

Abstract
Now that numerous high-quality complete genome sequences are available, many efforts are focusing on the “second genomic code”, namely the code that determines how the precise temporal and spatial expression of each gene in the genome is achieved. In this regard, the elucidation of transcription regulatory networks that describe combined transcriptional circuits for an organism of interest has become valuable to our understanding of gene expression at a systems level. Such networks describe physical and regulatory interactions between transcription factors (TFs) and the target genes they regulate under different developmental, physiological, or pathological conditions. The mapping of high-quality transcription regulatory networks depends not only on the accuracy of the experimental or computational method chosen, but also relies on the quality of TF predictions. Moreover, the total repertoire of TFs is not only determined by the protein-coding capacity of the genome, but also by different protein properties, including dimerization, co-factor interactions and post-translational modifications. Here, we discuss the factors that influence TF functionality and, hence, the functionality of the networks in which they operate.
1 Introduction
Transcription regulatory networks can be represented as graph models that combine physical and regulatory interactions between TFs and their target genes (reviewed in ref. 1). Several methods that can be used to identify physical interactions between TFs and their targets have been developed and applied to the study of both yeast and metazoan transcription regulatory networks. These include TF-centered methods such as chromatin immunoprecipitations,2–4 protein binding microarrays,5 DamID6,7 and bacterial one-hybrid assays,8 as well as gene-centered methods such as high-throughput yeast one-hybrid assays.9 The TF-DNA interaction data obtained by these methods are often visualized into network models. Fig. 1 depicts a hypothetical network that contains several of the architectural and topological network features described to date, including TF hubs (TFs that bind a large number of target genes), target gene hubs (genes bound by a large number of TFs), and TF modules (sets of TFs that share numerous target genes). Metazoan TF hubs were uniquely revealed in the nematode Caenorhabditis elegans by using gene-centered yeast one-hybrid assays.10,11 Specifically, we found that, whereas the majority of TFs bind only few promoters, a small subset of TFs bind up to 40% of all promoters tested. This suggests that these proteins play a more essential role in regulating transcription than most other TFs. By performing chromatin immunoprecipitation with eight TFs in the budding yeast Saccharomyces cerevisiae, Snyder and colleagues12 showed that the genes that code for two of these TFs, MGA1 and PHD1, were themselves targeted by all eight TFs, suggesting that these two TFs may be target gene hubs. Interestingly, MGA1 and PHD1 are master regulators of pseudohyphal growth. The authors suggest that genes regulated by large number of TFs may function as master regulators of biological processes. In agreement with this, we found that the promoter of the master regulator of D-type GABA-ergic motor neurons unc-30 in C. elegans can interact with 36 different TFs.11
Fig. 1.

The mapping of physical interactions between transcription factors (TFs) and their target genes has resulted in the discovery of several interesting network features, some of which are shown here. Some TFs (circles) target a disproportionately large number of genes (diamonds) and are referred to as TF hubs. Sets of TFs that share many target genes are referred to as TF modules. Target gene hubs interact with a disproportionately large number of TFs.
TF modules are another feature of transcription regulatory networks that have recently emerged.11 TF modules are distinct from more extensively studied general network modules that are defined as highly interconnected groups of nodes without regard for directionality or node type (i.e., TFs vs. target genes).13,14 We have defined TF modules as sets of TFs that share many of their target genes (Fig. 1).11 As such, these are uniquely found in bipartite transcription regulatory networks. Both general network modules and TF modules may reflect functionality, for instance within a cell or tissue type, or regarding a particular biological process. Finally, transcription regulatory networks are composed of recurring circuits, referred to as network motifs. For instance, feed forward loops are found frequently in networks from both pro- and eukaryotic organisms. Such motifs represent widely used regulatory mechanisms that can, for instance, be used to stabilize gene expression.15,16
The transcription regulatory networks that have been described to date represent compilations of multiple events that take place during the lifetime of an organism collapsed into a single model. However, in reality, only a subset of the network is active in particular cell types, under different developmental or physiological conditions, or at any given time.17–19 In addition, each TF in regulatory networks is represented as a single node, whereas it is known that TFs exist in many different functional forms that are determined by a variety of factors including post-translational modifications and dimerization. These factors themselves may depend on specific developmental or physiological conditions. Each TF form may interact with distinct target genes, or with the same target gene, but under different conditions. Here, we discuss the factors that need to be taken into account to determine how many functional TFs occur in an organism of interest, and how this information can be incorporated into transcription regulatory network models to study differential gene expression at a systems level.
