

Abstract
Two-photon fluorescence lifetime imaging microscopy (TPFLIM) enables the quantitative measurements of fluorescence resonance energy transfer (FRET) in small subcellular compartments in light scattering tissue. We evaluated and optimized the FRET pair of mEGFP (monomeric EGFP with the A206K mutation) and REACh (non-radiative YFP variants) for TPFLIM. We characterized several mutants of REACh in terms of their “darkness,” and their ability to act as a FRET acceptor for mEGFP in Hela cells and hippocampal neurons. Since the commonly used monomeric mutation A206K increases the brightness of REACh, we introduced a different monomeric mutation (F223R) which does not affect the brightness. Also, we found that the folding efficiency of original REACh, as measured by the fluorescence lifetime of a mEGFP-REACh tandem dimer, was low and variable from cell to cell. Introducing two folding mutations (F46L, Q69M) into REACh increased the folding efficiency by ∼50%, and reduced the variability of FRET signal. Pairing mEGFP with the new REACh (super-REACh, or sREACh) improved the signal-to-noise ratio compared to the mEGFP–mRFP or mEGFP–original REACh pair by ∼50 %. Using this new pair, we demonstrated that the fraction of actin monomers in filamentous and globular forms in single dendritic spines can be quantitatively measured with high sensitivity. Thus, the mEGFP-sREACh pair is suited for quantitative FRET measurement by TPFLIM, and enables us to measure protein-protein interactions in individual dendritic spines in brain slices with high sensitivity.
Introduction
In the central nervous system, most excitatory synapses are located in dendritic spines, tiny (volume 0.1 - 0.01 femtoliters) mushroom-shaped protrusions emanating from the dendritic surface. Biochemical signaling in spines is important for many forms of synaptic plasticity and structural plasticity of spines (Alvarez and Sabatini, 2007; Kennedy et al., 2005). Because signaling in each dendritic spine is regulated differently (Alvarez and Sabatini, 2007; Kennedy et al., 2005), the coupling between synaptic activity and intracellular signaling has to be studied ultimately at the level of individual spines. However, due to the small size of dendritic spines (∼femtoliter), and light scattering by brain tissue, it has been difficult to measure the molecular signaling in spines. Recently, protein-protein interactions in individual dendritic spines in brain slices have been successfully measured by combining FRET imaging techniques with 2-photon microscopy (Okamoto et al., 2004; Yasuda et al., 2006). Further improvements in the sensitivity of FRET imaging will be crucial for quantitative measurements of signaling processes in spines.
FRET is the process of non-radiative energy transfer from an excited donor fluorophore to an acceptor fluorophore. Because FRET strongly depends on the distance between the donor and acceptor, FRET can be used as a readout of protein-protein interactions for proteins that are fused to fluorophores (Lakowicz, 2006; Miyawaki, 2003). FRET is often measured by calculating the ratio between the donor and acceptor fluorescence. Alternatively, the fluorescence lifetime of the donor, which is the time between excitation of fluorophore and emission of photon, can be used as a readout of FRET, since the lifetime shortens as the FRET efficiency increases (Lakowicz, 2006).
Fluorescence lifetime imaging microscopy (FLIM) has many advantages over ratiometic fluorescence or other intensity based measurements of FRET(Yasuda, 2006). First, fluorescence lifetime is independent of local fluorophore concentration or wavelength dependent light scattering, unlike the intensity based measurements. Second, because the fluorescence lifetime is proportional to FRET efficiency, calculation of the FRET efficiency is straightforward. Finally, this technique allows us to deconvolve the FRET and non-FRET components to measure the binding fraction of the FRET population, or the fraction of donors bound to acceptors. As a FRET donor for TPFLIM, enhanced green fluorescent protein (EGFP) or its monomeric variant (EGFPA206K or mEGFP) is superior to other GFP color variants, because it is bright and photostable under 2-photon microscopy and has a mono-exponential fluorescence lifetime decay. For the FRET acceptor, monomeric red fluorescent protein (mRFP) (Campbell et al., 2002) has often been used for FLIM, because of its high extinction coefficient and good spectral separation with EGFP (Peter et al., 2005; Tramier et al., 2006; Yasuda et al., 2006). Although mRFP is not the brightest red fluorescent protein, the brightness of the acceptor is not important for FLIM, because FRET-FLIM typically measures only the donor fluorescence. However, a high acceptor absorption coefficient is required for high FRET efficiency (Yasuda, 2006). An acceptor with low quantum yield could provide a better signal-to-noise ratio, because of less bleed-through from the acceptor into the donor fluorescence detector. With respect to this, the recently developed fluorophore REACh, a YFP variants with extremely small quantum efficiency (QY ∼0.1) but with high absorbance, can also be used as a FLIM acceptor (Ganesan et al., 2006).
In this paper, we evaluated the mEGFP–REACh FRET pair for TPFLIM, and further optimized the properties of REACh by introducing several mutations. The resulting pair of mEGFP–REACh variant (sREACh) provides better signal-to-noise ratio compared to the mEGFP – REACh or mEGFP – mRFP pairs. Using this new FRET pair, we quantified the molar fraction of the actin monomers in filamentous form (F-actin) in spines and dendrites.
Results
The relative darkness of REACh variants measured in HeLa cells
Because the fluorescence spectrum of REACh overlaps with mEGFP, mEGFP fluorescence lifetimes could be underestimated due to contamination by the residual short lifetime of REACh (∼ 0.35 ns). Thus, to use REACh as an acceptor for mEGFP, REACh's “darkness” must be maximized. We have developed a system to quantify the darkness of REACh compared to mEGFP under the microscope. To achieve this, we expressed a tandem construct consisting of mKeima and REACh in HeLa cells, and excited both simultaneously with a 920 nm laser (Fig. 1a). We used the relative fluorescence intensities of REACh (green) to mKeima (red) as a measure of the darkness of REACh. This ratio was then normalized by the ratio between mEGFP and mKeima fluorescence (Fig. 1b, c). Bleed through of mEGFP into the red channel was corrected. We ignored FRET from mEGFP or REACh to mKeima, because mEGFP and REACh's emission spectra do not overlap much with mKeima's excitation spectrum (Kogure et al., 2006). The brightness of REACh1 (#2, EYFPY145W) and REACh2 (#3, EYFPY145W, V148V) was measured to be 2.8% and 1.3% of mEGFP, respectively (Fig. 1c).
