Scholar]
- 16.Mollnes TE, Song WC, Lambris JD. Complement in inflammatory tissue damage and disease. Trends Immunol. 2002;23:61–4. doi: 10.1016/s1471-4906(01)02129-9. [DOI] [PubMed] [Google Scholar]
- 17.Carroll MC. The complement system in regulation of adaptive immunity. Nat Immunol. 2004;5:981–6. doi: 10.1038/ni1113. [DOI] [PubMed] [Google Scholar]
- 18.Storrs SB, Kolb WP, Olson MS. C1q binding and C1 activation by various isolated cellular membranes. J Immunol. 1983;131:416–22. [PubMed] [Google Scholar]
- 19.Peitsch MC, Tschopp J, Kress A, Isliker H. Antibody-independent activation of the complement system by mitochondria is mediated by cardiolipin. Biochem J. 1988;249:495–500. doi: 10.1042/bj2490495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kovacsovics T, Tschopp J, Kress A, Isliker H. Antibody-independent activation of C1, the first component of complement, by cardiolipin. J Immunol. 1985;135:2695–700. [PubMed] [Google Scholar]
- 21.Ghebrehlwet B, Randazzo BP, Dunn JT, Sllverberg M, Kaplan AP. Mechanisms of activation of the classical pathway of complement by Hageman factor fragment. J Clin Invest. 1983;71:1450–6. doi: 10.1172/JCI110898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Garlanda C, Bottazzl B, Bastone A, Mantovani A. Pentraxins at the crossroads between innate immunity, inflammation, matrix deposition, and female fertility. Annu Rev Immunol. 2005;23:337–66. doi: 10.1146/annurev.immunol.23.021704.115756. [DOI] [PubMed] [Google Scholar]
- 23.Ogden CA, deCathellneau A, Hoffmann PR, Bratton D, Ghebrehlwet B, Fatlok VA, Henson PM. C1q and mannose binding lectin engagement of cell surface calretic-ulin and CD91 initiates macropinocytosis and uptake of apoptotic cells. J Exp Med. 2001;194:781–95. doi: 10.1084/jem.194.6.781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Takahashl M, Iwakl D, Kanno K, Ishitla Y, Xlong J, Matsushita M, Endo Y, Mi lira S, Ishii N, Sugamura K, Fujita T. Mannose-binding lectin (MBL)-associated serine protease (MASP)-1 contributes to activation of the lectin complement pathway. J Immunol. 2008;180:6132–8. doi: 10.4049/jimmunol.180.9.6132. [DOI] [PubMed] [Google Scholar]
- 25.Walport MJ. Complement. First of two parts. N Engl J Med. 2001;344:1058–66. doi: 10.1056/NEJM200104053441406. [DOI] [PubMed] [Google Scholar]
- 26.Kohl J. The role of complement in danger sensing and transmission. Immunol Res. 2006;34:157–76. doi: 10.1385/IR:34:2:157. [DOI] [PubMed] [Google Scholar]
- 27.Thurman JM, Holers VM. The central role of the alternative complement pathway in human disease. J Immunol. 2006;176:1305–10. doi: 10.4049/jimmunol.176.3.1305. [DOI] [PubMed] [Google Scholar]
- 28.Szalal AJ, Agrawal A, Greenhough TJ, Volanakls JE. C-reactive protein: structural biology, gene expression, and host defense function. Immunol Res. 1997;16:127–36. doi: 10.1007/BF02786357. [DOI] [PubMed] [Google Scholar]
- 29.Agrawal A. CRP after 2004. Mol Immunol. 2005;42:927–30. doi: 10.1016/j.molimm.2004.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kang YS, Do Y, Lee HK, Park SH, Cheong C, Lynch RM, Loeffler JM, Stelnman RM, Park CG. A dominant complement fixation pathway for pneumococcal polysaccharides initiated by SIGN-R1 interacting with C1q. Cell. 2006;125:47–58. doi: 10.1016/j.cell.2006.01.046. [DOI] [PubMed] [Google Scholar]
