

Abstract
Type I cGMP-dependent protein kinase (PKG-I) mediates nitric oxide (NO) and hormone dependent smooth muscle relaxation and stimulates smooth muscle cell-specific gene expression. Expression of PKG-I in cultured smooth muscle cells depends on culture conditions and is inhibited by inflammatory cytokines such as interleukin-I and tumor necrosis factor-α, which are known to stimulate Type II NO synthase (iNOS) expression. We report here that the suppression of PKG-I protein levels in smooth muscle cells is triggered by the ubiquitin/26S proteasome pathway. Incubation of vascular smooth muscle cells with phosphodiesterase-resistant cyclic GMP analogs (e.g., 8-bromo-cGMP) decreases PKG-I protein level in a time- and concentration-dependent manner. To study this process, we tested the effects of 8-Br-cGMP on PKG-I protein level in Cos7 cells, which do not express endogenous type I PKG mRNA. 8-Br-cGMP induced the ubiquitination and down-regulation of PKG-Iα, but not PKG-Iβ. Treatment of cells with the 26S proteasome inhibitor, MG-132, increased ubiquitination of PKG. Blocking PKG-I catalytic activity using the cell-permeant specific PKG-I inhibitor, DT-2, inhibited cGMP-induced PKG-I ubiquitination and down-regulation, suggesting that PKG catalytic activity and autophosphorylation were required for suppression of PKG-I level. Mutation of the known autophosphorylation sites of PKG-Iα to alanine uncovered a specific role for autophosphorylation of serine-64 in cGMP-dependent ubiquitination and suppression of PKG-I level. The results suggest that chronic elevation of cGMP, as seen in inflammatory conditions, triggers ubiquitination and degradation of PKG-Iα in smooth muscle.
Keywords: Nitric oxide, cGMP, PKG, Phosphorylation, Smooth muscle, Gene expression, Phenotype, Ubiquitin
1. Introduction
Cyclic GMP-dependent protein kinase (PKG) is a widely expressed serine/threonine protein kinase and is an important mediator of nitric oxide (NO) and hormone signaling in smooth muscle cells (SMC), neurons, and platelets [1-6]. PKG is expressed as two gene products in eukaryotic cells: PKG-I and PKG-II. PKG-I, in turn, is expressed as two alternatively spliced variants wherein the first or second exon is spliced into the initial coding sequences to yield PKG-Iα and PKG-Iβ, respectively [7-9]. The two isoforms of PKG-I (Mr=80 kDa) are identical except for the N-terminal ~100 amino acids, which comprise the autoinhibitory, autophosphorylation and dimerization subdomains.
In SMC, only PKG-I is expressed, and both α and β isoforms of PKG-I are believed to mediate relaxation through a variety of mechanisms. These include phosphorylation of proteins that regulate intracellular calcium homeostasis or calcium sensitivity [10-12], and stimulation of the expression of several SMC-specific gene products, as demonstrated in cell culture [13-15]. PKG-I null mice predictably exhibit disruptions in smooth muscle relaxation and are hypertensive, and the viable null animals die within 5 weeks due to gastrointestinal obstruction [16]. Conditional smooth muscle-specific PKG-I null animals have also been described, but these animals do not demonstrate abnormal cardiovascular or gastrointestinal phenotypes [17,18].
The level of PKG-I protein expression in vascular SMC is variable [15,19-23]. For example, rat aortic SMC express high levels of PKG-I in primary and early-passaged cultures, but with continuing passage the levels of PKG-I decline. Furthermore, PKG-I mRNA levels in SMC and other tissues are highly variable, often do not correspond with protein expression, and are difficult to detect using standard northern blot procedures [24,25]. Recently, we reported that inflammatory cytokines such as IL-1β and TNF-α inhibit PKG-I mRNA and protein expression in primary cultures of bovine aortic SMC using real time PCR and western blotting, respectively [25]. The reduction in expression of PKG-I in vascular SMC under these inflammatory conditions may contribute to phenotypic modulation of the SMC from differentiated, contractile cells to more proliferative, secretory cells that are characteristic of inflammatory vascular lesions including those seen in atherosclerosis [13,14,26]. In other studies, NO-cGMP signaling has been shown to inhibit vascular SMC proliferation and/or stimulate vascular SMC apoptosis [19,26-30]. Obviously, a better understanding of the mechanisms regulating PKG-I expression in SMC would be of great importance in defining the role of the NO-cGMP signaling pathway in inflammatory vascular disorders.
One of the principle actions of inflammatory cytokines (e.g., IL-1β and TNF-α) on cells is the induction of Type II NO synthase (iNOS) [30-35]. Cytokine-induced iNOS expression leads to activation of NO-sensitive guanylyl cyclase, commonly known as soluble guanylyl cyclase (sGC), marked elevations in cellular cGMP in SMC [31-36], and as mentioned above, down-regulation of PKG-I expression [25]. Studies in our laboratory demonstrated that cGMP analogs decreased PKG-I both mRNA and protein levels [24]. There have been only a few studies directed towards understanding the molecular mechanisms regulating PKG-I expression in cells, including vascular SMC. Our laboratory has examined the proximal human PKG-I promoter and has determined that the principle transcription factors that regulate PKG-I mRNA expression are members of the Sp family and the Upstream Stimulatory Factor (USF) family [37,38]. Likewise, Pilz's laboratory has suggested that Kruppel-like transcription factor-4 (KLF-4) interacts with the Sp1/Sp3 sites on the PKG-I promoter and appears to be necessary for gene transcription [23]. More recent studies have reported that a reciprocal relationship may exist between the expression of sGC α and β subunits and the expression of PKG-I protein in various SMC culture systems [39].
One of the major mechanisms regulating protein turnover in eukaryotic cells is the ubiquitin-26S proteasome system. With regard to cellular protein kinases, it has been shown that the binding of phorbol myristate acetate (PMA) to protein kinase C (PKC) leads to activation of the enzyme in the short term and down-regulation of PKC expression upon longer exposures due to ubiquitination of the enzyme [40-42]. Because the ligand for PKG (i.e., cGMP or its analogs) down-regulates PKG-I expression, we hypothesize that cGMP leads to down-regulation of PKG-I via the ubiquitin-proteasome pathway. The findings presented here may have major implications not only for the regulation of the NO-cGMP signaling pathway in cells, but also for strategies to use PDE-5 inhibitors and other drugs that affect the pathway.
2. Methods and materials
2.1. Materials
Cell culture sera (fetal bovine and calf) and Dulbecco's Modified Minimal Essential Medium (DMEM) were purchased from Invitrogen-Gibco (Grand Island, NY). 8-para-cholorophenylthio-cGMP (8-pCPT-cGMP) and 8-bromo-cyclic GMP (8-Br-cGMP) were purchased from Biolog (Bremen, Germany), and DETA-NONOate was obtained from Alexis Biochemicals (San Diego, CA). Polyclonal antibody to PKG-I was acquired from StressGen Biotechnology, Inc. (Victoria, British Columbia, Canada), monoclonal antibodies for ubiquitin and antibodies for total VASP and p-VASP(S239) were purchased from Cell Signaling, Inc. (Danvers, MA), and monoclonal antibody for β-actin was obtained from Sigma-Aldrich. Secondary antibodies used in these studies were purchased from Jackson Immuno Lab (West Grove, PA). Fugene-6 transfection reagents (Roche Applied Science, Indianapolis, IN) were used for all the transient transfections. The PKG peptide inhibitor, DT-2, and control peptides were synthesized by Dostmann and colleagues [43] and generously provided by Dr. Wolfgang Dostmann, University of Vermont. All other reagents were purchased from Sigma-Aldrich.