Fig. 1. Brightness of REACh mutants.
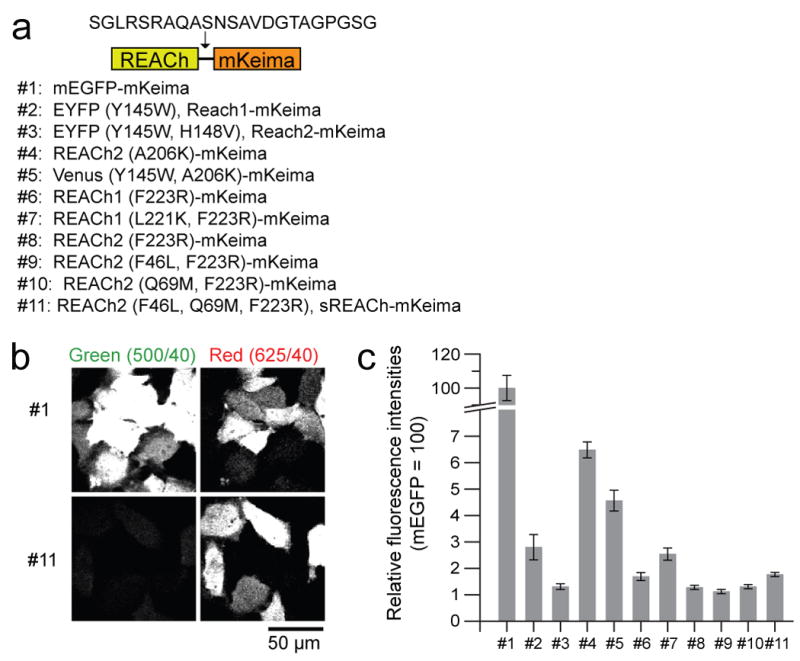
(a) A schematic representation of the structure of the tandem fluorescent protein made to evaluate the brightness of REACh mutants, and the list of mutants used for evaluation of brightness.
(b) Simultaneous capture of REACh mutant and mKeima fluorescences in the green and red channels, respectively. HeLa cells expressing mEGFP-mKeima (Top panels) or sREACh-mKeima (bottom panels) were simultaneously excited using a 920 nm excitation beam. Left panels show the fluorescence captured using a HQ500/40 filter. Right panels show the fluorescence captured using a HQ625/40 filter. Scale bar = 50 μm.
(c) Quantification of the relative brightness of REACh proteins with various mutations. In individual cells, the fluorescence intensities of mEGFP or REACh mutants were divided by fluorescence intensities of mKeima. The relative fluorescence intensity of mEGFP divided by mKeima fluorescence is normalized to 100. The number of cells is 279, 21, 97, 96, 71, 48, 29, 135, 120, 68, and 204, respectively. Error bars indicate ± s.e.m.
Since EGFP variants have a small tendency to dimerize (Zacharias et al., 2002), we introduced a monomerizing mutation, A206K (#4), to REACh2, and found that this greatly increased brightness, which could interfere with the fluorescence lifetime imaging of mEGFP. Adding the REACh1 mutation (Y145W) to monomeric VenusA206K (#5; Venus is brighter variant of YFP) also made Venus three times brighter than REACh2 (#3, EYFPY145W, V148V)(Kwok et al., 2008; Nagai et al., 2002). To avoid this brightening by the A206K mutation, we tried a different published monomerizing mutations (#6, #7, #8) (Zacharias et al., 2002), and found that the F223R (#6, #8) mutation did not affect the brightness (Fig. 1c). Since the fluorescence of this monomeric REACh is only ∼1% of mEGFP fluorescence, it will not have a major impact on the lifetime measurements of mEGFP.
Characterization of REACh mutants as FLIM acceptors
To evaluate the signal-to-noise ratio of the mEGFP-REACh pair under FLIM, we compared the fluorescence lifetime of mEGFP-REACh and mEGFP-mRFP fusion proteins under TPFLIM (Fig. 2). The fluorescence lifetime decay curve after a short laser pulse (Ti: Sapphire laser, pulse width ∼ 200 fs, repetition rate ∼ 80 MHz) was measured by time-correlated single photon counting (TCSPC). As reported previously, the fluorescence lifetime decay curve of mEGFP fits well with a single exponential function convolved with a Gaussian system response function, G(t) (Fig. 2b):
Fig. 2. FRET efficiency and maturity of REACh mutants.