2 Transcription factor predictions
There are two classes of TFs: basal TFs that are involved in transcription of most, if not all, genes, and regulatory TFs that control only subsets of genes.20 For the understanding of differential gene expression at a systems level, we only consider regulatory TFs (hereafter referred to as TFs). TFs interact with their target genes by binding specific cis-regulatory gene elements through a sequence-specific DNA binding domain. Different DNA binding domains are used to group TFs into TF families. Examples of DNA binding domains include basic region helix–loop–helix domains (bHLH), homeodomains and various types of zinc fingers. Computational tools have been developed both to define consensus DNA binding domains and to predict additional TFs of that family encoded by a genome of interest.21,22 We found that, although such computational tools are powerful, they do incorporate false predictions and miss many known TFs.23 For instance, by using a combination of computational tools and extensive manual curation we predicted a high-quality compendium of 934 TFs in the nematode Caenorhabditis elegans, which extended purely computational predictions by ∼50%.23 However, even though this compendium is more complete than previous collections, it is not yet comprehensive as algorithms and experimental assays continue to improve and, therefore, additional TFs continue to be discovered.24
3 DNA binding domains
Over the past decades, many different sequence-specific DNA binding domains have been uncovered. However, we propose that it is unlikely that all DNA binding domains are known. This is because, by applying yeast one-hybrid assays to only 112 C. elegans gene promoters, we have already discovered 11 C. elegans proteins that robustly interact with their target promoters in yeast, but that do not possess a known DNA binding domain. By using chromatin immunoprecipitation assays in yeast, we confirmed that these interactions are direct for nine of these proteins.10,11 We do not know yet whether these proteins directly bind to DNA or if they are recruited to their target promoters by interacting with other DNA binding proteins. Future structure–function analysis will provide insight into the mechanism of action of these novel putative TFs. Importantly, the cataloging of the DNA binding domain(s) of these proteins may enable the identification of additional proteins with similar domains in C. elegans, and perhaps in other organisms as well.
4 Alternative splicing
In metazoans, many gene transcripts, including those encoding TFs, are alternatively spliced, which often leads to multiple variants of a protein. Interestingly, it has been found that TF-encoding genes in mice undergo alternative splicing more frequently than other genes.25 Alternative splicing may lead to TFs with different functions. For instance, DNA binding domains or transcription regulatory domains may be included or excluded from the TF variant. At least 144 C. elegans TFs undergo alternative splicing, resulting in 379 different proteins, 30 of which lack a DNA-binding domain.23 The latter may function as regulators of TF function, for instance by titrating interaction partners of the corresponding TFs that do possess a DNA binding domain. Several C. elegans TFs contain more than one DNA binding domain and alternative splicing can affect which domains are present in the different protein products. For instance, several DAF-16 variants are generated as a result of alternative splicing, and each variant carries a unique combination of domains26 (Fig. 2(A)). DAF-16 is a critical regulator of various physiological processes including fat storage, aging, and the formation of dauers (an alternative larval stage of development, resistant to many forms of stress). It contains two potential forkhead DNA binding domains and is known to bind or regulate numerous target genes.27 It is tempting to speculate that each forkhead domain is responsible for the interaction with a distinct set of target genes, each of which may be involved in a particular biological process. A human example involves the homeodomain proteins hepatocyte nuclear factor HNF1 and vHNF1. There are three HNF1 splice variants, each of which encodes a protein with varying transcription activation properties. vHNF1 is also differentially spliced and one of the resulting protein products, vHNF1-C, functions as a trans-dominant repressor of all three HNF1 variants.28 TF variants may be expressed in different cell types or under particular conditions, leading to variable outputs of the transcriptional circuits in which they function. For instance, the HNF isoforms are differentially expressed in the human digestive tract, liver and kidney, where they may either regulate distinct target genes or, alternatively, the same target genes but at different levels. Genome-scale analyses of alternative splicing, for instance using whole genome or exon junction tiling arrays,29 will greatly facilitate the accurate identification of all TF variants that are produced in each cell and tissue type, and in various model organisms.