2.2. Vascular SMC isolation and culture
Rat aortic SMC were isolated from the thoracic and abdominal aortas of Sprague-Dawley male rats (150-200 g) as described previously [13,30]. Mouse aortic SMC were isolated from abdominal and thoracic aortas of Balb-c and C57 wild type mice. Briefly, the mice were sacrificed by CO2 inhalation and the aortas were excised and placed in a “wash medium” (DMEM containing 20 mM N-2-hydroxyethlypiperazine-N′2-ethanesulfonic acid [HEPES], 1 mg/ml bovine serum albumin (BSA), 5 μg/ml amphiteracin B, and 50 μg/ml gentamicin). After removing adhering fat and connective tissue, aortas were placed in a digestion medium consisting of wash medium plus 1 mg/ml elastase and 130 U/ml of collagenase for 10 min to dislodge endothelial and adventitial cells. The remaining tissue was minced and further digested for 1-2 h in the same medium except that collagenase was increased to 200 U/ml until a homogeneous smooth muscle cell suspension was obtained. After washing twice by low speed centrifugation in wash medium, cells were placed in tissue culture medium (DMEM, 10% fetal bovine serum (FBS) and 50 μg/ml gentamicin), plated in plastic tissue culture plates (60 mm or 100 mm) without any matrix coating, and incubated at 37 °C in a 95% air, 5% CO2 humidified incubator. For subculturing, cells were removed from the tissue culture plates using 0.05% tris-buffered trypsin and split at a ratio of 1:3.
2.3. Transfection of Cos7 with PKG-1 cDNAs
Bovine PKG-Iα cDNA and human PKG-Iβ cDNA were cloned into pcDNA-1 neo vector into the BamHI site and the pcDNA-3 vector at the EcoRI-XbaI site, respectively, amplified and purified using Endofree DNA maxi prep kit (Qiagen). Cos7 cells were plated in 60 mm dishes the day before transfection and grown to 70% confluency in culture medium. Fugene-6 transfection reagent was diluted 25-fold in serum-free DMEM, incubated for 5 min at room temperature, and combined with 2 μg of either empty pcDNA neo-1 vector, pcDNA/PKG-1α, empty pcDNA-3 vector or pcDNA-3/PKG-Iβ. The DNA transfection reagent mixture was incubated at room temperature for 15 min, added dropwise onto the Cos7 monolayers, evenly distributed, and incubated at 37 °C. Transfection efficiencies for pcDNA-1 were> 75%, while transfection efficiencies for pcDNA-3 were >90%.
2.4. Site-directed mutagenesis of PKG-Iα
pBluescript II KS+ (Stratagene) containing PKG-Iα nucleotides 1-1342 was used as the template for long-range amplification PCR site-directed mutagenesis according to the method of Kunkel [44]. Oligonucleotides (Operon) were constructed to be identical to wild type human PKG-Iα cDNA sequence except for nucleotides which serve to mutate the serines or threonine to alanine [45]. After amplifying the cDNAs and verifying the incorporation of the mutations by changes in restriction enzyme banding patterns and/or sequencing, the mutant PKG-Iα constructs were digested with EcoRI and NcoI. The 485-base pair EcoRI/NcoI fragments of PKG-Iα were subcloned into the pVL1392+hcGKIα vector. All vectors were propagated in E. coli, and all the plasmids were sequenced on an Automated Biosystems, Inc. DNA Sequencer 373A by the dideoxy chain termination method. The PKG-Iα mutants were expressed in a baculovirus system (Pharmingen, Orbigen), and PKG-Iα activity was verified according to the procedures provided in [45]. The cDNA sequences were subsequently subcloned into pcDNA-3/neo vectors.
2.5. Immunoprecipitation
Equal amounts (200-250 μg protein) of cell lysate were incubated for 2 h with protein-A-agarose treated with preimmune IgG in an immunoprecipitation solution (50 mM Tris-HCl, pH 7.4, 5 mM EDTA, 1% NP-40 and 0.02% NaN3) and centrifuged. Supernatants were transferred to new microcentrifuge tubes and polyclonal PKG-I antibody was added. The PKG-I antibody is directed towards the C-terminal amino acid sequence of the catalytic domain of the holoenzyme, and therefore recognizes both PKG-Iα and PKG-Iβ. After incubation at 4 °C from 2 h to overnight, 25 μl of 50% protein-A-agarose was added, and the mixture was incubated for 2-4 h. The agarose beads were centrifuged and washed 3 times in 100 mM Tris-HCl, pH 7.4, 150 mM NaCl, 0.1% NP-40 and 0.25% deoxycholate followed by two rinses with phosphate-buffered saline (PBS). The beads were then suspended in sample loading buffer, heated to 100 °C for 5 min, and loaded in 8% SDS polyacrylamide gels. Following electrophoresis, proteins were transferred to nitrocellulose and detected via western blotting.
2.6. Western blot analysis
Cells (SMC or Cos7) were harvested from 60 or 100 mm dishes in 0.5 ml or 1 ml, respectively, ice cold PEM buffer (20 mM sodium phosphate, pH 6.8, 2 mM EDTA, 15 mM β-mercaptoethanol, 0.15 M NaCl) in the presence of a cocktail of protease inhibitors (0.1 mM PMSF, 10 μg/ml pepstatin A, 10 μg/ml leupeptin, and 5 μg/ml aprotonin). The cells were homogenized 3 times for 10 s in a sonicator set at 50%, and the homogenates were centrifuged in a tabletop microfuge for 5 min at 4 °C. The supernatant was collected, and its proteins were separated on 8% SDS polyacrylamide gels (SDS-PAGE). The proteins were transferred for 1 h to nitrocellulose, blocked in 5% nonfat dried milk in TBS buffer at room temperature, and incubated overnight at 4 °C with primary antibody solutions as described previously [14]. The blots were then washed 3 times in Tris-buffered saline (TBS) buffer and incubated for at least 1 h with secondary antibody solutions at room temperature. The blots were developed using chemiluminescence, and the films were scanned by densitometry and quantitated using a computer-assisted program. For monitoring PKG expression and in order to quantitate expression by western blot analysis in the linear range of the standard curve, it was necessary to dilute the antibody obtained from Stress-Gen 1:5000 rather than the 1:1000 dilution suggested by the manufacturer.
2.7. Statistical analysis
In all experiments, the mean values for at least three experiments±SEM were calculated. The results were submitted to statistical analysis using the Student's t test, and p<0.05 was considered to be significant.
3. Results
3.1. Effects of cGMP analogs on PKG-I protein expression in VSMC
As previously reported by this laboratory, NO donors and inflammatory cytokines that elevate cellular cGMP suppressed PKG-I mRNA expression and inhibited PKG-I promoter activity in vascular SMC [24,25]. To determine whether or not cGMP itself regulated PKG-I expression, mouse aortic SMC were incubated with 8-Br-cGMP (1 mM) for various times. Protein was extracted and PKG-I levels were analyzed by western blotting. As shown in Fig. 1, 8-Br-cGMP decreased PKG-I levels at the earliest time point examined (24 h) and levels remained suppressed for up to 96 h in the presence of 8-BrcGMP. A similar finding was found using rat aortic and bovine aortic SMC (data not shown). Down-regulation of PKG-I protein was also observed in the presence of 8-parachlorophenothio-cGMP (8-pCPT-cGMP), but 8-Br-5′-GMP, which is inactive in the cGMP signaling pathway, did not cause down-regulation of PKG protein expression (data not shown).
Fig. 1.

Effect of 8-Br-cGMP on PKG-I protein expression in mouse aortic SMC. SMC in passage 3 were plated at 70% confluency and incubated with or without 1 mM 8-Br-cGMP for the times indicated. Protein (50 μg) was separated by SDS-PAGE and PKG-I expression was visualized by western blotting. This experiment is representative of experiments that were repeated at least six times.