- 31.Geljtenbeek TB, Groot PC, Nolte MA, van Vllet SJ, Gangaram-Panday ST, van Duljnhoven GC, Kraal G, van Oosterhout AJ, van KY. Marginal zone macrophages express a murine homologue of DC-SIGN that captures blood-borne antigens in vivo. Blood. 2002;100:2908–16. doi: 10.1182/blood-2002-04-1044. [DOI] [PubMed] [Google Scholar]
- 32.Kang YS, Yamazakl S, lyoda T, Pack M, Bruenlng SA, Kim JY, Takahara K, Inaba K, Stelnman RM, Park CG. SIGN-R1 a novel C-type lectin expressed by marginal zone macrophages in spleen, mediates uptake of the polysaccharide dextran. Int Immunol. 2003;15:177–86. doi: 10.1093/intimm/dxg019. [DOI] [PubMed] [Google Scholar]
- 33.Kang YS, Kim JY, Bruenlng SA, Pack M, Charalambous A, Prltsker A, Moran TM, Loeffler JM, Stelnman RM, Park CG. The C-type lectin SIGN-R1 mediates uptake of the capsular polysaccharide of Streptococcus pneumoniae in the marginal zone of mouse spleen. Proc Natl Acad Sci USA. 2004;101:215–20. doi: 10.1073/pnas.0307124101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lanoue A, Clatworthy MR, Smith P, Green S, Townsend MJ, John HE, Smith KG, Fallen PG, McKenzle AN. SIGN-R1 contributes to protection against lethal pneumococcal infection in mice. J Exp Med. 2004;200:1383–93. doi: 10.1084/jem.20040795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hourcade DE. The role of properdin in the assembly of the alternative pathway C3 convertases of complement. J Biol Chem. 2006;281:2128–32. doi: 10.1074/jbc.M508928200. [DOI] [PubMed] [Google Scholar]
- 36.Spltzer D, Mitchell LM, Atkinson JP, Hourcade DE. Properdin can initiate complement activation by binding specific target surfaces and providing a platform for de novo convertase assembly. J Immunol. 2007;179:2600–8. doi: 10.4049/jimmunol.179.4.2600. [DOI] [PubMed] [Google Scholar]
- 37.Klmura Y, Mlwa T, Zhou L, Song WC. Activator-specific requirement of properdin in the initiation and amplification of the alternative pathway complement. Blood. 2008;111:732–40. doi: 10.1182/blood-2007-05-089821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kemper C, Mitchell LM, Zhang L, Hourcade DE. The complement protein properdin binds apoptotic T cells and promotes complement activation and phagocytosis. Proc Natl Acad Sci USA. 2008;105:9023–8. doi: 10.1073/pnas.0801015105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Marklewskl MM, Nllsson B, Ekdahl KN, Mollnes TE, Lambrls JD. Complement and coagulation: strangers or partners in crime? Trends Immunol. 2007;28:184–92. doi: 10.1016/j.it.2007.02.006. [DOI] [PubMed] [Google Scholar]
- 40.Umme A, Rittirsch D, Fllerl MA, Bruckner U, Klos A, Gebhard F, Lambrls JD, Huber-Lang M. Interaction between the coagulation and complement system. Adv Exp Med Biol. 2008 doi: 10.1007/978-0-387-78952-1_6. ; (in press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hamad OA, Ekdahl KN, Nllsson PH, Andersson J, Magottl P, Lambrls JD, Nilsson B. Complement activation triggered by chondroitin sulfate released by thrombin receptor-activated platelets. J Thromb Haemost. 2008;6:1413–21. doi: 10.1111/j.1538-7836.2008.03034.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kalowskl S, Howes EL, Jr, Margaretten W, McKay DG. Effects of intravascular clotting on the activation of the complement system: the role of the platelet. Am J Pathol. 1975;78:525–36. [PMC free article] [PubMed] [Google Scholar]