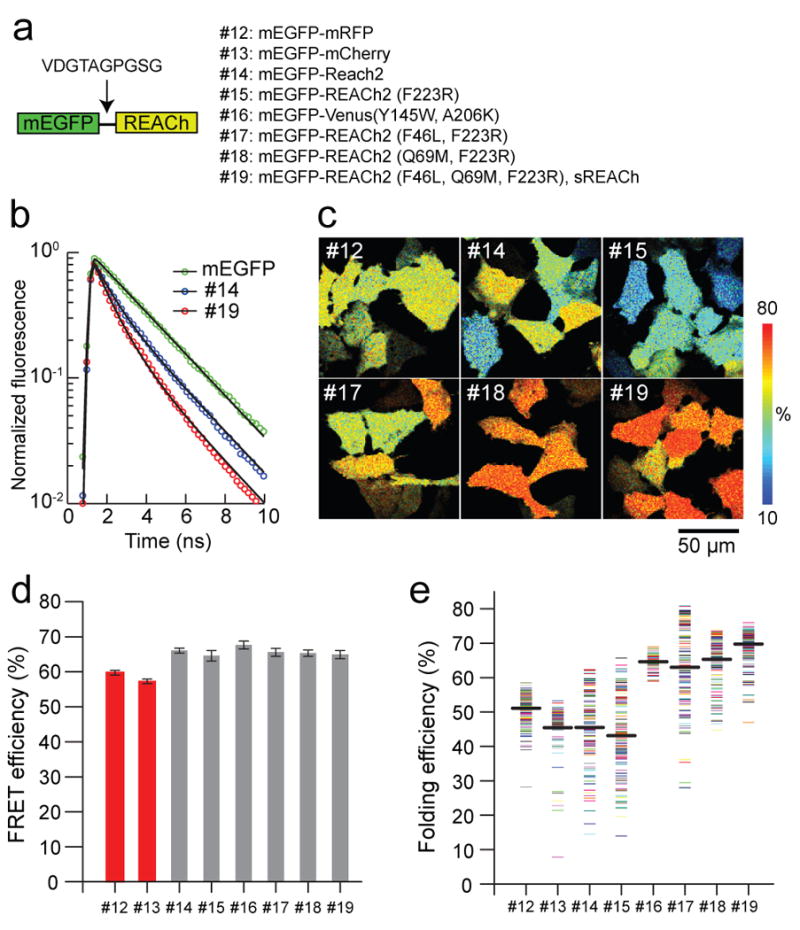
(a) A schematic representation of the structure of the tandem fluorescent protein, and the list of mutants used to evaluate the FRET efficiency and maturation level of REACh mutants.
(b) Fluorescence lifetime curves of mEGFP, mEGFP-REACh2 (#14), and mEGFP-sREACh (#19). Fluorescence lifetime curves were fitted with a double exponential function (Eq. 2), and normalized to F0 (Eq. 2).
(c) Representative folding efficiency images of the mEGFP-mRFP and mEGFP-REACh tandem mutants in HeLa cells. For mutant identities, see (a).
(d) Comparison of the FRET efficiency of the mEGFP-mRFP and mEGFP-REACh tandems. The FRET efficiency of each image was obtained from the fluorescence decay curve averaged over the whole image. The number of images is 29, 20, 20, 22, 25, 20, 22, and 58. Each image contains 6-12 cells. Error bars indicate ± s.d.
(e) Comparison of the binding fraction of mEGFP-mRFP and mEGFP-REACh tandems. Each point represents one cell. Average binding fractions (thick black horizontal bar) are #12, 51.2 ± 4.3; #13, 45.5 ± 7.8; #14, 45.5 ± 10.2; #15, 43.2 ± 11.7; #16,64.6 ± 2.2; #17, 63.0 ± 12.2; #18, 65.3 ± 7.0; and #19,68.6 ± 4.8 (mean ± s.d.). The number of cells is 147, 82, 119, 122, 79, 112, 137, and 159, and 230, respectively.
| (Eq. 1) |
where τmEGFP = 2.6 ns is the time constant of mEGFP. The fluorescence decay curve of mEGFP fused to REACh or mRFP showed a decreased fluorescence lifetime, indicating FRET from mEGFP to REACh or mRFP (Fig. 2b). The fluorescence lifetime curve of these tandem dimers showed multiple components. The time constant of the slowest component was similar to that of free mEGFP (2.6 ns), suggesting that this component is from proteins with mEGFP but an unfolded acceptor. To quantify the FRET efficiency and folding fraction of the acceptor separately, we fitted the fluorescence lifetime curve with a double exponential function, with the fluorescence lifetime of the second component fixed to that of mEGFP (2.6 ns):
| (Eq. 2) |
where P0 and P1 are the population of fast and slow components. Assuming that the fast and slow components are from mEGFP paired with folded and unfolded acceptors, respectively, the mean FRET efficiency (YFRET) between mEGFP and folded acceptor was calculated as
| (Eq. 3) |
and the folding efficiency (YFold) was calculated as
| (Eq. 4) |
The FRET efficiencies for mEGFP-REACh2 (#14) and the mEGFP-monomeric REACh2F223R pair (#15) were similar to those for mEGFP-mRFP and mEGFP-mCherry pairs (#12, #13) (Fig. 2c,d). This shows that, when folded, REACh is just as good of a FRET acceptor as mRFP. However, the folding efficiency of REACh2 and monomeric REACh2 was lower and more variable from cell to cell than mRFP (ranges 15-65%) (Fig. 2b, e). The variable folding efficiency of the acceptor is presumably caused by the environmental sensitivity of REACh2 folding.
To increase the folding efficiency of REACh2F223R, and decrease the variability of FRET signal between mEGFP and REACh2F223R, we introduced further mutations into REACh2F223R (Fig. 2a, e). Introducing the F46L mutation, which improves the folding of YFP (“Venus” mutation) (Nagai et al., 2002)), into monomeric REACh2 improved the folding efficiency by ∼1.5 times (Fig. 2e, #15 vs. #17). Venus with REACh1 mutation (Y145W) also showed high folding efficiency and small variability (Fig. 2e, #16). Introducing Q69M, which makes YFP less pH sensitive (“Citrin” mutation) (Griesbeck et al., 2001), increased the mean folding efficiency and decreased the variability (Fig. 2e, #15 vs. #18). This suggests that Q69M improves the folding efficiency as well as reducing the environmental sensitivity of REACh2. Finally, introducing both mutations (F46L and Q69M) caused the highest folding efficiency and lowered variability (Fig. 2e, #19).
As expected, these folding mutations did not change the FRET efficiency of the folded acceptor (Fig. 2d). Despite the improved folding efficiency, these mutations did not brighten REACh2 (Fig. 1c, #9, #10, #11). Thus, we concluded that REACh2 (F46L, Q69M, F223R) is the best FLIM acceptor because of its “darkness” and high folding efficiency, and named it sREACh (Fig. 1c, 2e).