3.2. Effects of NO and cyclic nucleotide phosphodiesterase (PDE) inhibition on PKG-I expression
To further explore the effects of cGMP on down-regulating PKG-I protein level, we examined the effect of the NO donor, DETANONOate (DETA-NO), on PKG-I levels in rat aortic SMC. As shown in Fig. 2A, DETA-NO (1 μM) alone had a relatively minor effect on PKG-I protein levels, despite the ability of DETA-NO to elevate cGMP levels by approximately 2-fold in the cells (Fig. 2B). However, when cells were treated with 1 μM DETA-NO in the presence of 0.2 mM of the general cyclic nucleotide PDE inhibitor, 3-isobutyl-1-methylxanthine (IBMX), PKG-I protein levels decreased by more than 90%. The cGMP level under these conditions was increased more than 10-fold (Fig. 2B). 8-Br-cGMP in the absence or presence of IBMX decreased PKG-I protein expression, as expected; IBMX itself had no significant effect on PKG-I protein expression with the NO donor present.
Fig. 2.
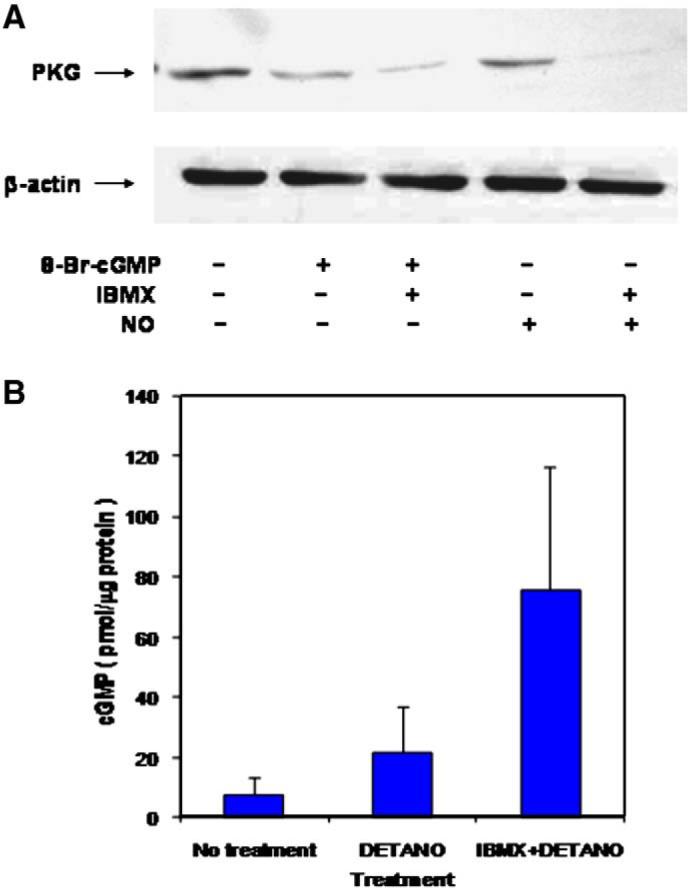
Effects of DETA-NONOate and IBMX on PKG-I protein expression in rat aortic SMC. Panel A. Rat aortic SMC in passage 3 were plated at 70% confluency and incubated for 24 h with 8-Br-cGMP (500 μM), IBMX (0.2 mM), and DETA-NONOate (1 μM) in the combinations shown. Protein (50 μg) was separated by SDS-PAGE and PKG-I expression was visualized by western blotting. Data are representative of experiments that were repeated three times. Panel B. Rat aortic SMC were incubated with 1 μM DETA-NONOate with or without 0.2 mM IBMX for 5 min, and cGMP concentrations were measured by immunoassay. Results are the mean ± standard error for n=4.
3.3. Effects of cGMP analogs on PKG-I protein expression in Cos7 cells
Vascular SMC from mouse, rat and bovine tissues express PKG-I mRNA, and therefore it was possible that the effects of 8-Br-cGMP and DETA-NO/IBMX were due to suppression of PKG-I gene expression [24,25]. Thus, to determine whether or not cGMP caused down-regulation of PKG-I at the level of protein production, the effect of 8-Br-cGMP on PKG-I expression in Cos7cells, which do not express measurable PKG-I mRNA or protein, was examined. Because PKG-Iα is the major isoform of PKG-I expressed in the vascular SMC examined above, the cDNA for this isoform of PKG-I was transfected into Cos7 cells. As shown in Fig. 3, Cos7 cells expressed abundant PKG-Iα protein following transfection. However, incubation with increasing concentrations of 8-Br-cGMP caused a down-regulation of PKG-Iα at 6 h, with the half-maximally effective concentration of 8-Br-cGMP observed at approximately 250 μM. This is consistent with numerous literature reports wherein maximal short-term effects of 8-Br-cGMP on cellular responses are generally observed between 100 and 500 μM. When a maximally-effective concentration of 500 μM 8-Br-cGMP was used, PKG-Iα protein level was decreased after only 2 h of treatment, after which no further down-regulation was observed up to 6 h (Fig. 4A and B). These experiments demonstrate that cGMP causes down-regulation of PKG-Iα protein expression in cells that do not express endogenous PKG-I at the genomic (i.e., mRNA) level.
Fig. 3.
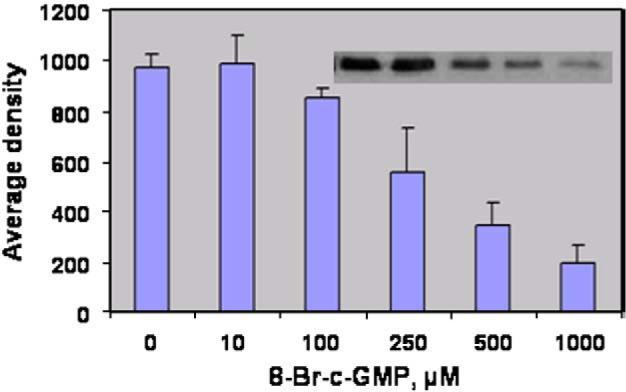
Effects of 8-Br-cGMP on PKG-Iα expression in transfected Cos7 cells. Cos7 cells were transfected with pcDNA1-neo/PKG-Iα vector and grown for 24 h. The cells were incubated for 6 h with the concentrations of 8-Br-cGMP indicated, protein (50 μg) was separated by SDS-PAGE, and PKG-Iα expression was visualized by western blotting. The blots were scanned, and optical density was determined for each value. The results are the mean±standard error of the mean for n=4. The insert shows a representative western blot for this experiment.
Fig. 4.
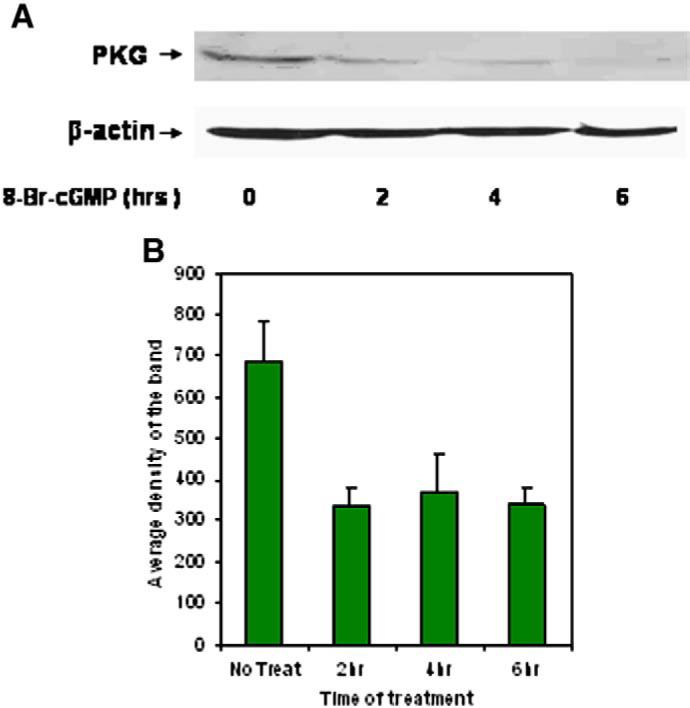
Panel A. Time course for the effect of 8-Br-cGMP on PKG-Iα expression in transfected Cos7 cells. Cos7 cells were transfected with pcDNA1-neo/PKG-Iα vector and grown for 24 h. The cells were incubated with 500 μM 8-Br-cGMP for the times indicated and protein expression was visualized by western blotting. Panel B. The immunoreactive band representing PKG-Iα was scanned, and the average densities of the bands for four experiments were plotted as the mean±standard error of the mean.