Fig. 2.
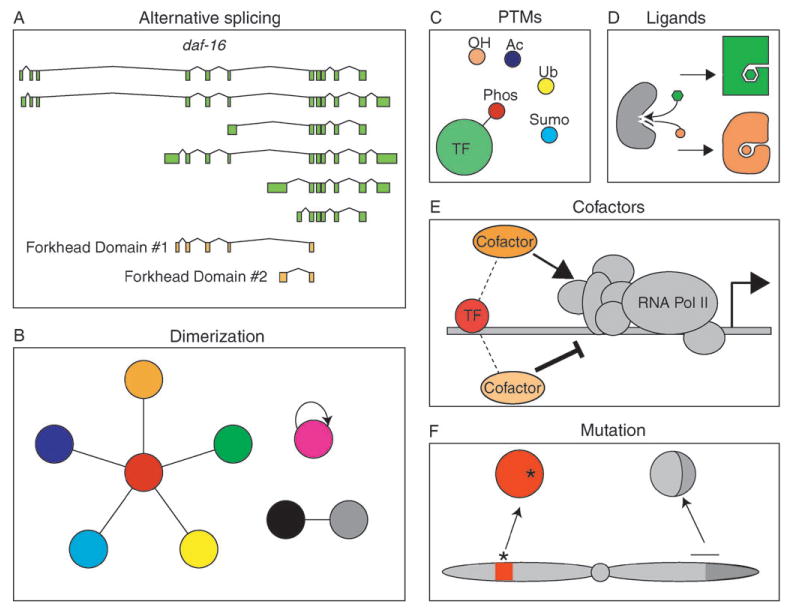
Various factors influence the functionality of TFs and impact transcription regulatory network modeling and analysis. Whether a TF binds to a promoter, and activates or represses transcription, depends on: (A) alternative splicing that may produce TF variants that carry unique combinations of functional domains involved in the regulation of gene expression. The example shows the C. elegans daf-16 gene that produces many splice variants, resulting in TFs that contain either of two possible forkhead DNA-binding domains, or neither. (B) Dimerizing TFs potentially combine in different ways to generate a large array of different hetero- or homodimers, each with its own function. (C, D) Some TFs may have an altered function after ligand binding or post-translational modifications (PTMs), such as phosphorylation (Phos), hydroxylation (OH), acetylation (Ac), ubiquitination (Ub), or sumoylation (Sumo). Such modifications can induce different conformational changes, thereby affecting TF functionality. (E) Co-factors can mediate varying affects of TF activity. (F) Mutation or translocation of TF-encoding genes can result in TFs with reduced, enhanced, or novel activity. The asterisk indicates a point mutation and the horizontal line depicts the breaking point of translocation within the chromosome that carries the TF-encoding gene.
5 Dimerization
Several TFs bind DNA as obligatory dimers, including members from the basic region leucine zipper (bZIP), bHLH and nuclear hormone receptor (NHR) families.30–32 Dimerization should be taken into account when considering the total complement of functional TFs because, if a particular TF only functions when it dimerizes with another TF, the dimer should be considered a single functional unit. Dimerization can affect the total number of functional TFs in different ways (Fig. 2(B)). In one model, each TF from a family can dimerize with itself and any other member of that family. If this would be the case, the number of functional TFs would be dramatically greater than the number of individually predicted TFs. For instance, dimerization between all 274 C. elegans NHRs23 would result in a total of 37675 NHR dimers. In another model, each TF dimerizes exclusively with one other TF of the same family. If this were the case for the C. elegans NHRs, this would result in 137 functional TF complexes, which would reduce the total number of functional TFs. In a third, intermediate model, one or more TFs from a family could serve as central dimerization partners that can interact with multiple members of the relevant TF family. Although no comprehensive data regarding TF dimerization is as yet available, our preliminary data indicate that this third model is likely most relevant (C. A. G. and A. J. M. W., in preparation). Systematic protein–protein interaction mapping efforts will be required to identify all functional TF dimers. High-throughput yeast two-hybrid assays33 are particularly well suited for this task as they identify binary interactions. For instance, we found by high-throughput yeast two-hybrid assays that NHR-49 serves as a dimerization hub, that can interact with at least 15 other NHRs.24 In the future, it will be important not only to identify all TF dimers, but to also determine the DNA-binding specificities of each dimer and where and when dimerization partners are co-expressed.
