

Abstract
Toll-like receptor (TLR)/MyD88 signalling has emerged as a major pathway of pathogen recognition in the innate immune system. Here, we review recent data that begin to show how this pathway controls the immune response to protozoan infection, with particular emphasis on the opportunistic pathogen Toxoplasma gondii. The various ways that the parasite activates and suppresses TLR/MyD88 signalling defines several key principals that illuminate the complexities of the host–pathogen interaction. We also speculate how TLR/MyD88 signalling might be exploited to provide protection against Toxoplasma, as well as other protozoa and infection in general.
Keywords: infection – protozoan, innate immunity, Toll-like receptor
Introduction
Detection of infectious non-self is the first step in the anti-microbial immune response that enables host protection against infection. This is achieved through recognition of pathogen-associated molecular patterns (PAMPs) by germ line-encoded pattern receptor molecules (PRR) expressed in diverse cell types, most prominently those that comprise the innate immune system [1]. Recognition triggers signal transduction leading to production of inflammatory cytokines and other anti-microbial mediators. Dysregulation in the system may lead to inflammatory pathology that can cause clinical disease or even death, as in the case of lethal septic shock. The Toll-like receptor (TLR)/MyD88 pathway has emerged as a major pattern recognition system in these responses [2]. In mice, extensive evidence underscores the importance of TLR/MyD88 in resistance to viral, bacterial and protozoan infection. Recent studies by us and others have revealed the important role of TLR/MyD88 in mouse immunity to the intracellular protozoan Toxoplasma gondii. The host response to this parasite highlights several key aspects of the TLR/MyD88 system, emphasizing the complexities and consequences of pathogen recognition by this and other PRR families.
The T. gondii is an opportunistic infection that, before the advent of highly active anti-retroviral therapy, was a major pathogen associated with acquired immune deficiency syndrome progression [3]. As an apicomplexan parasite, Toxoplasma is related closely to Plasmodium falciparum that kills approximately 2 million people every year [4]. Other less closely related protozoa, such as Trypanosoma and Leishmania spp., are also major causes of human sickness and death. Thus, protozoan pathogens exert a tremendous toll on human health, and control of these pathogens is a major priority. For Toxoplasma in immunocompromised patients, encephalitis is the most common manifestation of disease. This results from reactivation of the parasite in latently infected individuals, and is lethal if not treated appropriately. Toxoplasma may also cross the placenta and infect the fetus, an event that can result in fetal death, hydrocephalus, seizures, mental retardation and retinochoroiditis often emerging as sequelae of infection later in life [5]. The parasite is transmitted orally by ingestion of cysts in undercooked meat, as well as by accidental ingestion of infectious oocysts shed in the faeces of members of the cat family [6]. Tachyzoites disseminate rapidly from the intestinal mucosa, spreading widely throughout the body. Latent infection, marked by conversion to bradyzoites and formation of cysts, is established predominantly within the skeletal muscle and central nervous system and occurs concurrently with the rise of adaptive immunity. Cell-mediated immunity is required to contain the parasite and, in both mice and humans, infection is characterized by high levels of proinflammatory cytokines such as interleukin (IL)-12, tumour necrosis factor (TNF)-α and interferon (IFN)-γ[7,8]. There has been much interest in determining initial events in immunity to this parasite, because these early interactions are likely to play an important role in shaping the ultimate outcome of infection. In this regard, encounter with host TLR/MyD88 pathways is probably an important and possibly decisive force.
Brief overview of the TLR/MyD88 pathway
The TLR/MyD88 pathway has been reviewed extensively and expertly by others [9–11]. To date, 13 mouse TLR and 11 human TLR have been identified. The TLR recognize a wide range of microbial molecules, including bacterial lipopolysaccharide (LPS) (TLR-4), lipid and carbohydrate molecules expressed by Gram-positive bacteria (TLR-1, -2, -6), bacterial flagellin (TLR-5) and bacterial and viral nucleic acids (TLR-3, -7, -8 and -9). Most TLR molecules are expressed on the cell surface, but TLR-3, -7, -8 and -9 are found within cytoplasmic membrane-bound compartments. The intracellular localization of nucleic acid-specific TLR is thought to minimize potential reactivity with self-DNA and RNA [12].
The MyD88 molecule plays a central role in signalling through most TLR as well as signalling mediated through receptors for IL-1 and IL-18 [13,14]. TLR activation results in recruitment of the adaptor molecule MyD88, resulting in recruitment of IL-1 receptor-associated kinases (IRAK) 1 and 4. The IRAK molecules form a complex with TNF receptor-associated factor (TRAF)6, promoting interaction with Ubc13 and Uva1. This complex functions as an E3 ligase, leading to TRAF6 polyubiquitination, which induces activation of transforming growth factor-β-activated kinase-1 (TAK1). In combination with TAK1-binding proteins-1 and -2, TAK1 triggers mitogen-activated protein kinase (MAPK) and nuclear factor (NF)-κB signalling pathways that lead to induction of proinflammatory cytokines. While most TLR use this pathway, TLR-4 activates additionally an MyD88-independent pathway that utilizes the adaptor molecule Toll-IL-1 receptor domain containing adaptor-inducing interferon (TRIF) to convey signal transduction, resulting in induction of type 1 IFN and IFN-inducible genes. Signalling through TLR3 is dependent solely upon the TRIF molecule [15].