3.4. Mechanism of cGMP-dependent down-regulation on PKG-I: ubiquitination
The cell utilizes several mechanisms for down-regulating protein levels. Degradation by the ubiquitin-26S proteasome pathway is one such mechanism. Therefore, whether PKG-Iα undergoes ubiquitination, and if cGMP stimulates ubiquitination of PKG, was tested. The level of ubiquitin modification of PKG-Iα was determined using immunoprecipitation and western blot analysis. As shown in Fig. 5A, 8-Br-cGMP (500 μM) induced a time-dependent increase in ubiquitinated PKG-Iα as early as 1 h after the addition of 8-Br-cGMP. In this experiment, PKG-I was immunoprecipitated from Cos7 cell extracts and blotted using anti-ubiquitin. Ubiquitination of PKG-Iα was noted by the larger molecular weight species that were immunoprecipitated using the PKG antibody. In a separate experiment, Cos7 cells were treated for 24 h, and this blot was placed along side the earlier time course for comparison (Fig. 5A). As expected from the earlier experiments, the 8-Br-cGMP-induced ubiquitination was associated with a decrease in immunoprecipitable PKG-Iα from Cos7 cells (Fig. 5B). Treatment of Cos7 cells with the 26S proteasome inhibitor, MG-132, increased the level of PKG-Iα-ubiquitin complex but only if the cells were also treated with 8-Br-cGMP (Fig. 5C). This would be the predicted effect of the drug since degradation, but not ubiquitination, was blocked. Therefore, the decrease in PKG-Iα expression occurred in response to cGMP, presumably as a result of cGMP binding to the enzyme, and down-regulation appeared to be correlated with ubiquitination and subsequent degradation of PKG-Iα by the 26S proteasome.
Fig. 5.
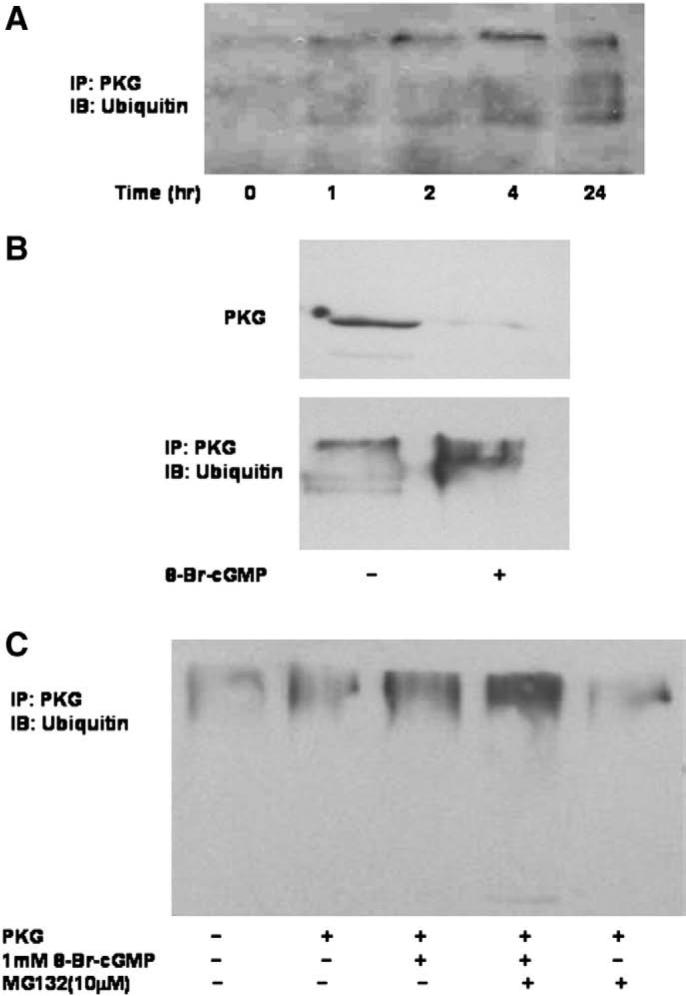
Effects of 8-Br-cGMP on PKG-Iα ubiquitination and expression in Cos7 cells. Panel A. Cos7 cells were transfected with pcDNA1-neo/PKG-Iα vector and grown for 24 h. After the cells were incubated with 500 μM 8-Br-cGMP for the times indicated, cells were lysed and PKG-Iα was immunoprecipitated with a polyclonal anti-PKG-I antibody. Protein was separated by SDS-PAGE and ubiquitin was visualized using anti-ubiquitin. This experiment is representative of more than five different experiments each yielding similar results. Panel B. Cos7 cells were transfected with pcDNA1-neo/PKG-Iα vector and grown for 24 h followed by treatment with 500 μM 8-Br-cGMP for 2 h. PKG-Iα was immunoprecipitated with a polyclonal anti-PKG-I antibody. Protein was separated by SDS-PAGE and ubiquitin was visualized using anti-ubiquitin (bottom panel). PKG-Iα protein expression was examined in cell extracts from a parallel experiment (upper panel). Panel C. Cos7 cells were transfected with pcDNA1-neo/PKG-Iα vector and grown for 24 h followed by treatment with 500 μM 8-Br-cGMP for 2 h in the presence or absence of MG-132 (10 μM). PKG-Iα was immunoprecipitated with a polyclonal anti-PKG-I antibody. Protein was separated by SDS-PAGE and ubiquitin was visualized using anti-ubiquitin. This experiment is representative of experiments that were repeated six times.
3.5. Role of PKG-dependent phosphotransferase activity on ubiquitination
Several protein kinases are ubiquitinated and degraded by the 26S proteasome in vivo. In some of these instances, phosphorylation or autophosphorylation of the kinase triggers ubiquitination and degradation [40-42,46,47]. For PKG-Iα, it is well-established that binding of cGMP induces autophosphorylation on serine and threonine residues in the autoinhibitory domain [48-50]. To determine whether cGMP-induced PKG-Iα autophosphorylation was necessary for ubiquitination and degradation, a specific and potent PKG-I inhibitor peptide, DT-2, was used to block PKG catalytic activity. To ensure that the PKG inhibitor was effectively blocking PKG phosphotransferase activity in Cos7 cells, PKG-I-dependent phosphorylation of the specific substrate, VAsodilator Stimulated Phosphoprotein (VASP) was measured in the absence and presence of DT-2. As shown in Fig. 6A, 5 μM DT-2 inhibited cGMP-induced VASP phosphorylation on the cGMP selective site, serine-239. The effects of 5 μM DT-2 on 8-Br-cGMP-dependent ubiquitination and down-regulation of PKG-Iα are shown in Fig. 6B. Whereas 8-BrcGMP suppressed PKG-Iα protein level and induced ubiquitination in the Cos7 cells, DT-2 inhibited cGMP-induced down-regulation and ubiquitination. By itself, DT-2 had no effect on ubiquitination (not shown). These results suggest that catalytic activity of PKG-Iα is necessary for the down-regulation process.
Fig. 6.