6 Post-translational modifications
Many proteins, including TFs, are post-translationally modified under different conditions and by different modifiers. Several post-translational modifications of TFs have been reported, including phosphorylation, hydroxylation, acetylation, ubiquitination and sumoylation (Fig. 2(C)).34 Such modifications often result from the activation of signal transduction pathways in response to environmental stimuli or developmental cues. Post-translational modifications can affect the regulatory activity of a TF, as well as its localization or stability. For instance, estrogen receptor β (ERβ), normally activated by ligand, can be phosphorylated by MAP kinase (MAPK) which leads to recruitment of the steroid receptor coactivator-1 (SRC-1) and ligand-independent activation of target genes.35 ERα can be acetylated by p300 at conserved lysine residues resulting in enhanced DNA-binding activity and, perhaps, ligand-dependent transcriptional activation.36 For most TFs it is not clear which modified forms exist and how these forms function to regulate gene expression. For a thorough understanding of gene regulatory networks, it will be important to determine which modification each TF is subjected to, under which circumstances, and how these modifications affect TF functionality.
7 Ligands
Many TFs become activated or inactivated as a result of ligand binding (Fig. 2(D)). One of the most prominent classes of ligand-dependent TFs is the NHR family, which includes ER, androgen receptor (AR), peroxisome proliferator-activated receptors (PPARs), retinoic acid receptor (RAR) and others. NHR ligands are hydrophobic molecules that can freely diffuse into the nucleus where they specifically interact with their target receptors. Most NHRs become potent transcriptional activators upon ligand binding. Human ER has been studied extensively because of its association with the development of breast cancer.37 A number of ER ligands (endogenous and exogenous) have been identified, some of which are non-steroidal compounds that are referred to as selective ER modulators (SERMs). Whereas steroidal compounds such as estrogen function to naturally modulate ER activity, SERMs such as tamoxifen are used in cancer treatment. Different ligands bind to ER with varying affinities and have different effects. Depending on tissue and cell-type context, ligands induce conformational changes in ER that promote transcriptional activation, whereas others promote transcriptional repression.38 The ligand(s) for most NHRs remain to be identified, and, therefore, such NHRs are referred to as “orphan” receptors. For instance, the nematode C. elegans has 274 predicted NHRs,23 but a ligand (dafachronic acid) has only been identified for a single NHR, DAF-12.39
Another class of ligand-binding TFs is the bHLH-PAS sub-family that includes the aryl hydrocarbon receptor (AHR). AHR can interact with a variety of exogenous compounds or toxins such as dioxin, and mediate a biological response (for a review see ref. 40). The range of compounds that can activate AHR is still under investigation, and although most appear to be exogenous in origin, it has been proposed that endogenous AHR ligands may play a role in organism development or homeostasis.41 Indeed, the C. elegans ortholog of AHR, ahr-1, is required for the proper development and specification of touch-receptor neurons, interneurons, and motor neurons.42,43
8 Co-factors
Regulatory TFs often activate or repress transcription, either by recruiting the RNA polymerase II machinery, or by preventing its access to the transcription start site. While many TFs interact directly with general TFs or components of RNA polymerase II, others function by interacting with intermediate proteins called co-factors (Fig. 2(E)).44–46 Depending on environmental or developmental circumstances, the same TF can interact with different coactivators or corepressors, illustrating the versatility of TFs in carrying out opposing regulatory effects in different contexts. RAR, for instance, can recruit the NCoR/SMRT corepressor complexes thereby repressing transcriptional activity of its target genes. Upon binding its ligand retinoic acid, however, RAR changes conformation and adopts a form capable of recruiting coactivator complexes and subsequently activates transcription.45 Interestingly, cofactor interactions can also affect DNA binding specificity,47 implying that different TF-cofactor pairs may interact with different sets of target genes. Future large-scale genomic and proteomic experiments are needed to identify the full spectrum of ligands and co-factors each TF in an organism can interact with and to unravel how these interactions affect the biochemical and biological function of each TF.