Actin polymerization quantified with the mEGFP–sREACh pair
Next, we investigated whether the mEGFP-sREACh pair would give better FRET signal when fused to β-actin to measure the molar fraction of polymerized β-actin in dendritic spines (Okamoto et al., 2004). β-actin is highly enriched in filopodia and spines (Bassell et al., 1998; Eom et al., 2003), and regulates the shape of dendritic spines and filopodia during development and structural plasticity at mature synapses (Fischer et al., 2004; Honkura et al., 2008; Krucker et al., 2000; Lamprecht and LeDoux, 2004; Matus, 2000; Okamoto et al., 2004; Sekino et al., 2007). The equilibrium between the filamentous form of actin (F-actin) and globular form (G-actin) is thought to be important for the regulation of the spine structure (Okamoto et al., 2004).
Using the mEGFP–sREACh pair, we measured the molar fraction of F-actin and G-actin in dendritic spines using a similar strategy developed previously for the ECFP–EYFP pair (Okamoto et al., 2004). Neurons in cultured hippocampal slices were sparsely transfected with mEGFP-actin and sREACh-actin by ballistic gene transfer (McAllister, 2000). When mEGFP-actin and sREACh-actin monomers are incorporated into F-actin next to each other, they should produce FRET because the distance between actin monomers in F-actin (5.5 nm) (Holmes et al., 1990) is similar to the Förster distance of the EGFP-REACh pair (5.4-5.9 nm) (Ganesan et al., 2006) (Fig. 3a). Using TPFLIM, we quantified the fluorescence lifetime decay of mEGFP to measure FRET. The fraction of mEGFP-actin monomers that are located next to sREACh -actin monomers on actin filaments (the binding fraction) can be measured by fitting with a double exponential (Eq. 2). Because endogenous actin and unfolded sREACh dilute the signal, the binding fraction depends on the overexpression level. However, the binding fraction should be proportional to the fraction of mEGFP-actin monomer incorporated in F-actin within the same cell (Fig. 3a). Thus, this value can be used to quantify the relative F-actin molar fraction between different subcellular compartments (e.g. spines vs. dendrites) and their time course.
Fig. 3. Comparison between mEGFP- / sREACh-actin and mEGFP- /mRFP-actin FRET sensors.
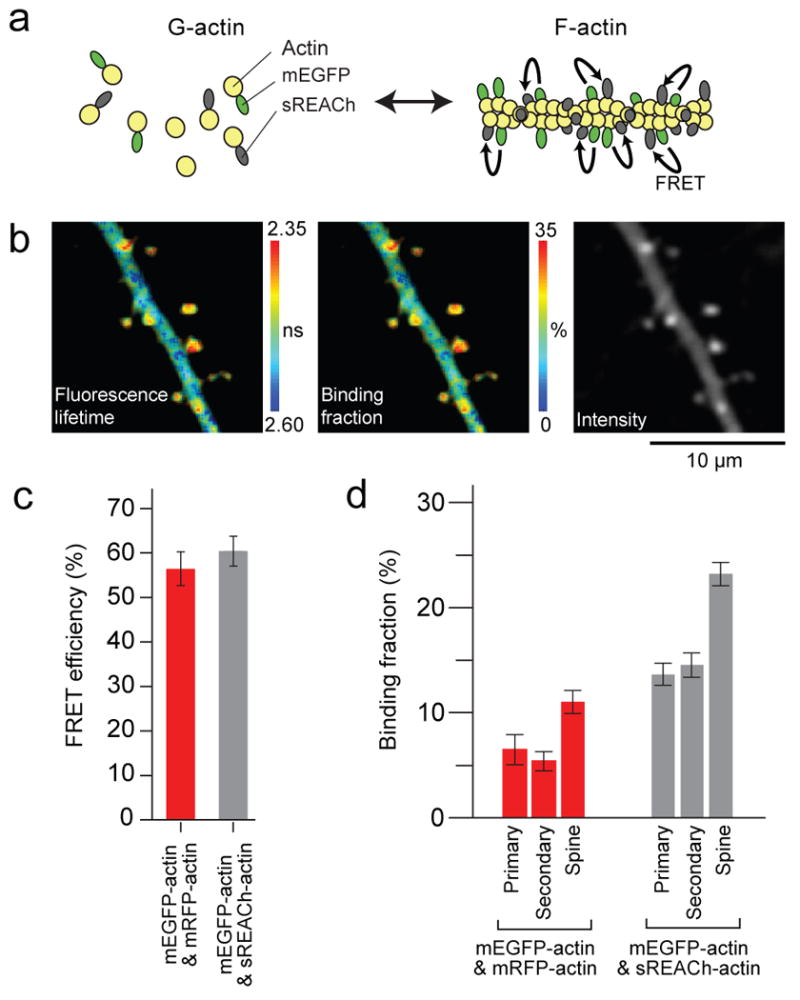
(a) Schematic drawing of the experimental design.
(b) Representative images of a primary dendrite expressing mEGFP- /sREACh-actin sensor. From left, fluorescence lifetime image (measured in nanoseconds), binding fraction image (% donors near acceptors), and fluorescence intensity image of mEGFP-actin.
(c) Comparison of the FRET efficiency of the mEGFP-actin/sREACh-actin (gray bar) and mEGFP-actin/mRFP-actin (red bar). The number of images is 13 and 25, respectively. Each image contains over 10 spines. Error bars indicate ± S.D.
(d) Comparison of the binding fraction of the mEGFP-actin/mRFP-actin (red bar) and mEGFP-actin/sREACh-actin (gray bar). Binding fractions in Primary dendrite, Secondary dendrite, and spines on a secondary dendrite were analyzed. The number of dendrites and spines analyzed is 6, 22, 146 for mRFP, 9, 24, 161 for sREACh, respectively. Error bars indicate ± s.e.m.
The binding fraction of the mEGFP-sREACh pair (transfected with the donor/acceptor cDNA ratio of 1:2) was 23 ± 1% and 14 ± 1% in spines and the dendritic shaft, respectively (Fig. 3b-d). This indicates that spines have ∼1.7 times higher molar fraction of F-actin than in dendrites. Similar ratios were observed for different cDNA transfection ratios (1.7 and 1.5 for mEGFP : sREACh = 1 : 3 and 1 : 4, respectively; data not shown) although the absolute binding fraction was higher for higher ratio. This indicates the linearity of the binding fraction to the molar fraction of F-actin. The higher F-actin fraction in spines than in dendritic shafts is consistent with previous studies (Okamoto et al., 2004; Star et al., 2002). We did not observe a significant difference between primary and secondary dendrites (Fig. 3d).