The TLR signalling pathway is subject to negative regulation at multiple points [16]. This includes negative interaction with IRAK and TRAF6 molecules to shut down signalling [as in the case of the single immunoglobulin IL-1R-related molecule][17], Triad3-dependent down-regulation of TLR4 and 9, expression of dominant negative variants of components of the TLR signalling pathway (examples include IRAK-M and MyD88s) [18,19] and suppressor of cytokine signalling-1-dependent down-regulation of Toll/Il-1 receptor-domain containing adaptor protein [20]. The presence of negative regulatory circuits emphasizes the importance of avoiding excessive proinflammatory cytokine production by innate immune cells that can lead to pathology and death in the host.
The TLR ligands of Toxoplasma and other protozoans
Ligands of TLR were characterized initially in bacteria and viruses. However, a growing number of protozoan TLR ligands have now been identified [4]. Genomic DNA from Trypanosoma brucei and T. cruzi contains unmethylated cytosine phosphate guanosine (CpG) motifs that trigger TLR-9 [21,22]. Similarly, DNA from L. major, L. infantum and Entamoeba histolytica has been reported to activate macrophages and dendritic cells (DC) through TLR-9 [23,24]. Complexes of haemozoin and DNA from P. falciparum also activate host TLR-9, although controversy surrounds whether this is due to haemozoin itself or closely associated parasite DNA [21,25]. Glycosyphosphatidylinositol (GPI) anchors from several protozoa, including L. major, L. donovani, T. cruzi, T. brucei, and P. falciparum, have emerged as major PAMPs that are recognized by TLR-2, and, in some instances, TLR-4 [26–30].
For the case of Toxoplasma, the existence of parasite TLR ligands was inferred by the initial observation that mice lacking MyD88 are highly susceptible to infection [31]. The existence of multiple parasite TLR ligands was suggested by the identification of distinct MyD88-dependent IL-12 and MyD88-dependent CCL2/MCP1 inducing fractions obtained from tachyzoite lysates [32]. Biochemical fractionation of tachyzoite lysate antigen led to identification of a parasite profilin-like molecule (TGPRF) that is capable of inducing DC IL-12 production through TLR-11, a TLR that is expressed by mice but not humans [33,34]. While TLR-11 was implicated originally in recognition of uropathogenic bacteria [35], Toxoplasma profilin remains the only molecule identified as a ligand of this particular TLR to date. Reverse genetic studies using conditional knock-out parasites revealed that T. gondii TGPRF plays a non-redundant role in host cell invasion [36]. It has been suggested that apicomplexan profilins are a major class of PAMP recognized by TLR-11 [34], a hypothesis that is supported by evidence that a profilin-like molecule derived from Eimeria spp. elicits high-level IL-12 production from splenic CD11c+ DC [37]. Nevertheless, this cannot be universally true for all apicomplexan profilins, because P. falciparum profilin is, at most, a weak inducer of TLR-11-dependent IL-12 [36].
The Toxoplasma tachyzoite surface is dominated by GPI-anchored proteins, and indeed the ability to synthesize GPI is essential for parasite viability [38]. Biochemical studies using Chinese hamster ovary cells co-transfected with TLR and NF-κB reporter plasmids have shown that Toxoplasma GPI molecules are recognized by TLR-4 [39,40]. Interestingly, core glycans and lipid moieties obtained from parasite GPI were recognized by both TLR-2 and TLR-4. In mouse macrophages, both TLR-2 and TLR-4 were found to mediate recognition of Toxoplasma GPI, as measured by induction of TNF-α synthesis [40]. The involvement of both TLR-2 and TLR-4 in GPI recognition by macrophages may indicate that parasite GPI is cleaved by phospholipases expressed at the macrophage surface.
Studies carried out in MyD88−/− mice show unambiguously that this adaptor molecule is required to survive T. gondii infection [31,41,42]. Mice that do not produce IL-1 or IL-18 display normal resistance to infection [41]. This is an important result, because it argues that increased susceptibility in the absence of MyD88 reflects defective TLR signalling per se rather than absence of IL-1/IL-18 signalling that also involves signalling through MyD88. Nevertheless, defects in individual TLR have less dramatic effects during in vivo infection relative to MyD88−/− animals. Mice deficient in TLR-11 survive infection, although brain cyst numbers are increased relative to wild-type counterparts [33]. TLR-2−/− mice were reported to display increased susceptibility to Toxoplasma infection, but here the effect was seen only under conditions of high parasite dose [43]. Under low-dose infection conditions, it has been reported that genetic deletion of both TLR-2 and TLR-4 in the same host leads to an increase in cyst burden, whereas knock-out of individual TLR has no effect [40]. Taken together, the data suggest that optimal resistance to Toxoplasma involves responses to multiple TLR, and that inactivation of any single TLR, therefore, has only modest effects on resistance. Similar conclusions have been made with regard to the cooperative protective activity of TLR-2 and TLR-9 during infection with T. cruzi, as well as in models of Mycobacterium tuberculosis infection [44,45].