9 Transcription factor variants and disease
TFs play a crucial role in numerous diseases, including congenital disorders and cancer. Mutations in TF-encoding genes can result in loss-of-function, gain-of-function or neomorph TFs that attain a function not shared by the original TF. One of the best-studied TFs mutated in cancer is p53, a tumor suppressor gene that is inactivated by mutation in most human cancers. Interestingly, it appears that some mutations can also convert p53 into an oncogene.48 p53 regulates the expression of various cell cycle inhibitors and proteins involved in apoptosis. It will be interesting to see how the different forms of mutant p53 are affected in their biochemical and biological functions.
Several mutated TFs have been found to result in a variety of human congenital disorders. For instance, altered dimerization between the bHLH TFs Twist1 and Hand2 was found in patients with Saethre–Chotzen syndrome.49 Common neomorph TF variants that are found in instances of leukemia are fusion proteins resulting from chromosomal translocation/inversion (Fig. 2(F)). For example, an inversion on murine chromosome 16 leads to an aberrant Cbfb-MYH11 fusion protein, resulting in the development of acute myeloid leukemia.50 It will be important to understand the variety of mutant forms of TFs that exist in different diseases and how they perturb the regulatory networks that contribute to a disease state.
10 Conclusions
Although complete genome sequences have provided a great first step toward the comprehensive identification of the compendium of TFs that function in an organism of interest, we are far from having a complete picture of all the protein variants that may exist for each predicted TF. As we discussed here, there are numerous factors that affect the functional states of TFs throughout development, homeostasis, and in disease. Since the gene count is strikingly similar between organisms of widely different complexity, a larger number of TF permutations may contribute to more intricate regulatory networks in higher eukaryotes such as humans. In the future, different TF forms need to be incorporated as individual nodes in transcription regulatory networks to facilitate network modeling and hypothesis derivation (Fig. 3).
Fig. 3.

Whereas traditional transcription regulatory networks have been visualized using a single node for a TF protein, the use of individual nodes for each functional TF state may help to depict the regulatory capacity of each TF. TF (blue circle) variant “a” forms heterodimers, variant “b” is post-translationally modified and variant “c” is a result of a mutation in the gene encoding the TF. Each variant has different target genes (diamonds) and/or different effects on those targets (arrows = transcriptional activation, flat arrows = repression).
Acknowledgments
We thank members of the Walhout lab and Job Dekker for discussions and critical reading of the manuscript. Research in the Walhout lab is sponsored by NIH grants DK068429 and DK071713.
Biographies

Christian Grove received an interdisciplinary BS in biology and physics from Worcester Polytechnic Institute in 2001. Currently, he is a PhD student of the Interdisciplinary Graduate Program at the University of Massachusetts Medical School in Worcester, MA. His current thesis work is focused on the study of the C. elegans basic helix–loop–helix (bHLH) transcription factors, particularly their dimerization preferences, in vivo localization, and DNA-binding specificities.

A. J. Marian Walhout is an Associated Professor in the Program of Gene Function and Expression and the Program in Molecular Medicine at the University of Massachusetts Medical School. She obtained her BS (Biology, 1992) and PhD (Physiological Chemistry, 1997) degrees from Utrecht University, The Netherlands. She did her postdoctoral work at Harvard Medical School in the lab of Dr Marc Vidal, first at the MGH cancer center and later at the Dana-Farber Cancer Institute in the department of Cancer Biology.