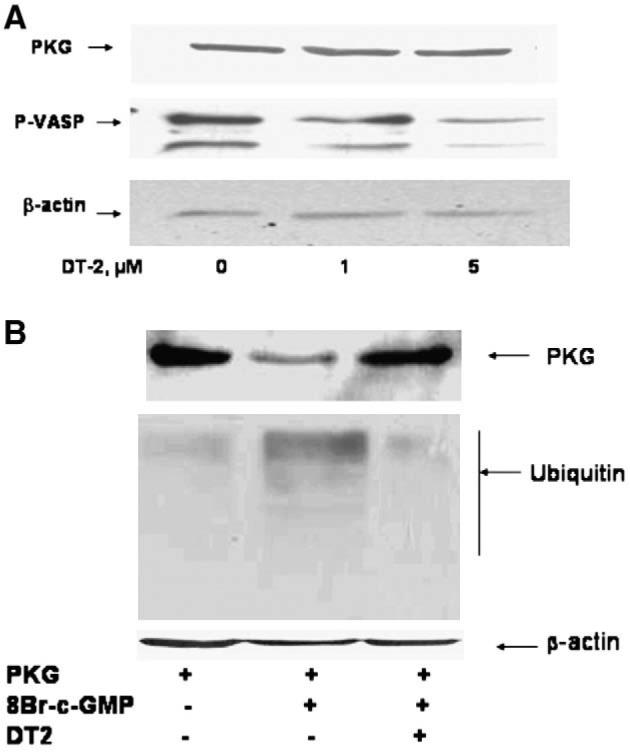
Effects of the PKG-I inhibitor, DT-2, on 8-Br-cGMP-induced PKG-Iα activation (Panel A) and ubiquitination and down-regulation (Panel B) in Cos7 cells. Panel A: Cos7 cells were transfected with pcDNA1-neo/PKG-Iα vector and grown for 24 h. Cells were treated for 10 min with 500 μM 8-Br-cGMP in the absence or presence of DT-2 (1 μM and 5 μM). Protein (50 μg) was separated by SDS-PAGE and VASP serine-239 phosphorylation (P-VASP) was visualized by western blotting using a specific phosphoserine-239 antibody (middle bands). The upper band demonstrates expression of PKG-Iα in the Cos7 cells and the lower band is the β-actin control for protein loading. Panel B: Cells were grown as described in Panel A and treated with or without 500 μM 8-Br-cGMP in the presence or absence of 5 μM DT-2. PKG-Iα was immunoprecipitated with a polyclonal anti-PKG-I antibody. Protein was separated by SDS-PAGE and ubiquitin was visualized using anti-ubiquitin antibodies. This experiment is representative of experiments that were repeated at least three times.
3.6. Autophosphorylation of PKG-Iα on serine-64 is required for down-regulation
Previous studies have shown that PKG-Iα is autophosphorylated on Ser-50, Ser-64, and Thr-84, and to a lesser extent on Thr-58 and Ser-72 [45]. In particular, Ser-64 phosphorylation was found to alter conformation of the enzyme sufficiently to promote activation [45]. We examined whether PKG-Iα was ubiquitinated and down-regulated when Ser-50, Ser-64, and Thr-84 were separately mutated to non-phosphorylated alanine. As shown in Fig. 7A, 8-Br-cGMP effectively down-regulated wild type PKG-Iα, the S50A mutant, and the T84A mutant when expressed in Cos7 cells. On the other hand, 8-Br-cGMP did not down-regulate the S64A mutant when expressed in the cells. Not shown are the data demonstrating that alanine mutation at the minor autophosphorylation sites (positions Thr-58, Ser-72) also did not impair 8-Br-cGMP induced down-regulation of PKG-Iα in Cos7. Therefore, of the mutants tested, only the ser-64 to alanine mutant (S64A) was not down-regulated in response to 8-Br-cGMP. When PKG-Iα was immunoprecipitated from the 8-Br-cGMP-treated Cos7 cells and probed with an anti-ubiquitin antibody, significant ubiquitination of wild type PKG-Iα was observed; little, if any, ubiquitination of the S64A mutant was observed (Fig. 7B). These results demonstrate that autophosphorylation of Ser-64 in response to 8-Br-cGMP was necessary for both ubiquitination and down-regulation of PKG-Iα in response to 8-Br-cGMP.
Fig. 7.

Effects of 8-Br-cGMP on wild-type (WT) and mutant PKG-Iα expression in Cos7 cells. Panel A. Cells were transfected with wild-type (WT) pcDNA-1/PKG-Iα vector or pcDNA-3/S50A/, S64A/, or T84A/PKG-Iα vector and grown for 24 h. The cells were treated for 6 h with 500 μM 8-Br-cGMP. Protein was extracted, and PKG-Iα visualized by western blotting. Panel B. Cos7 cells were transfected with either wild-type (WT) pcDNA-1/PKG-Iα vector or pcDNA-1/S64A-PKG-Iα vector and grown for 24 h. The cells were treated with 500 mM 8-Br-cGMP for 6 h, and PKG-Iα (WT or S64A) was immunoprecipitated with a polyclonal anti-PKG-I antibody. Protein was separated by SDS-PAGE and ubiquitin was visualized using anti-ubiquitin (bottom panel). PKG-Iα protein expression was examined in cell extracts from a parallel experiment (upper panel). This experiment is representative of experiments that were repeated four times.
3.7. Isoform-specific down-regulation of PKG-I in response to 8-Br-cGMP
PKG-I is expressed as two alternately spliced gene products, PKG-Iα and PKG-Iβ, which differ in the first ~100 amino acids that comprise the autoinhibitory/autophosphorylation domain. To investigate whether PKG-Iβ was also down-regulated in response to cGMP, Cos7 cells were transfected with PKG-Iβ cDNA and treated with varying concentrations of 8-Br-cGMP. As shown in Fig. 8, PKG-Iβ was not down-regulated in response to 8-Br-cGMP concentrations as high as 1 mM. In this set of experiments, the vector into which PKG-Iβ was cloned expressed higher levels of recombinant protein than did the vector into which PKG-Iα was cloned. Nevertheless, it is clear that 8-Br-cGMP only produced down-regulation of the Iα isoform. These results suggest that cGMP induces down-regulation of PKG-Iα but not PKG-Iβ.
Fig. 8.
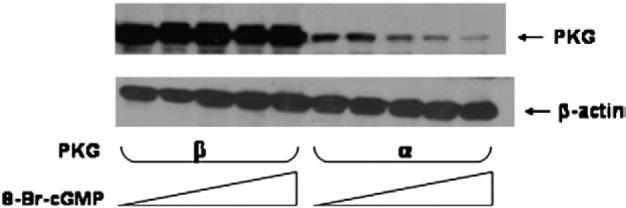
Effects of 8-Br-cGMP on expression levels of PKG-Iβ and PKG-Iα in Cos7 cells. Cells were transfected with wild-type pcDNA-1/PKG-Iα vector or pcDNA-3/PKG-Iβ vector and grown for 24 h. Cells were treated for 6 h with varying concentrations of 8-Br-cGMP (0, 100, 250, 500, 1000 μM). PKG-Iα or Iβ expression in the cell extracts was visualized by western blotting. This experiment is representative of experiments that were repeated four times.
4. Discussion
Activation of PKG-I in SMC mediates NO-dependent and hormone-induced relaxation, and more recent studies have shown that PKG-I is an important regulator of vascular SMC gene expression [13-15]. Restoration of PKG-I expression to PKG-deficient vascular SMC or overexpression of PKG-I in vascular SMC increases the expression of various SMC-specific marker proteins such as smooth muscle myosin heavy chain and calponin, and decreases the expression of extra-cellular matrix proteins. Endogenous PKG-I activity in vascular SMC may be needed to maintain the quiescent, contractile phenotype that is characteristic of normal, non-proliferating SMC. Inflammatory and atherogenic cytokines such as IL-1β and TNF-α suppress PKG-I mRNA and protein expression in vascular SMC [25,37]. This effect may contribute to vascular SMC phenotypic modulation characteristic of inflammatory vascular disorders.