Function of MyD88/TLR in adaptive immunity and anti-Toxoplasma effector activity
Effective control of T. gondii infection depends upon a robust IL-12 response early after infection. Without IL-12, parasite replication remains unchecked because of diminished IFN-γ responses, resulting in early death of the host [46]. IFN-γ exerts its anti-Toxoplasma effects through phagocyte activation and up-regulation of anti-microbial molecules such as p47 guanosine triphosphate family members and the iNOS (NOS2) enzyme [47,48]. In an intraperitoneal (i.p.) infection model, absence of MyD88 results in impaired IL-12 and IFN-γ, and the animals succumb rapidly because of high levels of parasites [31]. Along similar lines, MyD88−/− mice undergoing oral infection display increased susceptibility associated with elevated parasite levels in the lamina propria and lymphoid tissues of the intestinal mucosa, and decreased IL-12 production in spleen and mesenteric lymph nodes [42]. Absence of MyD88 also results in defective infiltration of neutrophils to the lamina propria during early oral infection, as well as to the peritoneal cavity during i.p. infection. A requirement for MyD88 for neutrophil recruitment to infected tissues has also been established in several other models of inflammation induced by microbes [49,50].
Signalling through the TLR/MyD88 pathway has been suggested to be a major orchestrator of the adaptive immune response [51]. This is because MyD88-transduced signalling leads to upregulation of co-stimulatory molecules and T cell polarizing factors such as IL-12 that are required to activate T cells and drive effector cell differentiation. The ability of professional antigen-presenting cells such as DC to present antigen and transduce TLR signals simultaneously suggests that TLR ligands can control T cell immunodominance during infection (Fig. 1a) [52,53]. Indeed, the CD4+ T cell response to i.p. injection of soluble tachyzoite lysate is directed largely to TGPRF in a manner dependent upon TLR-11 signalling in splenic DC [54]. A role for T cell intrinsic MyD88 signalling has also been suggested. Bone marrow chimera experiments generating mice deficient in T cell MyD88 expression showed that although mice survived acute infection, the T cell IFN-γ response was diminished, leading to toxoplasmic encephalitis during chronic infection [55].
Fig. 1.
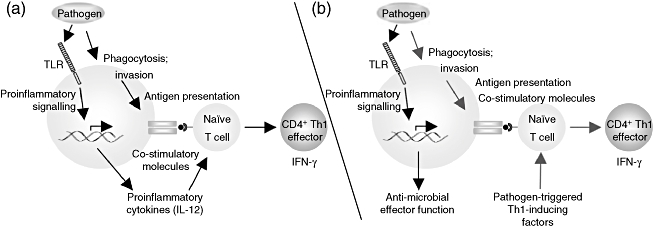
Alternative models for the role of MyD88 in immunity to infection. (a) MyD88 signalling as a bridge to acquired immunity. After pathogen internalization through either active invasion or phagocytosis by a dendritic cell, microbial peptides are displayed on the cell surface in association with major histocompatibility complex (MHC) class II molecules. At the same time, pathogen-associated Toll-like receptor (TLR) ligands trigger proinflammatory cytokine and co-stimulatory expression on the dendritic cell. The combined stimuli trigger activation and differentiation of T cells specific for the antigenic peptide, resulting in some cases to an immunodominant T cell response. (b) Some studies suggest that T lymphocyte activation proceeds independently of MyD88, and that other pathogen-triggered factors provide signals for T cell differentiation (blue arrows). MyD88 is still important in the response to infection (black arrows), but its function is relegated to signalling pathways that lead to innate anti-microbial control of infection, allowing host survival until development of adaptive immunity. Which of these models predominates may depend on the nature of the pathogen, the particular cell type involved or the route of infection. In principle, it is possible that both pathways operate simultaneously in the host response to infection.
However, several studies suggest that TLR/MyD88 signalling is non-essential in generation of T cell immunity during microbial infection and, indeed, a recent study found that humans lacking MyD88 are surprisingly resistant to infection [56–59]. We examined this issue in detail in mice infected orally with Toxoplasma[42]. Although emergence of T helper Type 1 (Th1) effector cells was delayed in the absence of MyD88 signalling, IFN-γ producing CD4+ T lymphocytes were generated independently of this TLR adaptor. Vaccination of MyD88−/− mice with an avirulent T. gondii strain elicited fully functional protective immunity to both subcutaneous and oral challenge with a lethal dose of the parasite. Because TLR/MyD88 signal transduction is required to control Toxoplasma infection, this signalling axis is clearly essential in the innate anti-microbial response to the parasite. None the less, MyD88 does not appear to be absolutely necessary for adaptive immunity to T. gondii, suggesting that recognition independent of TLR/MyD88 can drive T cell effector differentiation (Fig. 1b). In this model, Th1-inducing factors would be induced independently of MyD88. For Toxoplasma, the identity of these factors is not known currently. However, the finding that mouse bone marrow-derived macrophages can produce MyD88-independent IL-12 raises the possibility that this can also occur in vivo[60].