References
- 1.Walhout AJM. Genome Res. 2006;16(12):1445–1454. doi: 10.1101/gr.5321506. [DOI] [PubMed] [Google Scholar]
- 2.Harbison CT, Gordon DB, Lee TI, Rinaldi NJ, Macisaac KD, Danford TW, Hannett NM, Tagne JB, Reynolds DB, Yoo J, Jennings EG, Zeitlinger J, Pokholok DK, Kellis M, Rolfe PA, Takusagawa KT, Lander ES, Gifford DK, Fraenkel E, Young RA. Nature. 2004;431(7004):99–104. doi: 10.1038/nature02800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Boyer LA, Lee TI, Cole MF, Johnstone SE, Levine SS, Zucker JP, Guenther MG, Kumar RM, Murray HL, Jenner RG, Gifford DK, Melton DA, Jaenisch R, Young RA. Cell. 2005;122(6):947–956. doi: 10.1016/j.cell.2005.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sandmann T, Jensen LJ, Jakobsen JS, Karzynski MM, Eichenlaub MP, Bork P, Furlong EE. Dev Cell. 2006;10(6):797–807. doi: 10.1016/j.devcel.2006.04.009. [DOI] [PubMed] [Google Scholar]
- 5.Mukherjee S, Berger MF, Jona G, Wang XS, Muzzey D, Snyder M, Young RA, Bulyk ML. Nat Genet. 2004;36(12):1331–1339. doi: 10.1038/ng1473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.van Steensel B, Henikoff S. Nat Biotechnol. 2000;18(4):424–428. doi: 10.1038/74487. [DOI] [PubMed] [Google Scholar]
- 7.Moorman C, Sun LV, Wang J, de Wit E, Talhout W, Ward LD, Greil F, Lu XJ, White KP, Bussemaker HJ, van Steensel B. Proc Natl Acad Sci USA. 2006;103(32):12027–12032. doi: 10.1073/pnas.0605003103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Meng X, Brodsky MH, Wolfe SA. Nat Biotechnol. 2005;23(8):988–994. doi: 10.1038/nbt1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Deplancke B, Dupuy D, Vidal M, Walhout AJM. Genome Res. 2004;14(10B):2093–2101. doi: 10.1101/gr.2445504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Deplancke B, Mukhopadhyay A, Ao W, Elewa AM, Grove CA, Martinez NJ, Sequerra R, Doucette-Stamm L, Reece-Hoyes JS, Hope IA, Tissenbaum HA, Mango SE, Walhout AJM. Cell. 2006;125(6):1193–1205. doi: 10.1016/j.cell.2006.04.038. [DOI] [PubMed] [Google Scholar]
- 11.Vermeirssen V, Barrasa MI, Hidalgo CA, Babon JA, Sequerra R, Doucette-Stamm L, Barabási AL, Walhout AJM. Genome Res. 2007;17(7):1061–1071. doi: 10.1101/gr.6148107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Borneman AR, Leigh-Bell JA, Yu H, Bertone P, Gerstein M, Snyder M. Genes Dev. 2006;20(4):435–448. doi: 10.1101/gad.1389306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Babu MM, Luscombe NM, Aravind L, Gerstein M, Teichmann SA. Curr Opin Struct Biol. 2004;14(3):283–291. doi: 10.1016/j.sbi.2004.05.004. [DOI] [PubMed] [Google Scholar]
- 14.Guelzim N, Bottani S, Bourgine P, Képès F. Nat Genet. 2002;31(1):60–63. doi: 10.1038/ng873. [DOI] [PubMed] [Google Scholar]
- 15.Shen-Orr SS, Milo R, Mangan S, Alon U. Nat Genet. 2002;31(1):64–68. doi: 10.1038/ng881. [DOI] [PubMed] [Google Scholar]