- 43.del CI, Cruz MA, Zhang H, Lopez JA, Afshar-Kharghan V. Platelet activation leads to activation and propagation of the complement system. J Exp Med. 2005;201:871–9. doi: 10.1084/jem.20041497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Peerschke El, Yin W, Grlgg SE, Ghebrehlwet B. Blood platelets activate the classical pathway of human complement. J Thromb Haemost. 2006;4:2035–42. doi: 10.1111/j.1538-7836.2006.02065.x. [DOI] [PubMed] [Google Scholar]
- 45.Ekdahl KN, Nilsson B. Phosphorylation of complement component C3 and C3 fragments by a human platelet protein kinase. Inhibition of factor l-mediated cleavage of C3b. J Immunol. 1995;154:6502–10. [PubMed] [Google Scholar]
- 46.Ricklin D, Lambris JD. Complement-targeted therapeutics. Nat Biotechnol. 2007;25:1265–75. doi: 10.1038/nbt1342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wiggins RC, Giclas PC, Henson PM. Chemotactic activity generated from the fifth component of complement by plasma kallikrein of the rabbit. J Exp Med. 1981;153:1391–404. doi: 10.1084/jem.153.6.1391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lachmann PJ, Pangburn MK, Oldroyd RG. Breakdown of C3 after complement activation Identification of a new fragment, C3g, using monoclonal antibodies. J Exp Med. 1982;156:205–16. doi: 10.1084/jem.156.1.205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Spath P, Gabl F. Critical role of the conversion of the third complement component C3 (beta 1 C/beta 1A) for its immunochemical quantitation. Clin ChimActa. 1976;73:171–5. doi: 10.1016/0009-8981(76)90319-3. [DOI] [PubMed] [Google Scholar]
- 50.Huber-Lang M, Sarma JV, Zetoune FS, Rittirsch D, Neff TA, McGuire SR, Lambris JD, Warner RL, Flierl MA, Hoesel LM, Gebhard F, Younger JG, Drouin SM, Wetsel RA, Ward PA. Generation of C5a in the absence of C3: a new complement activation pathway. Nat Med. 2006;12:682–7. doi: 10.1038/nm1419. [DOI] [PubMed] [Google Scholar]
- 51.Quezado ZM, Hoffman WD, Winkelstein JA, Yatsiv I, Koev CA, Cork LC, Elin RJ, Eichacker PQ, Natanson C. The third component of complement protects against Escherichia coli endotoxin-induced shock and multiple organ failure. J Exp Meof. 1994;179:569–78. doi: 10.1084/jem.179.2.569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wessels MR, Butko P, Ma M, Warren HB, Lage AL, Carroll MC. Studies of group B streptococcal infection in mice deficient in complement component C3 or C4 demonstrate an essential role for complement in both innate and acquired immunity. Proc Natl Acad Sci USA. 1995;92:11490–4. doi: 10.1073/pnas.92.25.11490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Fischer MB, Prodeus AP, Nicholson-Weller A, Ma MH, Murrow J, Reid RR, Warren HB, Lage AL, Moore FDJ, Rosen FS, Carroll MC. Increased susceptibility to endotoxin shock in complement C3- and C4-deficient mice is corrected by C1 inhibitor replacement. J Immunol. 1997;159:976–82. [PubMed] [Google Scholar]
- 54.Ward PA. Role of the complement in experimental sepsis. J Leukoc Blol. 2008;83:467–70. doi: 10.1189/jlb.0607376. [DOI] [PubMed] [Google Scholar]
- 55.Kim DD, Song WC. Membrane complement regulatory proteins. Clin Immunol. 2006;118:127–36. doi: 10.1016/j.clim.2005.10.014. [DOI] [PubMed] [Google Scholar]