In comparison, using the actin sensor made of the mEGFP-mRFP pair, we observed 6% and 11% binding fraction in primary dendrites and spines. The lower fraction compared to the mEGFP-sREACh pair is presumably due to lower folding efficiency of mRFP (Fig. 2). The fraction of F-actin was again ∼1.8 times larger in spines than in dendrites, consistent with the measurements using mEGFP-sREACh pair. Thus, the actin polymerization sensor made of the mEGFP-sREACh pair has a higher signal-to-noise ratio compared to the mEGFP-mRFP pair likely due to higher folding efficiency of sREACh (Fig. 2e). In contrast, the FRET efficiency of the mEGFP-sREACh and mEGFP-mRFP pairs was similar, consistent with the results obtained from the tandem dimer made of these pairs (Fig. 2d).
To test the specificity of the actin sensor, we shifted the balance between G- and F-actin by applying Latrunculin A and Jasplakinolide, which prevents and stabilizes polymerization of actin filament, respectively. To monitor both polymerization and the spine volume simultaneously, mCherry, a brighter variant of mRFP (Shaner et al., 2004), was co-expressed with the actin sensor. Latrunculin A decreased F-actin in spines and dendritic shafts by 55 ± 9 and 30 ± 5 % respectively (Fig. 4a, c), and transformed spines into filopodia-like structures (Fig. 4d), implying that presence of F-actin is essential for maintaining the morphology of dendritic spines (Zhang and Benson, 2001). Also, Jasplakinolide increased F-actin by 34 ± 4 and 75 ± 21 % in spines and in dendritic shafts, respectively (Fig. 4b, c). The volume of spines increased over 100% after Jasplakinolide treatment (Fig. 4b, d) (Hering and Sheng, 2003; Wang et al., 2007). Taken together, these results indicate that the fluorescence lifetime of the actin FRET sensor is a sensitive, selective and quantitative measure of actin polymerization.
Fig. 4. Effects of Latrunculin A and Jasplakinolide on actin polymerization.
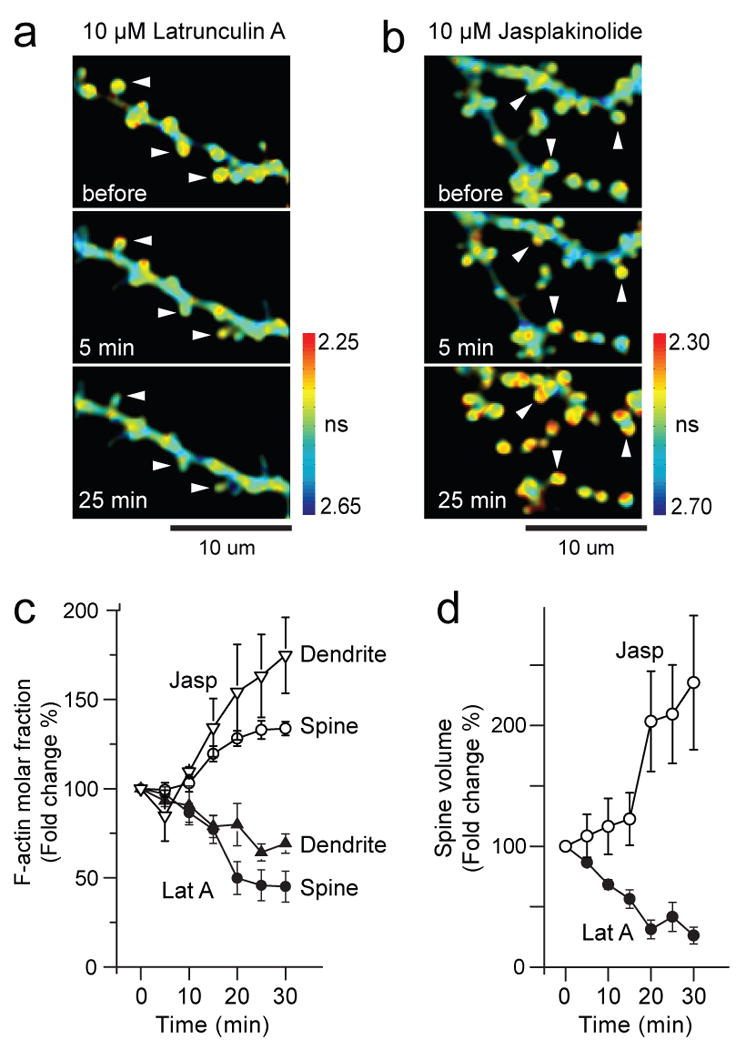
(a-b) Time lapsed fluorescence lifetime images of single dendritic spines on secondary dendrite in the presence of 10μM Latrunculin A (a), and Jasplakinolide (b). Arrowheads point at spines that shrank or transformed into filopodia-like structures. Colorbar shows fluorescence lifetime in nanoseconds.
(c) Time course of polymerization of actin in dendrites (opened triangle, n=3) and spines (opened circle, n=38) caused by treatment with Jasplakinolide, and depolymerization of actin in dendrite (closed triangle, n=3) and spines (closed circle, n=26) caused by treatment with Latrunculin A.
(d) Volume changes of spines caused by treatment with Jasplakinolide (opened circle) or Latrunculin A (closed circle) for the experiment shown in (c).