- 16.Milo R, Shen-Orr S, Itzkovitz S, Kashtan N, Chklovskii D, Alon U. Science. 2002;298(5594):824–827. doi: 10.1126/science.298.5594.824. [DOI] [PubMed] [Google Scholar]
- 17.Davidson EH, Rast JP, Oliveri P, Ransick A, Calestani C, Yuh CH, Minokawa T, Amore G, Hinman V, Arenas-Mena C, Otim O, Brown CT, Livi CB, Lee PY, Revilla R, Rust AG, Pan Z, Schilstra MJ, Clarke PJ, Arnone MI, Rowen L, Cameron RA, McClay DR, Hood L, Bolouri H. Science. 2002;295(5560):1669–1678. doi: 10.1126/science.1069883. [DOI] [PubMed] [Google Scholar]
- 18.Luscombe NM, Babu MM, Yu H, Snyder M, Teichmann SA, Gerstein M. Nature. 2004;431(7006):308–312. doi: 10.1038/nature02782. [DOI] [PubMed] [Google Scholar]
- 19.Smith J, Theodoris C, Davidson EH. Science. 2007;318(5851):794–797. doi: 10.1126/science.1146524. [DOI] [PubMed] [Google Scholar]
- 20.Lemon B, Tjian R. Genes Dev. 2000;14(20):2551–2569. doi: 10.1101/gad.831000. [DOI] [PubMed] [Google Scholar]
- 21.Kummerfeld SK, Teichmann SA. Nucleic Acids Res. 2006;34(Database issue):D74–D81. doi: 10.1093/nar/gkj131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Messina DN, Glasscock J, Gish W, Lovett M. Genome Res. 2004;14(10B):2041–2052. doi: 10.1101/gr.2584104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Reece-Hoyes JS, Deplancke B, Shingles J, Grove CA, Hope IA, Walhout AJM. Genome Biol. 2005;6(13):R110. doi: 10.1186/gb-2005-6-13-r110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vermeirssen V, Deplancke B, Barrasa MI, Reece-Hoyes JS, Arda HE, Grove CA, Martinez NJ, Sequerra R, Doucette-Stamm L, Brent MR, Walhout AJM. Nat Methods. 2007;4(8):659–664. doi: 10.1038/nmeth1063. [DOI] [PubMed] [Google Scholar]
- 25.Taneri B, Snyder B, Novoradovsky A, Gaasterland T. Genome Biol. 2004;5(10):R75. doi: 10.1186/gb-2004-5-10-r75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ogg S, Paradis S, Gottlieb S, Patterson GI, Lee L, Tissenbaum HA, Ruvkun G. Nature. 1997;389(6654):994–999. doi: 10.1038/40194. [DOI] [PubMed] [Google Scholar]
- 27.Oh SW, Mukhopadhyay A, Dixit BL, Raha T, Green MR, Tissenbaum HA. Nat Genet. 2005;38(2):251–257. doi: 10.1038/ng1723. [DOI] [PubMed] [Google Scholar]
- 28.Bach I, Yaniv M. EMBO J. 1993;12(11):4229–4242. doi: 10.1002/j.1460-2075.1993.tb06107.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Johnson JM, Castle J, Garrett-Engele P, Kan Z, Loerch PM, Armour CD, Santos R, Schadt EE, Stoughton R, Shoemaker DD. Science. 2003;302(5653):2141–2144. doi: 10.1126/science.1090100. [DOI] [PubMed] [Google Scholar]
- 30.Wolberger C. Annu Rev Biophys Biomol Struct. 1999;28:29–56. doi: 10.1146/annurev.biophys.28.1.29. [DOI] [PubMed] [Google Scholar]
- 31.Newman JR, Keating AE. Science. 2003;300(5628):2097–2101. doi: 10.1126/science.1084648. [DOI] [PubMed] [Google Scholar]
- 32.Lamb P, McKnight SL. Trends Biochem Sci. 1991;16(11):417–422. doi: 10.1016/0968-0004(91)90167-t. [DOI] [PubMed] [Google Scholar]