Based on this information, mechanisms for regulating PKG-I expression in vascular SMC is of interest for understanding the role of PKG-I in SMC phenotypic modulation and vascular pathology. Previously, our laboratory reported that PKG-I transcription is under the control of ubiquitous transcription factors such as USF-1/USF-2 and Sp1/Sp3 [37,38]. Zeng et al. have found that KLF proteins, which interact with Sp1/Sp3 sites on the proximal PKG-I promoter, play an important role in endogenous PKG-I expression [23]. Furthermore, NO inhibited Sp1/Sp3-dependent activation of the PKG-I promoter in vascular SMC, possibly by elevating intracellular cGMP levels that then cross-activated PKA [37]. However, cGMP analogs (e.g., 8-Br-cGMP, 8-pCPT-cGMP) were more potent than cAMP analogs in reducing PKG-I protein levels but not steady-state mRNA levels, according to unpublished observations in our laboratory (Browner et al.). Because USF, Sp, and KLF transcription factors operate under many growth conditions (i.e., perform a “housekeeping” function), these findings suggest that the control of PKG-I protein levels by posttranscriptional/posttranslational mechanisms is likely to be important. This study suggests that one such mechanism is cGMP-stimulated ubiquitination of PKG-I.
Cyclic GMP suppresses the expression of PKG-Iα in vascular SMC. In transfected Cos7 cells, which do not express endogenous PKG-I mRNA, PKG-Iα protein expression was inhibited by cGMP analogs, implying that a non-genomic, post-translational mechanism controls PKG-Iα expression. Typically, modest, transient elevations of cGMP which activate PKG. However, down-regulation of PKG-I by cGMP appears to require a more persistent increase in cGMP elevation since an NO donor by itself did not suppress PKG protein expression. When combined with a PDE inhibitor to enhance cGMP elevation, NONOate was effective in down-regulating of PKG expression. Furthermore, the time course of down-regulation suggests that cGMP begins to affect PKG-I levels after approximately 30 to 60 min. Normally, cGMP levels are maintained within a narrow concentration range due in part to the activation of cGMP-specific PDE-5 by PKG-dependent phosphorylation. However, cGMP levels may be raised to high enough levels, e.g., 10-fold [30], by iNOS induction. These high cGMP levels induce ubiquitination.
PKG-Iα down-regulation is dependent upon ubiquitination. Higher molecular weight ubiquitinated species of PKG-Iα were observed in the presence, but not in the absence, of 8-Br-cGMP. Ubiquitination was accompanied by down-regulation of PKG-Iα protein in the cells, and the 26S proteasome inhibitor, MG-132, significantly increased the presence of ubiquitinated PKG species in the western blots. Collectively, these findings suggest that cGMP-induced down-regulation of PKG-I may be an important physiological feedback mechanism to blunt signaling in the face of robust NO/cGMP production. Ubiquitination, rather than control of mRNA expression, may play a significant role in controlling PKG-Iα levels in vascular SMC. A model for the control of PKG-I expression is shown in Fig. 9.
Fig. 9.

Model for regulation of PKG-Iα expression by the NO/cGMP pathway. Persistently high levels of NO and ANP activate soluble and particulate guanylyl cyclase (sGC and pGC), respectively, and cause prolonged elevation of intracellular cGMP levels. These high concentrations of cGMP induce autophosphorylation and ubiquitination of PKG-Iα. Inhibition of cGMP-specific phosphodiesterases (PDE) also maintain elevated intracellular cGMP levels particularly in the presence of a GC activator. This persistent high level of cGMP provides a mechanism by which PKG-Iα autophosphorylation and ubiquitination may be stimulated. Proteasome degradation of PKG-Iα reduces expression and blunts NO signaling in cells.
The trigger for cGMP-induced ubiquitination is autophosphorylation of the kinase, since ubiquitination and down-regulation of PKG-Iα were prevented by blocking autophosphorylation with an inhibitor of PKG-I catalytic activity. It has been known for approximately four decades that PKG-I undergoes autophosphorylation [45,48-50]. Autophosphorylation of serine and threonine residues within the N-terminal autoinhibitory domain of PKG-I is activated by cGMP binding. Because the N-terminal autoinhibitory domain differs between PKG-Iα and PKG-Iβ, investigators have suggested that autophosphorylation may explain the different sensitivity of the two isoforms to activation by cGMP. Also, since the autophosphorylation sites on the two isoforms are distinct, autophosphorylation may perform different roles for the two isoforms. At least five autophosphorylated residues have been identified in PKG-Iα (Ser-50, Thr-58, Ser-64, Ser-72, and Thr-84), and two autophosphorylated residues (Ser-63 and Ser-79) exist in PKG-Iβ. Studies by Busch et al. [45] have shown that autophosphorylation of Ser-64 is likely to be involved in the activation of PKG-Iα; similarly, autophosphorylation of the analogous site in PKG-Iβ, Ser-79, activates this isoform [48]. Thus, at least one common role for autophosphorylation of the autoinhibitory domain is to participate in the activation process. Other roles for autophosphorylation, particularly in the non-homologous autoinhibitory domains of the two isoforms, include targeting the isoforms to distinct proteins or sites within the cell [11,12,51], and increasing cAMP binding affinity to promote PKG-I cross-activation by cAMP [52].
This study provides a new role for autophosphorylation of PKG-Iα: regulation of ubiquitination and protein degradation. Using single alanine mutants of the PKG-Iα autophosphorylation sites, it was observed that the S64A mutant was not down-regulated by 8-BrcGMP when expressed in Cos7 cells. This suggests that autophosphorylation of Ser-64 is sufficient to trigger ubiquitination of PKG-Iα. The S50A and T84A autophosphorylation mutants behaved like the wild type PKG-Iα (Fig. 7A). The PKG-Iβ isoform was not down-regulation (Fig. 8). Thus, one distinct role for autophosphorylation of PKG-I may be to selectively target the down-regulation of PKG-Iα.
The different responses of the two PKG-I isoforms to down-regulation by cGMP is of interest. Some investigators proposed that PKG-Iβ acts as a “sink” for excess cGMP accumulation in the cell [53]. PKG Iβ is expressed more robustly in non-vascular SMC such as uterine and gastrointestinal SMC and PKG-Iα protein is expressed more highly in vascular SMC. Furthermore, PKG-Iβ has a higher KAct (i.e., lower affinity) for cGMP compared with PKG-Iα due to the higher affinity of the Iβ autoinhibitory domain for the common catalytic domain [54].
However, a more likely hypothesis is that the different responses of the two PKG-I isoforms allow for differential expression in cells. Platelets express mainly PKG-Iβ protein and express no PKG-I mRNA for the life span of the cell (approx. 60 days). At the protein level, but not at the mRNA level, PKG-Iα is more highly expressed in vascular SMC [7,8,12,15,53], and more actively down-regulated than PKG-Iβ. Indeed, it is generally observed that vascular SMC (aortic, pulmonary, and venous) demonstrate a loss of PKG-I protein expression when passaged multiple times compared with non-vascular SMC (e.g., trachea, uterus), which retain PKG-I protein expression. Thus, the presence of an autoinhibitory domain that does not induce ubiquiti-nation upon autophosphorylation may be an adaptive mechanism to allow PKG-Iβ expression in cells where mRNA levels encoding the enzyme are very low or absent; stated another way, the presence of an autoinhibitory domain that does induce ubiquitination upon autophosphorylation allows rapid loss of PKG-Iα during physiological conditions where excess cGMP is produced (e.g., inflammation) regardless of the level of PKG-I mRNA. These observations suggest that it is cGMP itself (whose levels reflect the activity of guanylyl cyclases and phosphodiesterases) that controls the expression of the responding pathway through PKG-Iα/Iβ protein expression. Notably, guanylyl cyclases, especially the NO-sensitive sGC subunits, are highly regulated at the transcriptional level in cells [55-60].