Discussion
Improvement of REACh for FLIM
The FRET pair of mEGFP-REACh has many favorable properties for TPFLIM over the mEGFP-RFP pair. First, mEGFP-REACh's Förster distance is larger than the mEGFP-mRFP pair, making it easier to design protein-interaction sensors (Ganesan et al., 2006). Second, since REACh does not emit fluorescence, we can use red fluorescent proteins such as mCherry in the same experiment. In this study we further improved REACh as a FRET acceptor by introducing several mutations, and characterized its properties by using simple methods. First, we identified a monomerizing mutation (F223R) that does not affect the darkness of REACh. In contrast, the commonly used A206K mutation made the fluorophore 6-fold brighter. These results suggest that there is some interaction between “dark” mutations at amino acids 145 and 148 and “monomeric” mutations at amino acids 206, 221, and 223. Second, presumably due to the unstable folding and environmental sensitivity of REACh, the mean FRET efficiency of tandem mEGFP-REACh was low, and differed from cell to cell. This folding problem was solved by introducing F46L and Q69M mutations to REACh2. Because unfolded fluorophores do not contribute to FRET signal, the 50% higher folding efficiency of sREACh compared to original REACh should improve FRET signal by a similar percentage, indicating that mEGFP-sREACh pair is suitable for quantitative measurements of protein-protein interactions. We also demonstrated that the signal-to-noise ratio of the actin sensor made with the mEGFP-sREACh pair is double that of the mEGFP–mRFP pair. Furthermore, we also showed that FRET signal in spines and the volume of the same spines can be simultaneously monitored by using the mEGFP-sREACh pair together with cytosolic mCherry (Fig. 4). This was not possible with mEGFP-mRFP pair, because mRFP fluorescence from the FRET pair interferes with the fluorescence measurements of mCherry.
Quantitative measurements of actin polymerization
We demonstrated that the equilibrium between F-actin and G-actin in spines and in dendritic shafts in brain slices can be measured quantitatively by measuring FRET between mEGFP-actin and sREACh-actin with TPFLIM. Previously, it has been demonstrated that changes in the equilibrium between F-actin and G-actin in spines can be measured by FRET between ECFP-actin and Venus-actin using ratiometric imaging. However, because the ratio depends on the relative expression levels of these proteins as well as light scattering of slices, only qualitative analyses of the equilibration were shown. We overcome this limitation by using TPFLIM. By deconvoluting the double-exponential curve of the fluorescence lifetime decay curve, we can measure the fraction of mEGFP-actin monomer with adjacent sREACh-labeled monomer in F-actin. Thus, our method provides a value that is proportional to the fraction of F-actin within the same cell. Using this method, we found that F-actin molar fraction in spines is 1.5 - 1.8 times larger than in dendrites. This ratio was independent of the ratio of expression level between mEGFP and REACh. Furthermore, we obtained the similar value from the mEGFP-mRFP pair. Assuming that the F-actin molar fraction in spines is ∼85 % (Star et al., 2002), the dendritic F-actin in dendrites is 45 -55 %.
In summary, we have demonstrated that the mEGFP-sREACh pair provides quantitative and sensitive FRET measurement under TPFLIM. Using this FRET pair, we were able to measure protein-protein interactions in single dendritic spines in brain slices with high sensitivity
Methods
DNA plasmids
cDNA of mRFP, Venus/mKeima, pECFP-actin, and pCAGGS are kind gift of R.Tsien, A. Miyawaki Y. Hayashi, and M.Matsuda, respectively. Plasmids containing cDNAs of mEGFP (EGFPA206K), mVenus (VenusA206K) and mCherry were prepared as described previously(Zacharias et al., 2002). Plasmid containing cDNA of REACh variants were prepared from pEYFP-C1(Clontech) by a Site-Directed Mutagenesis kit (Stratagene). cDNA of mKeima was subcloned into pEGFP-N3 plasmid by replacing EGFP cDNA with mKeima cDNA. mEGFP-mKeima and REACh variants-mKeima (Fig. 1a) were prepared by mEGFP and REACh variants into the mKeima plasmid. mEGFP-mRFP and mEGFP-REACh variants (Fig. 2) were made by subcloning mRFP and REACh variants into mEGFP plasmids. Plasmids containing cDNA of mEGFP-actin, REACh-actin and mRFP-actin were prepared from pECFP-actin by replacing ECFP with mEGFP, REACh, and mRFP, respectively. The promoters of these constructs (CMV) were then replaced by chicken β-actin promoter from pCAGGS.
HeLa cell culture and transfection
HeLa cells were cultured on 35 mm dishes in Dulbecco's modified eagle medium supplemented with 10% fetal calf serum at 37°C in a 5% CO2 atmosphere. We transfected cells with plasmids using Lipofectamine transfection reagent (Invitrogen Corporation), according to the manufacturer's instructions. Imaging was carried out 18-24 hours after transfection.
Cultured hippocampal slices and transfection
Cultured hippocampal slices were prepared from postnatal day 6 or 7 rats, as described (Stoppini et al., 1991), in accordance with the animal care and use guidelines of Duke University Medical Center. After 1-2 weeks in culture, CA1 cells were transfected with ballistic gene transfer (McAllister, 2000) using gold beads (8-12 mg) coated with pCAGGS-mEGFP, pCAGGS-sREACh-βactin, and mCherry-C1 at the ratio 1:2:1. Imaging was carried out 2-3 days after transfection.
2-photon fluorescence lifetime imaging
The details of FRET imaging using 2-photon FLIM in hippocampal slice culture were described previously (Yasuda et al., 2006). We used a custom-built two-photon microscope with two Ti:sapphire lasers. One laser was tuned to 920 nm to excite both mEGFP-actin for fluorescence lifetime measurements and mCherry for morphology data. The intensity of the laser beam was controlled using electro-optical modulators (Pockels cells, Conoptics). The laser scanning was controlled using scan mirrors and the fluorescence was acquired by the objective lens (60×, 0.9 NA, Olympus). The mEGFP and mCherry fluorescence were separated using a dichroic mirror (565 nm) and band-pass filters (500/40, 620/40; Chroma).
TPFLIM was performed as described previously (Yasuda et al., 2006). Briefly, fluorescence decay curves were measured by comparing the times of laser pulses (80 MHz) detected by a photodiode (FDS010, Thorlabs) and photon pulses from a fast photomultiplier tube (PMT; H7422-40, Hamamatsu) using a time-correlated single photon counting board (SPC-730, Becker-Hickl) (Lakowicz, 2006; Yasuda et al., 2006). Data acquisition and analysis was performed by custom software on Matlab 7.0. Red fluorescence signals from PMTs (R3896, Hamamatsu) were acquired using a separate data acquisition board (PCI-6110).