Role of TLR/MyD88 in the mucosal immune response during Toxoplasma infection
Oral infection of C57BL/6 mice with high doses of T. gondii triggers acute lethal ileal inflammation [61]. Resistance to inflammatory gut pathology is controlled by IL-10 production, and studies in i.p.-infected mice suggest that this may derive from conventional T-bet+, forkhead box P3− CD4+ T cells [62]. In many ways the pathology resembles human inflammatory bowel disease, in particular Crohn's disease, in that it is driven by a dysregulated Th1 cytokine response in which CD4+ T cells play a prominent role [63,64]. Also like Crohn's disease, intestinal inflammation is associated with alterations in populations of commensal gut flora, and there is evidence that during Toxoplasma infection, pathology is exacerbated by TLR recognition of intestinal bacteria [65,66].
Following high-dose T. gondii infection, Gram-negative bacteria accumulate at sites of mucosal damage, and this is associated with translocation of bacteria into intestinal tissue. Mice treated with broad-spectrum antibiotics to eliminate endogenous gut flora prior to T. gondii infection are protected from ileitis [65]. Furthermore, TLR-4−/−, but not TLR-2−/−, mice are protected against development of intestinal pathology and subsequent mortality [66]. In addition, absence of TLR-4 reduces concentrations of nitric oxide and IFN-γ, two proinflammatory mediators implicated in parasite-induced gut pathology [67]. Nevertheless, another study reported that C3H/HeJ (TLR-4 defective) displayed exacerbated pathology and increased mortality compared with C3H/HeN (normal TLR-4) during oral Toxoplasma infection [68]. Possibly, the discrepancy lies in the fact that genetic background (C57BL/6 versus C3H) differed in these studies. Regardless, an independent group reported that absence of TLR-9 also ameliorates Toxoplasma-induced inflammatory ileitis and decreases infection-induced Th1 cytokine responses [69]. Although there are some differences, the emerging picture is that Toxoplasma serves as an inflammatory trigger by causing damage to the intestinal epithelium. In turn, infiltration of bacteria results in uncontrolled TLR/MyD88 signalling, resulting in type 1 cytokine pathology in the gut that resembles human Crohn's disease. Under these conditions, the parasite's own TLR ligands may play a subsidiary role to TLR expressed by commensal bacteria.
Suppression of TLR signalling pathways by Toxoplasma
While activation of TLR/MyD88 signalling pathways clearly has a major impact on Toxoplasma infection, the parasite itself is at the same time capable of suppressing TLR signal transduction in infected cells. The molecular details of how this occurs remain to be elucidated, and indeed multiple mechanisms appear to be involved [70,71]. Thus, the parasite blocks nuclear accumulation of NF-κB, although this is only a short-term effect [72–74]. Blocking of NF-κB may be the result of abnormal phosphorylation of the p65 subunit of this proinflammatory transcription factor [75]. Furthermore, tachyzoite infection renders macrophages defective in MAPK activation, in particular the p38 MAPK [76]. In addition to LPS-induced TNF-α and IL-12p40, many other proinflammatory cytokines are suppressed [77]. However, inhibition of proinflammatory cytokine synthesis is not the result of a general shut-down in cell responses as intracellular infection progresses, because TLR-induction of a subset of cytokines, including IL-10, is not affected by T. gondii[77]. A possible unifying explanation for the parasite's ability to simultaneously suppress a large panel of proinflammatory mediators is that the parasite targets the chromatin modification machinery rather than gene-specific transcription factors [78]. Although there is not yet a clear mechanistic picture of how Toxoplasma achieves these suppressive effects, a rhoptry-derived molecule, termed ROP16, has recently been shown to be an important mediator that targets host cell signalling pathways [79]. This molecule is injected into the host cell cytoplasm during parasite invasion, where it targets signalling machinery involved in down-regulation of IL-12 (described further below).
How can Toxoplasma trigger TLR/MyD88 signal transduction, yet at the same time suppress these very same pathways? There are several models to explain this apparent paradox. First, whether Toxoplasma activates or suppresses TLR signalling may depend upon cell type. Secondly, while intracellular infection with T. gondii clearly suppresses TLR signalling leading to proinflammatory cytokine production, bystander non-infected cells may be a source of infection-induced cytokine production. In this regard, we recently found that in populations of macrophages recruited to the peritoneal cavity as a result of i.p. infection, only infected cells are suppressed in TNF-α production [78]. Along similar lines, splenic plasmacytoid DC have been found recently to serve as an IL-12 source during Toxoplasma infection [80]. Among these cells, those infected are suppressed in IL-12-inducing activity, the non-infected population produces IL-12 [81]. Deactivation of TLR signalling pathways in infected cells may indicate the necessity of avoiding anti-microbial effector mechanisms triggered by the parasite's own TLR ligands that would otherwise result in elimination of the parasite. Alternatively, it is possible that Toxoplasma-induced blocking of TLR signalling may be a way to avoid uncontrolled inflammatory responses that would otherwise be triggered by parasite-triggered exposure to bacterial TLR ligands during oral infection.