- 33.Walhout AJM, Sordella R, Lu X, Hartley JL, Temple GF, Brasch MA, Thierry-Mieg N, Vidal M. Science. 2000;287(5450):116–122. doi: 10.1126/science.287.5450.116. [DOI] [PubMed] [Google Scholar]
- 34.Berk AJ. Biochim Biophys Acta. 1989;1009(2):103–109. doi: 10.1016/0167-4781(89)90087-0. [DOI] [PubMed] [Google Scholar]
- 35.Tremblay A, Tremblay GB, Labrie F, Giguère V. Mol Cell. 1999;3(4):513–519. doi: 10.1016/s1097-2765(00)80479-7. [DOI] [PubMed] [Google Scholar]
- 36.Kim MY, Woo EM, Chong YT, Homenko DR, Kraus WL. Mol Endocrinol. 2006;20(7):1479–1493. doi: 10.1210/me.2005-0531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dahlman-Wright K, Cavailles V, Fuqua SA, Jordan VC, Katzenellenbogen JA, Korach KS, Maggi A, Muramatsu M, Parker MG, Gustafsson JA. Pharmacol Rev. 2006;58(4):773–781. doi: 10.1124/pr.58.4.8. [DOI] [PubMed] [Google Scholar]
- 38.Smith CL, O'Malley BW. Endocr Rev. 2004;25(1):45–71. doi: 10.1210/er.2003-0023. [DOI] [PubMed] [Google Scholar]
- 39.Motola DL, Cummins CL, Rottiers V, Sharma KK, Li T, Li Y, Suino-Powell K, Xu HE, Auchus RJ, Antebi A, Mangelsdorf DJ. Cell. 2006;124(6):1209–1223. doi: 10.1016/j.cell.2006.01.037. [DOI] [PubMed] [Google Scholar]
- 40.Denison MS, Nagy SR. Annu Rev Pharmacol Toxicol. 2003;43:309–334. doi: 10.1146/annurev.pharmtox.43.100901.135828. [DOI] [PubMed] [Google Scholar]
- 41.Denison MS, Heath-Pagliuso S. Bull Environ Contam Toxicol. 1998;61(5):557–568. doi: 10.1007/pl00002973. [DOI] [PubMed] [Google Scholar]
- 42.Qin H, Powell-Coffman JA. Dev Biol. 2004;270(1):64–75. doi: 10.1016/j.ydbio.2004.02.004. [DOI] [PubMed] [Google Scholar]
- 43.Huang X, Powell-Coffman JA, Jin Y. Development. 2004;131(4):819–828. doi: 10.1242/dev.00959. [DOI] [PubMed] [Google Scholar]
- 44.Roeder RG. FEBS Lett. 2005;579(4):909–915. doi: 10.1016/j.febslet.2004.12.007. [DOI] [PubMed] [Google Scholar]
- 45.Rosenfeld MG, Lunyak VV, Glass CK. Genes Dev. 2006;20(11):1405–1428. doi: 10.1101/gad.1424806. [DOI] [PubMed] [Google Scholar]
- 46.Perissi V, Rosenfeld MG. Nat Rev Mol Cell Biol. 2005;6(7):542–554. doi: 10.1038/nrm1680. [DOI] [PubMed] [Google Scholar]
- 47.Yu Y, Li W, Su K, Yussa M, Han W, Perrimon N, Pick L. Nature. 1997;385(6616):552–555. doi: 10.1038/385552a0. [DOI] [PubMed] [Google Scholar]
- 48.Strano S, Dell'Orso S, Di Agostino S, Fontemaggi G, Sacchi A, Blandino G. Oncogene. 2007;26(15):2212–2219. doi: 10.1038/sj.onc.1210296. [DOI] [PubMed] [Google Scholar]
- 49.Firulli BA, Krawchuk D, Centonze VE, Vargesson N, Virshup DM, Conway SJ, Cserjesi P, Laufer E, Firulli AB. Nat Genet. 2005;37(4):373–381. doi: 10.1038/ng1525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Castilla LH, Garrett L, Adya N, Orlic D, Dutra A, Anderson S, Owens J, Eckhaus M, Bodine D, Liu PP. Nat Genet. 1999;23(2):144–146. doi: 10.1038/13776. [DOI] [PubMed] [Google Scholar]