Hela cells were imaged in a solution containing HEPES(30 mM, pH7.3) buffered ACSF (130 mM NaCl, 2.5 mM KCl, 1 mM CaCl2, 1 mM MgCl2, 2 mM NaHCO3, 1.25 mM NaH2PO4 and 25 mM glucose).
CA1 pyramidal neurons in cultured hippocampal slices were imaged in ACSF (127 mM NaCl, 2.5 mM KCl, 2 mM CaCl2, 2 mM MgCl2, 25 mM NaHCO3, 1.25 mM NaH2PO4 and 25 mM glucose) aerated with 95% O2 and 5% CO2 at 25-26°C. In some experiments, 10 μM Jasplakinolide and 10 μM Latrunculin A (Calbiochem) were added to the ACSF with indicated concentrations.
Data analysis
To obtain mEGFP fluorescence lifetime, we summed over all pixels in an image of several HeLa cells expressing mEGFP, and fit a fluorescence lifetime curve with a single exponential function convolved with the Gaussian pulse response function:
where F0 is the constant, and
in which τD is the fluorescence lifetime of the free donor (mEGFP), τG is the width of the Guassian pulse response function, F0 is the peak fluorescence before convolution and t0 is the time offset, and erf is the error function.
To measure the fraction of donor that is bound to acceptor, we fit a fluorescence lifetime curve summed over a whole image with a double exponential function convolved with the Gaussian pulse response function:
where τAD is the fluorescence lifetime of donor bound with acceptor, PD and PAD are the fraction of free donor and donor bound with acceptor. We fixed τD to the fluorescence lifetime obtained from free mEGFP (2.6 ns). To obtain PD and PAD from regions-of-interest (ROIs) in an image (cells, spines, or dendrites), we fixed τD, τAD, τG, and t0 were fixed to the values obtained from the whole image.
To generate the fluorescence lifetime image, we calculated the mean photon arrival time, <t>, in each pixel as:
Then, the mean photon arrival time is related to the mean fluorescence lifetime, <τ>, by an offset arrival time, to, which is obtained by fitting the whole image:
To generate image of the binding fraction (such as Fig. 2c and 3d), the binding fraction in each pixel (PAD) was obtained as:
where τD and τAD were obtained by fitting the whole image.
Acknowledgments
We thank Drs. Y. Hayashi, M. Matsuda, and A. Miyawaki for plasmids, K. Svoboda and H. Zhong for discussion, and C. Harvey, M. Patterson for comments on the manuscript. We also thank A. Wan for preparing cultured slices and T. Zimmerman for laboratory management. This study was supported by the Burroughs Wellcome Fund, Alfred P. Sloan foundation, Dana foundation, National Aliance of Autism Research, National Institute of Health / National Institute of Mental Health, National Science Foundation, and the Japan Society for the Promotion of Science (HM).
References
- Alvarez VA, Sabatini BL. Anatomical and physiological plasticity of dendritic spines. Annu Rev Neurosci. 2007;30:79–97. doi: 10.1146/annurev.neuro.30.051606.094222. [DOI] [PubMed] [Google Scholar]
- Bassell GJ, Zhang H, Byrd AL, Femino AM, Singer RH, Taneja KL, Lifshitz LM, Herman IM, Kosik KS. Sorting of beta-actin mRNA and protein to neurites and growth cones in culture. J Neurosci. 1998;18:251–265. doi: 10.1523/JNEUROSCI.18-01-00251.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell RE, Tour O, Palmer AE, Steinbach PA, Baird GS, Zacharias DA, Tsien RY. A monomeric red fluorescent protein. Proc Natl Acad Sci U S A. 2002;99:7877–7882. doi: 10.1073/pnas.082243699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eom T, Antar LN, Singer RH, Bassell GJ. Localization of a beta-actin messenger ribonucleoprotein complex with zipcode-binding protein modulates the density of dendritic filopodia and filopodial synapses. J Neurosci. 2003;23:10433–10444. doi: 10.1523/JNEUROSCI.23-32-10433.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer A, Sananbenesi F, Schrick C, Spiess J, Radulovic J. Distinct roles of hippocampal de novo protein synthesis and actin rearrangement in extinction of contextual fear. J Neurosci. 2004;24:1962–1966. doi: 10.1523/JNEUROSCI.5112-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganesan S, Ameer-Beg SM, Ng TT, Vojnovic B, Wouters FS. A dark yellow fluorescent protein (YFP)-based Resonance Energy-Accepting Chromoprotein (REACh) for Forster resonance energy transfer with GFP. Proc Natl Acad Sci U S A. 2006;103:4089–4094. doi: 10.1073/pnas.0509922103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griesbeck O, Baird GS, Campbell RE, Zacharias DA, Tsien RY. Reducing the environmental sensitivity of yellow fluorescent protein. Mechanism and applications. J Biol Chem. 2001;276:29188–29194. doi: 10.1074/jbc.M102815200. [DOI] [PubMed] [Google Scholar]
- Hering H, Sheng M. Activity-dependent redistribution and essential role of cortactin in dendritic spine morphogenesis. J Neurosci. 2003;23:11759–11769. doi: 10.1523/JNEUROSCI.23-37-11759.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes KC, Popp D, Gebhard W, Kabsch W. Atomic model of the actin filament. Nature. 1990;347:44–49. doi: 10.1038/347044a0. [DOI] [PubMed] [Google Scholar]