The TLR/MyD88-independent recognition of Toxoplasma
The immune system can clearly sense and respond to Toxoplasma infection in the absence of MyD88 signalling, even generating a strong protective response [42]. Infection of mouse bone marrow-derived macrophages results in IL-12 production that is wholly or partially (depending on the parasite strain type) independent of MyD88 [60]. There is currently little information on MyD88-independent receptors and signalling pathways involved in recognition, although in mouse splenic DC a pathway involving parasite cyclophilin-18 and the chemokine receptor CCR5 appears to contribute to IL-12 production [82,83].
In addition, Gi protein-coupled receptors (GiPCR) other than CCR5 seem to play a role in macrophage recognition of Toxoplasma, leading to phosphatidylinositol (PI) 3-kinase-dependent extracellular-regulated kinase 1/2 and protein kinase B activation [60,84]. Rather than being required for IL-12 production, this cascade is involved in the now well-established ability of Toxoplasma to confer an anti-apoptotic state in infected cells [84,85]. An MyD88-independent, PI-3 kinase-dependent pathway is also involved in parasite-triggered macrophage release of several chemokines, including CCL17 and CCL22 [86]. Therefore, while the identity of the GiPCR involved is not yet known, this general pathway appears to be an important mechanism for MyD88-independent Toxoplasma recognition.
Toxoplasma is also able to activate directly Janus kinase (Jak)-Signal transducer and activator of transcription (Stat) signalling pathways that are initiated normally through cytokine receptors [87]. Rather than activation through host cell cytokines, T. gondii appears to inject the rhoptry kinase ROP16 directly into the host cell cytoplasm resulting in Stat3 activation [79]. Activation of Stat3, in turn, mediates suppression of IL-12 [73,79,87]. Because the ROP16 molecule is not a predicted tyrosine kinase, it is unlikely to activate Stat or Jak molecules directly. Thus, the primary recognition event that triggers ROP16-mediated activation of the Stat3 signalling cascade is currently unknown.
Clinical implications
Harnessing the power of TLR and their ligands has potential value with regard to anti-pathology and anti-infection vaccines, as well as to prevention of autoimmunity and allergy. As such, identification and isolation of protozoan TLR ligands, including those expressed by T. gondii, increases the collection of tools that may be employed as prophylaxis and in the clinic. Use of protozoan TLR ligands for these purposes remains largely undiscovered territory. Because protozoan parasites such as T. gondii are so exquisitely well adapted to survive in the host without adverse effect, we speculate that TLR ligands from these eukaryotic pathogens may possess unique properties that could possibly prove of increased benefit relative to prokaryotic and viral counterparts. For the specific case of Toxoplasma, TGPRF may be of limited potential as TLR-11 is non-functional in humans. Nevertheless, the ability of Eimeria profilin to stimulate DC IL-12 suggests that this molecule could be a useful tool in avian immunology [37]. Also, T. gondii and additional protozoan GPI molecules as well as other undiscovered TLR ligands could prove useful.
The most obvious use for protozoan TLR ligands is in adjuvant formulations for vaccines. Many chemically defined immunostimulatory molecules in use as adjuvants are TLR ligands (these include lipid A; TLR-4, flagellin; TLR-5, CpG DNA; TLR-9) [88]. Because these molecules possess potent proinflammatory activity that may be harmful to the host, developing new reagents is an important area of investigation. Protozoan TLR may also be of benefit for use in anti-pathology vaccines. For example, synthetic P. falciparum GPI has been employed as an anti-toxin vaccine to protect against inflammatory pathology associated with experimental malaria infection [89].
Another potential use for new TLR ligands is based on evidence that early childhood exposure to infection results in resistance to autoimmunity and allergy, possibly through induction of immunoregulatory networks in which regulatory T cells play a prominent role [90,91]. Evidence for TLR involvement in prevention of autoimmunity and allergy comes from several studies, including data showing that TLR-2 and TLR-10 polymorphisms influence atopic disease risk in children [92,93]. In addition, administration of CpG DNA can mitigate allergen-induced inflammation by modulating the Th2 response [94]. Nevertheless, vaccination in these situations will need to be controlled carefully, because it is also clear that under some conditions TLR ligands promote autoimmune inflammation [95]. Regardless, development of protozoan ligands as therapeutic tools has the potential to ultimately contribute significantly to the armamentarium of weapons to combat infection, allergy and autoimmunity.
Conclusions and future directions
The discovery of mammalian TLR by Janeway in the late 1990s heralded a new era in our understanding of innate immune recognition of infection. We are still working towards an understanding of how TLR/MyD88 signalling impacts protozoan infection, and how this pathway can be exploited for the patient in human medicine. At the same time, it seems likely that other equally important innate immune recognition systems play a role in recognition of infection by protozoans and other microbial pathogens. Finding the basis for MyD88-independent recognition of protozoans and other pathogens is an important priority. Continued investigation of the molecular basis for innate immune sensing of T. gondii can be expected to reveal new insights into host defence against this and other infectious disease agents.
Acknowledgments
Our work is supported by PHS grants AI47888 and AI50617.