- 56.Ross SC, Densen P. Complement deficiency states and infection: epidemiology pathogenesis and consequences of neisse-rial and other infections in an immune deficiency. Medicine. 1984;63:243–73. [PubMed] [Google Scholar]
- 57.Nathan C. Points of control in inflammation. Nature. 2002;420:846–52. doi: 10.1038/nature01320. [DOI] [PubMed] [Google Scholar]
- 58.Cohen J. The immunopathogenesis of sepsis. Nature. 2002;420:885–91. doi: 10.1038/nature01326. [DOI] [PubMed] [Google Scholar]
- 59.Sriskandan S, Altmann DM. The immunology of sepsis. J Pathol. 2008;214:211–23. doi: 10.1002/path.2274. [DOI] [PubMed] [Google Scholar]
- 60.Dumestre-Perard C, Doerr E, Colomb MG, Loos M. Involvement of complement pathways in patients with bacterial sep-ticemia. Mol Immunol. 2007;44:1631–8. doi: 10.1016/j.molimm.2006.08.008. [DOI] [PubMed] [Google Scholar]
- 61.Neth O, Jack DL, Johnson M, Klein NJ, Turner MW. Enhancement of complement activation and opsonophagocytosis by complexes of mannose-binding lectin with mannose-binding lectin-associated serine protease after binding to Staphylococcus aureus. J Immunol. 2002;169:4430–6. doi: 10.4049/jimmunol.169.8.4430. [DOI] [PubMed] [Google Scholar]
- 62.Lynch NJ, Roscher S, Hartung T, Morath S, Matsushita M, Maennel DN, Kuraya M, Fujita T, Schwaeble WJ. L-ficolin specifically binds to lipoteichoic acid, a cell wall constituent of Gram-positive bacteria, and activates the lectin pathway of complement. J Immunol. 2004;172:1198–202. doi: 10.4049/jimmunol.172.2.1198. [DOI] [PubMed] [Google Scholar]
- 63.Brouwer N, Dolman KM, van HM, Sta M, Roos D, Kuijpers TW. Mannose-binding lectin (MBL) facilitates opsonophagocytosis of yeasts but not of bacteria despite MBL binding. J Immunol. 2008;180:4124–32. doi: 10.4049/jimmunol.180.6.4124. [DOI] [PubMed] [Google Scholar]
- 64.Brown JS, Hussell T, Gilliland SM, Holden DW, Paton JC, Ehrenstein MR, Walport MJ, Botto M. The classical pathway is the dominant complement pathway required for innate immunity to Streptococcus pneumoniae infection in mice. Proc Natl Acad Sci USA. 2002;99:16969–74. doi: 10.1073/pnas.012669199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Taylor FB, Jr, Hack E, Lupu F. Observations on complement activity in the two-stage inflammatory/hemostatic response in the baboon and human models of E. coli sepsis and endotoxemia. Adv Exp Med Blol. 2006;586:203–16. doi: 10.1007/0-387-34134-X_14. [DOI] [PubMed] [Google Scholar]
- 66.Dhainaut JF, Shorr AF, Macias WL, Kollef MJ, Levi M, Reinhart K, Nelson DR. Dynamic evolution of coagulopathy in the first day of severe sepsis: relationship with mortality and organ failure. Crit Care Med. 2005;33:341–8. doi: 10.1097/01.ccm.0000153520.31562.48. [DOI] [PubMed] [Google Scholar]
- 67.Levi M. Disseminated intravascular coagulation. Grit Care Med. 2007;35:2191–5. doi: 10.1097/01.ccm.0000281468.94108.4b. [DOI] [PubMed] [Google Scholar]
- 68.Kienast J, Juers M, Wiedermann CJ, Hoffmann JN, Ostermann H, Strauss R, Keinecke HO, Warren BL, Opal SM. Treatment effects of high-dose antithrombin without concomitant heparin in patients with severe sepsis with or without disseminated intravascular coagulation. J Thromb Haemost. 2006;4:90–7. doi: 10.1111/j.1538-7836.2005.01697.x. [DOI] [PubMed] [Google Scholar]