Vascular SMC assume the role of a fibroproliferative cell phenotype for vascular repair, facilitating cell growth and dedifferentiation in response to vascular injury and inflammation. Our laboratory has suggested that PKG-I plays an important role in attenuating modulation of phenotypically contractile vascular SMC to fibroproliferative cells. Perhaps not coincidently then, vascular SMC express higher levels of the more strictly regulated PKG-Iα compared with PKG-Iβ. Its expression can be attenuated by inflammatory cytokines and growth factors that up-regulate NO production, sGC activity and hence cGMP levels. The decrease in PKG-Iα expression may be an important component of the response to injury, allowing the SMC to modulate to the fibroproliferative, synthetic phenotype — at least during wound-repair and response to injury, as alluded to in the model shown in Fig. 9. It is tempting to speculate that the up-regulation of cGMP synthesis, perhaps by increases in NO synthase and guanylyl cyclase expression, is a key mechanism for PKG-I down-regulation.
The studies reported here provide a molecular mechanism for the regulation of PKG-I under physiological/pathophysiological conditions and a cautionary note for investigators studying the physiological role of PKG in cells. In many experiments, high concentrations (N100 μM) of PDE-resistant cGMP analogs are added to cells in order to activate endogenous PKG. In many cases, these studies are carried out over a period of many hours to days, especially in studies designed to study the effects of PKG on cell growth or gene expression. Based on the results reported here, however, it is likely that PKG-Iα would be down-regulated as a result of ubiquitination-26S proteasome degradation in the cell. Our laboratory has examined PKG-I expression in colonic and lung cancer cells and found a similar down-regulation by cGMP elevation, suggesting that this mechanism of down-regulation is widespread (unpublished observations). Thus, like PMA-dependent activation and long-term down-regulation of PKC, cGMP-dependent activation of PKG-I may be followed by its more long-term down-regulation. In cell culture and perhaps in vivo, sustained elevations in cGMP either by activation of guanylyl cyclase or the addition of cGMP analogs may lead to suppression of PKG-I, and the physiologic parameters measured in response to cGMP in these studies may not be mediated by PKG-I due to its loss from the cells.
Acknowledgements
This work was supported by NIH grants HL66164 (T.M.L.) and DK40029 (J.D.C.).
References
- [1].Lincoln TM, Cornwell TL. FASEB J. 1993;7:728. doi: 10.1096/fasebj.7.2.7680013. [DOI] [PubMed] [Google Scholar]
- [2].Lohmann SM, Vaandrager AB, Smolenski A, Walter U, DeJonge HR. Trends Biochem. Sci. 1997;22:307. doi: 10.1016/s0968-0004(97)01086-4. [DOI] [PubMed] [Google Scholar]
- [3].Wang X, Robinson PJ. J. Neurochem. 1997;68:443. doi: 10.1046/j.1471-4159.1997.68020443.x. [DOI] [PubMed] [Google Scholar]
- [4].Lincoln TM, Dey N, Sellak H. J. Appl. Physiol. 2001;91:1421. doi: 10.1152/jappl.2001.91.3.1421. [DOI] [PubMed] [Google Scholar]
- [5].Hofmann F, Ammendola A, Schlossmann J. J. Cell Sci. 2000;113:1671. doi: 10.1242/jcs.113.10.1671. [DOI] [PubMed] [Google Scholar]
- [6].Hofmann F, Feil R, Kleppisch T, Schlossmann J. Physiol. Rev. 2006;86:1. doi: 10.1152/physrev.00015.2005. [DOI] [PubMed] [Google Scholar]
- [7].Lincoln TM, Thompson M, Cornwell TL. J. Biol. Chem. 1988;263:17632. [PubMed] [Google Scholar]
- [8].Francis SH, Woodford TA, Wolfe L, Corbin JD. Second Messengers Phosphoprot. 1988;12:301. [PubMed] [Google Scholar]
- [9].Orstavik S, Natarajan V, Tasken K, Jahnsen T, Sandberg M. Genomics. 1997;42:311. doi: 10.1006/geno.1997.4743. [DOI] [PubMed] [Google Scholar]
- [10].Sausbier M, Schubert R, Voigt V, Hirneiss C, Pfeifer A, Korth M, Kleppisch T, Ruth P, Hofmann F. Circ. Res. 2000;87:825. doi: 10.1161/01.res.87.9.825. [DOI] [PubMed] [Google Scholar]
- [11].Schlossmann J, Ammendola A, Ashman K, Zong X, Huber A, Neubauer G, Wang GX, Allescher HD, Korth M, Wilm M, Hofmann F, Ruth P. Nature. 2000;404:197. doi: 10.1038/35004606. [DOI] [PubMed] [Google Scholar]
- [12].Surks HK, Mochizuki N, Kasai Y, Georgescu SP, Tang KM, Ito M, Lincoln TM, Mendelsohn ME. Science. 1999;286:1583. doi: 10.1126/science.286.5444.1583. [DOI] [PubMed] [Google Scholar]
- [13].Boerth NJ, Dey N, Cornwell TL, Lincoln TM. J. Vasc. Res. 1997;34:245. doi: 10.1159/000159231. [DOI] [PubMed] [Google Scholar]
- [14].Dey NB, Boerth NJ, Murphy-Ullrich JE, Chang PL, Prince CW, Lincoln TM. Circ. Res. 1998;82:139. doi: 10.1161/01.res.82.2.139. [DOI] [PubMed] [Google Scholar]
- [15].Zhang T, Zhuang S, Casteel DE, Looney DJ, Boss GR, Pilz RB. J. Biol. Chem. 2007;282:33367. doi: 10.1074/jbc.M707186200. [DOI] [PubMed] [Google Scholar]
- [16].Pfeifer A, Klatt P, Massberg S, Ny L, Sausbier M, Hirneiss C, Wang GX, Korth M, Aszodi A, Andersson KE, Krombach F, Mayerhofer A, Ruth P, Fassler R, Hofmann F. EMBO J. 1998;17:3045. doi: 10.1093/emboj/17.11.3045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Wolfsgruber W, Feil S, Brummer S, Kuppinger O, Hofmann F, Feil R. Proc. Natl. Acad. Sci. U. S. A. 2003;100:13519. doi: 10.1073/pnas.1936024100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Feil R, Lohmann SM, deJonge HR, Walter U, Hofmann F. Circ. Res. 2003;93:907. doi: 10.1161/01.RES.0000100390.68771.CC. [DOI] [PubMed] [Google Scholar]
- [19].Chiche JD, Schlutsmeyer SM, Bloch DB, de la Monte SM, Roberts JD, Filippov G, Janssens SP, Rosenzweig A, Bloch KD. J. Biol. Chem. 1998;273:24263. doi: 10.1074/jbc.273.51.34263. [DOI] [PubMed] [Google Scholar]
- [20].Cornwell TL, Arnold E, Boerth NJ, Lincoln TM. Am. J. Physiol. 1994;267:C1405. doi: 10.1152/ajpcell.1994.267.5.C1405. [DOI] [PubMed] [Google Scholar]
- [21].Wyatt TA, Naftilan AJ, Francis SH, Corbin JD. Am. J. Physiol. 1998;274:H448. doi: 10.1152/ajpheart.1998.274.2.H448. [DOI] [PubMed] [Google Scholar]