References
- 1.Janeway CA, Medzhitov R. Innate immune recognition. Annu Rev Immunol. 2002;20:197–216. doi: 10.1146/annurev.immunol.20.083001.084359. [DOI] [PubMed] [Google Scholar]
- 2.Akira S, Uematsu S, Takeuchi O. Pathogen recognition and innate immunity. Cell. 2006;124:783–801. doi: 10.1016/j.cell.2006.02.015. [DOI] [PubMed] [Google Scholar]
- 3.Peterson E, Liesenfeld O. Clinical disease and diagnostics. In: Weiss LM, Kim K, editors. Toxoplasma gondii. The model apicomplexan: perspectives and methods. Amsterdam: Academic Press; 2007. pp. 81–100. [Google Scholar]
- 4.Gazzinelli RT, Denkers EY. Protozoan encounters with Toll-like receptor signalling pathways: implications for host parasitism. Nat Rev Immunol. 2006;6:895–906. doi: 10.1038/nri1978. [DOI] [PubMed] [Google Scholar]
- 5.Pfaff AW, Liesenfeld O, Candolfi E. Congenital toxoplasmosis. In: Ajioka JW, Soldati D, editors. Toxoplasma: molecular and cellular biology. Norfolk: Horizon Bioscience; 2007. pp. 93–110. [Google Scholar]
- 6.Hill DE, Chirukandoth S, Dubey JP. Biology and epidemiology of Toxoplasma gondii in man and animals. Anim Health Res Rev. 2005;6:41–61. doi: 10.1079/ahr2005100. [DOI] [PubMed] [Google Scholar]
- 7.Denkers EY, Gazzinelli RT. Regulation and function of T cell-mediated immunity during Toxoplasma gondii infection. Clin Microbiol Rev. 1998;11:569–88. doi: 10.1128/cmr.11.4.569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pepper M, Hunter CA. Innate immune recognition and regulation of protective immunity to Toxoplasma gondii. In: Ajioka JW, Soldati D, editors. Toxoplasma: molecular and cellular biology. Norfolk: Horizon Bioscience; 2007. pp. 111–26. [Google Scholar]
- 9.Kawai T, Akira S. TLR signaling. Semin Immunol. 2007;19:24–32. doi: 10.1016/j.smim.2006.12.004. [DOI] [PubMed] [Google Scholar]
- 10.Akira S, Takeda T. Toll-like receptor signaling. Nat Rev Immunol. 2004;4:499–511. doi: 10.1038/nri1391. [DOI] [PubMed] [Google Scholar]
- 11.Akira S. Toll-like receptor signaling. J Biol Chem. 2003;278:38105–8. doi: 10.1074/jbc.R300028200. [DOI] [PubMed] [Google Scholar]
- 12.Barton GM, Kagan JC, Medzhitov R. Intracellular localization of Toll-like receptor 9 prevents recognition of self DNA but facilitates access to viral DNA. Nat Immunol. 2006;7:49–56. doi: 10.1038/ni1280. [DOI] [PubMed] [Google Scholar]
- 13.Kawai T, Adachi O, Ogawa T, Takeda K, Akira S. Unresponsiveness of MyD88-deficient mice to endotoxin. Immunity. 1999;11:115–22. doi: 10.1016/s1074-7613(00)80086-2. [DOI] [PubMed] [Google Scholar]
- 14.O'Neill LA, Bowie AG. The family of five: TIR-domain-containing adaptors in Toll-like receptor signalling. Nat Rev Immunol. 2007;7:353–64. doi: 10.1038/nri2079. [DOI] [PubMed] [Google Scholar]
- 15.Yamamoto M, Sato S, Hemmi H, et al. Role of adaptor TRIF in the MyD88-independent Toll-like receptor pathway. Science. 2003;301:640–3. doi: 10.1126/science.1087262. [DOI] [PubMed] [Google Scholar]
- 16.O'Neill LA. When signaling pathways collide: positive and negative regulation of Toll-like receptor signal transduction. Immunity. 2008;29:12–20. doi: 10.1016/j.immuni.2008.06.004. [DOI] [PubMed] [Google Scholar]
- 17.Wald D, Qin J, Zhao Z, et al. SIGIRR, a negative regulator of Toll-like receptor-interleukin 1 receptor signaling. Nat Immunol. 2003;4:920–7. doi: 10.1038/ni968. [DOI] [PubMed] [Google Scholar]
- 18.Janssens S, Burns K, Vercammen E, Tschopp J, Beyaert R. MyD88S, a splice variant of MyD88, differentially modulates NF-kappaB- and AP-1-dependent gene expression. FEBS Lett. 2003;548:103–7. doi: 10.1016/s0014-5793(03)00747-6. [DOI] [PubMed] [Google Scholar]
- 19.Kobayashi K, Hernandez LD, Galan JE, Janeway CA, Jr, Medzhitov R, Flavell RA. IRAK-M is a negative regulator of Toll-like receptor signaling. Cell. 2002;110:191–202. doi: 10.1016/s0092-8674(02)00827-9. [DOI] [PubMed] [Google Scholar]
- 20.Mansell A, Smith R, Doyle SL, et al. Suppressor of cytokine signaling 1 negatively regulates Toll-like receptor signaling by mediating Mal degradation. Nat Immunol. 2006;7:148–55. doi: 10.1038/ni1299. [DOI] [PubMed] [Google Scholar]