- Honkura N, Matsuzaki M, Noguchi J, Ellis-Davies GC, Kasai H. The Subspine Organization of Actin Fibers Regulates the Structure and Plasticity of Dendritic Spines. Neuron. 2008;57:719–729. doi: 10.1016/j.neuron.2008.01.013. [DOI] [PubMed] [Google Scholar]
- Kennedy MB, Beale HC, Carlisle HJ, Washburn LR. Integration of biochemical signalling in spines. Nat Rev Neurosci. 2005;6:423–34. doi: 10.1038/nrn1685. [DOI] [PubMed] [Google Scholar]
- Kogure T, Karasawa S, Araki T, Saito K, Kinjo M, Miyawaki A. A fluorescent variant of a protein from the stony coral Montipora facilitates dual-color single-laser fluorescence cross-correlation spectroscopy. Nat Biotechnol. 2006;24:577–581. doi: 10.1038/nbt1207. [DOI] [PubMed] [Google Scholar]
- Krucker T, Siggins GR, Halpain S. Dynamic actin filaments are required for stable long-term potentiation (LTP) in area CA1 of the hippocampus. Proc Natl Acad Sci U S A. 2000;97:6856–6861. doi: 10.1073/pnas.100139797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwok S, Lee C, Sanchez SA, Hazlett TL, Gratton E, Hayashi Y. Genetically encoded probe for fluorescence lifetime imaging of CaMKII activity. Biochem Biophys Res Commun. 2008;369:519–525. doi: 10.1016/j.bbrc.2008.02.070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lakowicz JR. Principles of Fluorescence Spectroscopy. NY, USA: Plenum; 2006. [Google Scholar]
- Lamprecht R, LeDoux J. Structural plasticity and memory. Nat Rev Neurosci. 2004;5:45–54. doi: 10.1038/nrn1301. [DOI] [PubMed] [Google Scholar]
- Matus A. Actin-based plasticity in dendritic spines. Science. 2000;290:754–758. doi: 10.1126/science.290.5492.754. [DOI] [PubMed] [Google Scholar]
- McAllister AK. Biolistic transfection of neurons. Sci STKE. 2000;2000:PL1. doi: 10.1126/stke.2000.51.pl1. [DOI] [PubMed] [Google Scholar]
- Miyawaki A. Visualization of the spatial and temporal dynamics of intracellular signaling. Dev Cell. 2003;4:295–305. doi: 10.1016/s1534-5807(03)00060-1. [DOI] [PubMed] [Google Scholar]
- Nagai T, Ibata K, Park ES, Kubota M, Mikoshiba K, Miyawaki A. A variant of yellow fluorescent protein with fast and efficient maturation for cell-biological applications. Nat Biotechnol. 2002;20:87–90. doi: 10.1038/nbt0102-87. [DOI] [PubMed] [Google Scholar]
- Okamoto K, Nagai T, Miyawaki A, Hayashi Y. Rapid and persistent modulation of actin dynamics regulates postsynaptic reorganization underlying bidirectional plasticity. Nat Neurosci. 2004;7:1104–1112. doi: 10.1038/nn1311. [DOI] [PubMed] [Google Scholar]
- Peter M, Ameer-Beg SM, Hughes MK, Keppler MD, Prag S, Marsh M, Vojnovic B, Ng T. Multiphoton-FLIM quantification of the EGFP-mRFP1 FRET pair for localization of membrane receptor-kinase interactions. Biophys J. 2005;88:1224–1237. doi: 10.1529/biophysj.104.050153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sekino Y, Kojima N, Shirao T. Role of actin cytoskeleton in dendritic spine morphogenesis. Neurochem Int. 2007;51:92–104. doi: 10.1016/j.neuint.2007.04.029. [DOI] [PubMed] [Google Scholar]
- Shaner NC, Campbell RE, Steinbach PA, Giepmans BN, Palmer AE, Tsien RY. Improved monomeric red, orange and yellow fluorescent proteins derived from Discosoma sp. red fluorescent protein. Nat Biotechnol. 2004;12:1567–1572. doi: 10.1038/nbt1037. [DOI] [PubMed] [Google Scholar]
- Star EN, Kwiatkowski DJ, Murthy VN. Rapid turnover of actin in dendritic spines and its regulation by activity. Nat Neurosci. 2002;5:239–246. doi: 10.1038/nn811. [DOI] [PubMed] [Google Scholar]
- Stoppini L, Buchs PA, Muller DA. A simple method for organotypic cultures of nervous tissue. J Neurosci Methods. 1991;37:173–182. doi: 10.1016/0165-0270(91)90128-m. [DOI] [PubMed] [Google Scholar]
- Tramier M, Zahid M, Mevel JC, Masse MJ, Coppey-Moisan M. Sensitivity of CFP/YFP and GFP/mCherry pairs to donor photobleaching on FRET determination by fluorescence lifetime imaging microscopy in living cells. Microsc Res Tech. 2006;69:933–939. doi: 10.1002/jemt.20370. [DOI] [PubMed] [Google Scholar]
- Wang XB, Yang Y, Zhou Q. Independent expression of synaptic and morphological plasticity associated with long-term depression. J Neurosci. 2007;27:12419–12429. doi: 10.1523/JNEUROSCI.2015-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasuda R. Imaging spatiotemporal dynamics of neuronal signaling using fluorescence resonance energy transfer and fluorescence lifetime imaging microscopy. Curr Opin Neurobiol. 2006;16:551–561. doi: 10.1016/j.conb.2006.08.012. [DOI] [PubMed] [Google Scholar]
- Yasuda R, Harvey CD, Zhong H, Sobczyk A, van Aelst L, Svoboda K. Super-sensitive Ras activation in dendrites and spines revealed by 2-photon fluorescence lifetime imaging. Nat Neurosci. 2006;9:283–291. doi: 10.1038/nn1635. [DOI] [PubMed] [Google Scholar]
- Zacharias DA, Violin JD, Newton AC, Tsien RY. Partitioning of lipid-modified monomeric GFPs into membrane microdomains of live cells. Science. 2002;296:913–916. doi: 10.1126/science.1068539. [DOI] [PubMed] [Google Scholar]
- Zhang W, Benson DL. Stages of synapse development defined by dependence on F-actin. J Neurosci. 2001;21:5169–5181. doi: 10.1523/JNEUROSCI.21-14-05169.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]