- [22].Taylor RM, Okwuchukwuasanya C, Nickl CK, Tegge W, Brayden JE, Dostmann WRG. Mol. Pharmacol. 2004;65:1111. doi: 10.1124/mol.65.5.1111. [DOI] [PubMed] [Google Scholar]
- [23].Zeng Y, Zhuang S, Gloddek J, Tseng CC, Boss BR, Pilz RB. J. Biol. Chem. 2007;281:16951. doi: 10.1074/jbc.M602099200. [DOI] [PubMed] [Google Scholar]
- [24].Soff GA, Cornwell TL, Cundiff DL, Gately S, Lincoln TM. J. Clin. Invest. 1997;100:2580. doi: 10.1172/JCI119801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Browner NC, Sellak H, Lincoln TM. Am. J. Physiol. 2004;287:C88. doi: 10.1152/ajpcell.00039.2004. [DOI] [PubMed] [Google Scholar]
- [26].Anderson PG, Boerth NJ, Liu M, McNamara DB, Cornwell TL, Lincoln TM. Arterioscler. Thromb. Vasc. Biol. 2000;20:2192. doi: 10.1161/01.atv.20.10.2192. [DOI] [PubMed] [Google Scholar]
- [27].Atsuko D, Thompson WJ, Weinstein B. Cancer Res. 2004;64:3966. doi: 10.1158/0008-5472.CAN-03-3740. [DOI] [PubMed] [Google Scholar]
- [28].Pollman MJ, Yamada T, Horiuchi M, Gibbons GH. Circ. Res. 1996;79:748. doi: 10.1161/01.res.79.4.748. [DOI] [PubMed] [Google Scholar]
- [29].Garg UC, Hassid A. J. Clin. Invest. 1989;83:1774. doi: 10.1172/JCI114081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Cornwell TL, Soff GA, Traynor AE, Lincoln TM. J. Vasc. Res. 1994;31:330. doi: 10.1159/000159061. [DOI] [PubMed] [Google Scholar]
- [31].Beasley D, Eldridge M. Am. J. Physiol. 1994;266:R1197. doi: 10.1152/ajpregu.1994.266.4.R1197. [DOI] [PubMed] [Google Scholar]
- [32].Joly GA, Schini VR, Vanhoutte PM. Circ. Res. 1992;71:331. doi: 10.1161/01.res.71.2.331. [DOI] [PubMed] [Google Scholar]
- [33].Depre C, Havaux X, Renkin J, Vanoverschelde JLJ, Wijns W. Cardiovasc. Res. 1999;41:465. doi: 10.1016/s0008-6363(98)00304-6. [DOI] [PubMed] [Google Scholar]
- [34].Wilcox JN, Subramanian RR, Sundell CL, Ross TW, Pollock JS, Harrison DG, Marsden PA. Arterioscler. Thromb. Vasc. Biol. 1997;17:2479. doi: 10.1161/01.atv.17.11.2479. [DOI] [PubMed] [Google Scholar]
- [35].Yan ZQ, Hansson GK. Circ. Res. 1998;82:21. doi: 10.1161/01.res.82.1.21. [DOI] [PubMed] [Google Scholar]
- [36].Rosenberg RB, Bronere CW, O'Dorisio S. Biochem. Med. Metab. Biol. 1994;51:149. doi: 10.1006/bmmb.1994.1019. [DOI] [PubMed] [Google Scholar]
- [37].Sellak H, Yang X, Cao X, Cornwell TL, Soff GA, Lincoln TM. Circ. Res. 2002;90:405. doi: 10.1161/hh0402.105898. [DOI] [PubMed] [Google Scholar]
- [38].Sellak H, Choi C, Browner NC, Lincoln TM. J. Biol. Chem. 2005;280:18425. doi: 10.1074/jbc.M500775200. [DOI] [PubMed] [Google Scholar]
- [39].Browner NC, Dey NB, Bloch KD, Lincoln TM. J. Biol. Chem. 2004;279:46631. doi: 10.1074/jbc.M408518200. [DOI] [PubMed] [Google Scholar]
- [40].Lu Z, Liu D, Hornia A, Devonigh W, Pagano M, Foster DA. Mol. Cell. Biol. 1998;18:839. doi: 10.1128/mcb.18.2.839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Lee HW, Smith L, Pettit GR, Vinitsky A, Smith JB. J. Biol. Chem. 1996;271:20973. [PubMed] [Google Scholar]
- [42].Lee HW, Smith L, Pettit GR, Smith JB. Mol. Pharmacol. 1997;51:439. [PubMed] [Google Scholar]
- [43].Dostmann WRG, Taylor MS, Nickl CK, Brayden JE, Frank R, Tegge WJ. Proc. Natl. Acad. Sci. U. S. A. 2000;97:14772. doi: 10.1073/pnas.97.26.14772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Kunkel TA. In: Current Protocols in Molecular Biology. Ausubel FM, Brent R, Kingston RE, Moore DD, Seidman JB, Smith JA, Struhl K, editors. John Wiley & Sons, Inc.; NY: 1994. p. 8.1.1. [Google Scholar]
- [45].Busch JL, Bessay EP, Francis SH, Corbin JD. J. Biol. Chem. 2002;277:34048. doi: 10.1074/jbc.M202761200. [DOI] [PubMed] [Google Scholar]
- [46].Kuo WL, Duke CJ, Abe MK, Kaplan EL, Gomes S, Rosner MR. J. Biol. Chem. 2004;279:23073. doi: 10.1074/jbc.M313696200. [DOI] [PubMed] [Google Scholar]
- [47].Weisz Hubsman M, Volinsky N, Manser E, Yablonski D, Aronheim A. Biochem. J. 2007;404:487. doi: 10.1042/BJ20061696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Smith JA, Francis SH, Walsh KA, Kumar S, Corbin JD. J. Biol. Chem. 1996;271:20756. doi: 10.1074/jbc.271.34.20756. [DOI] [PubMed] [Google Scholar]
- [49].Lincoln TM, Flockhart DA, Corbin JD. J. Biol. Chem. 1978;253:6002. [PubMed] [Google Scholar]
- [50].Landgraf W, Hofmann F. Eur. J. Biochem. 1989;181:643. doi: 10.1111/j.1432-1033.1989.tb14771.x. [DOI] [PubMed] [Google Scholar]
- [51].Feil R, Gappa N, Rutz M, Schlossmann J, Rose CR, Konnerth A, Brummer S, Kuhbandner S, Hofmann F. Circ. Res. 2002;90:1080. doi: 10.1161/01.res.0000019586.95768.40. [DOI] [PubMed] [Google Scholar]
- [52].Landgraf W, Hullin R, Gobel C, Hofmann F. Eur. J. Biochem. 1986;154:113. doi: 10.1111/j.1432-1033.1986.tb09365.x. [DOI] [PubMed] [Google Scholar]
- [53].Keilbach A, Ruth P, Hofmann F. Eur. J. Biochem. 1992;208:467. doi: 10.1111/j.1432-1033.1992.tb17209.x. [DOI] [PubMed] [Google Scholar]
- [54].Francis SH, Corbin JD. Annu. Rev. Physiol. 1994;56:237. doi: 10.1146/annurev.ph.56.030194.001321. [DOI] [PubMed] [Google Scholar]
- [55].Papapetropooulos A, Marczin N, Mora G, Milici A, Murad F, Catravas JD. Hypertension. 1995;26:696. doi: 10.1161/01.hyp.26.4.696. [DOI] [PubMed] [Google Scholar]
- [56].Oka M, Karoor V, Homma N, Nagaoka T, Sakao E, Golembeski SM, Limbird J, Imamura M, Gebb SA, Fagan KA, McMurtry IF. Cardiovasc. Res. 2007;74:377. doi: 10.1016/j.cardiores.2007.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Li D, Laubach VE, Johns RA. Am. J. Physiol. 2001;281:L369. doi: 10.1152/ajplung.2001.281.2.L369. [DOI] [PubMed] [Google Scholar]
- [58].Liu H, Force T, Bloch KD. J. Biol. Chem. 1997;272:6038. doi: 10.1074/jbc.272.9.6038. [DOI] [PubMed] [Google Scholar]
- [59].Shimouchi A, Janssens SP, Bloch DB, Zapol WM, Bloch KD. Am. J. Physiol. 1993;265:L456. doi: 10.1152/ajplung.1993.265.5.L456. [DOI] [PubMed] [Google Scholar]
- [60].Takata M, Filippov G, Liu H, Ichinose F, Janssens S, Bloch DB, Bloch KD. Am. J. Physiol. 2001;280:L272. doi: 10.1152/ajplung.2001.280.2.L272. [DOI] [PubMed] [Google Scholar]