- 21.Bartholomeu DC, Ropert C, Melo MB, et al. Recruitment and endo-lysosomal activation of TLR9 in dendritic cells infected with Trypanosoma cruzi. J Immunol. 2008;181:1333–44. doi: 10.4049/jimmunol.181.2.1333. [DOI] [PubMed] [Google Scholar]
- 22.Harris TH, Cooney NM, Mansfield JM, Paulnock DM. Signal transduction, gene transcription, and cytokine production triggered in macrophages by exposure to trypanosome DNA. Infect Immun. 2006;74:4530–7. doi: 10.1128/IAI.01938-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ivory CP, Prystajecky M, Jobin C, Chadee K. Toll-like receptor 9-dependent macrophage activation by Entamoeba histolytica DNA. Infect Immun. 2008;76:289–97. doi: 10.1128/IAI.01217-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liese J, Schleicher U, Bogdan C. TLR9 signaling is essential for the innate NK cell response in murine cutaneous leishmaniasis. Eur J Immunol. 2007;37:3424–34. doi: 10.1002/eji.200737182. [DOI] [PubMed] [Google Scholar]
- 25.Parroche P, Lauw FN, Goutagny N, et al. Malaria hemozoin is immunologically inert but radically enhances innate responses by presenting malaria DNA to Toll-like receptor 9. Proc Natl Acad Sci USA. 2007;104:1919–24. doi: 10.1073/pnas.0608745104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Campos MAS, Almeida IC, Takeuchi O, et al. Activation of Toll-like receptor-2 by glycosylphosphatidylinositol anchors from a protozoan parasite. J Immunol. 2001;167:416–23. doi: 10.4049/jimmunol.167.1.416. [DOI] [PubMed] [Google Scholar]
- 27.de Veer MJ, Curtis JM, Baldwin TM, et al. MyD88 is essential for clearance of Leishmania major: possible role for lipophosphoglycan and Toll-like receptor 2 signaling. Eur J Immunol. 2003;33:2822–31. doi: 10.1002/eji.200324128. [DOI] [PubMed] [Google Scholar]
- 28.Gazzinelli RT, Ropert C, Campos MA. Role of the Toll/interleukin-1 receptor signaling pathway in host resistance and pathogenesis during infection with protozoan parasites. Immunol Rev. 2004;201:9–25. doi: 10.1111/j.0105-2896.2004.00174.x. [DOI] [PubMed] [Google Scholar]
- 29.Krishnegowda G, Hajjar AM, Zhu J, et al. Induction of proinflammatory responses in macrophages by the glycosylphosphatidylinositols of Plasmodium falciparum: cell signaling receptors, glycosylphosphatidylinositol (GPI) structural requirement, and regulation of GPI activity. J Biol Chem. 2005;280:8606–16. doi: 10.1074/jbc.M413541200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Oliveira AC, Peixoto JR, de Arruda LB, et al. Expression of functional TLR4 confers proinflammatory responsiveness to Trypanosoma cruzi glycoinositolphospholipids and higher resistance to infection with T. cruzi. J Immunol. 2004;173:5688–96. doi: 10.4049/jimmunol.173.9.5688. [DOI] [PubMed] [Google Scholar]
- 31.Scanga CA, Aliberti J, Jankovic D, et al. Cutting edge: MyD88 is required for resistance to Toxoplasma gondii infection and regulates parasite-induced IL-12 production by dendritic cells. J Immunol. 2002;168:5997–6001. doi: 10.4049/jimmunol.168.12.5997. [DOI] [PubMed] [Google Scholar]
- 32.Del Rio L, Butcher BA, Bennouna S, Hieny S, Sher A, Denkers EY. Toxoplasma gondii triggers MyD88-dependent and CCL2(MCP-1) responses using distinct parasite molecules and host receptors. J Immunol. 2004;172:6954–60. doi: 10.4049/jimmunol.172.11.6954. [DOI] [PubMed] [Google Scholar]
- 33.Yarovinsky F, Zhang D, Anderson JF, et al. TLR11 activation of dendritic cells by a protozoan profilin-like protein. Science. 2005;308:1626–9. doi: 10.1126/science.1109893. [DOI] [PubMed] [Google Scholar]
- 34.Yarovinsky F. Toll-like receptors and their role in host resistance to Toxoplasma gondii. Immunol Lett. 2008;119:17–21. doi: 10.1016/j.imlet.2008.05.007. [DOI] [PubMed] [Google Scholar]
- 35.Zhang D, Zhang G, Hayden MS, et al. A Toll-like receptor that prevents infection by uropathogenic bacteria. Science. 2004;303:1522–6. doi: 10.1126/science.1094351. [DOI] [PubMed] [Google Scholar]
- 36.Plattner F, Yarovinsky F, Romero S, et al. Toxoplasma profilin is essential for host cell invasion and TLR dependent induction of interleukin-12. Cell Host Microbe. 2008;14:77–87. doi: 10.1016/j.chom.2008.01.001. [DOI] [PubMed] [Google Scholar]
- 37.Rosenberg B, Juckett DA, Aylsworth CF, et al. Protein from intestinal Eimeria protozoan stimulates IL-12 release from dendritic cells, exhibits antitumor properties in vivo and is correlated with low intestinal tumorigenicity. Int J Cancer. 2005;114:756–65. doi: 10.1002/ijc.20801. [DOI] [PubMed] [Google Scholar]
- 38.Wichroski MJ, Ward GE. Biosynthesis of glycosylphosphatidylinositol is essential to the survival of the protozoan parasite Toxoplasma gondii. Eukaryot Cell. 2003;2:1132–6. doi: 10.1128/EC.2.5.1132-1136.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Debierre-Grockiego F, Azzouz N, Schmidt J, et al. Roles of glycosylphosphatidylinositols of Toxoplasma gondii. Induction of TNF-α production in macrophages. J Biol Chem. 2003;278:32987–93. doi: 10.1074/jbc.M304791200. [DOI] [PubMed] [Google Scholar]
- 40.Debierre-Grockiego F, Campos MA, Azzouz N, et al. Activation of TLR2 and TLR4 by glycosylphosphatidylinositols derived from Toxoplasma gondii. J Immunol. 2007;179:1129–37. doi: 10.4049/jimmunol.179.2.1129. [DOI] [PubMed] [Google Scholar]
- 41.Hitziger N, Dellacasa I, Albiger B, Barragan A. Dissemination of Toxoplasma gondii to immunoprivileged organs and role of Toll/interleukin-1 receptor signalling for host resistance assessed by in vivo bioluminsecence imaging. Cell Microbiol. 2005;6:837–48. doi: 10.1111/j.1462-5822.2005.00517.x. [DOI] [PubMed] [Google Scholar]
- 42.Sukhumavasi W, Egan CE, Warren AL, et al. TLR adaptor MyD88 is essential for pathogen control during oral Toxoplasma gondii infection but not adaptive immunity induced by a vaccine strain of the parasite. J Immunol. 2008;181:3464–73. doi: 10.4049/jimmunol.181.5.3464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mun H-S, Aosai F, Norose K, et al. TLR2 as an essential molecule for protective immunity against Toxoplasma gondii infection. Int Immunol. 2003;15:1081–7. doi: 10.1093/intimm/dxg108. [DOI] [PubMed] [Google Scholar]
- 44.Bafica A, Santiago HC, Goldszmid R, Ropert C, Gazzinelli RT, Sher A. Cutting edge: TLR9 and TLR2 signaling together account for MyD88-dependent control of parasitemia in Trypanosoma cruzi infection. J Immunol. 2006;177:3515–19. doi: 10.4049/jimmunol.177.6.3515. [DOI] [PubMed] [Google Scholar]
- 45.Bafica A, Scanga CA, Feng CG, Leifer C, Cheever A, Sher A. TLR9 regulates Th1 responses and cooperates with TLR2 in mediating optimal resistance to Mycobacterium tuberculosis. J Exp Med. 2005;202:1715–24. doi: 10.1084/jem.20051782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gazzinelli RT, Hieny S, Wynn T, Wolf S, Sher A. IL-12 is required for the T-cell independent induction of IFN-γ by an intracellular parasite and induces resistance in T-cell-deficient hosts. Proc Natl Acad Sci USA. 1993;90:6115–19. doi: 10.1073/pnas.90.13.6115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Scharton-Kersten T, Yap G, Magram J, Sher A. Inducible nitric oxide is essential for host control of persistent but not acute infection with the intracellular pathogen Toxoplasma gondii. J Exp Med. 1997;185:1–13. doi: 10.1084/jem.185.7.1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Taylor GA. IRG proteins key mediators of interferon-regulated host resistance to intracellular pathogens. Cell Microbiol. 2007;9:1099–107. doi: 10.1111/j.1462-5822.2007.00916.x. [DOI] [PubMed] [Google Scholar]
- 49.Lebeis SL, Bommarius B, Parkos CA, Sherman MA, Kalman D. TLR signaling mediated by MyD88 is required for a protective innate immune response by neutrophils to Citrobacter rodentium. J Immunol. 2007;179:566–77. doi: 10.4049/jimmunol.179.1.566. [DOI] [PubMed] [Google Scholar]
- 50.Rodriguez N, Fend F, Jennen L, et al. Polymorphonuclear neutrophils improve replication of Chlamydia pneumoniae in vivo upon MyD88-dependent attraction. J Immunol. 2005;174:4836–44. doi: 10.4049/jimmunol.174.8.4836. [DOI] [PubMed] [Google Scholar]
- 51.Kabelitz D, Medzhitov R. Innate immunity – cross-talk with adaptive immunity through pattern recognition receptors and cytokines. Curr Opin Immunol. 2007;19:1–3. doi: 10.1016/j.coi.2006.11.018. [DOI] [PubMed] [Google Scholar]
- 52.Pasare C, Medzhitov R. Toll-dependent control mechanisms of CD4 T cell activation. Immunity. 2004;21:733–41. doi: 10.1016/j.immuni.2004.10.006. [DOI] [PubMed] [Google Scholar]
- 53.Schnare M, Barton GM, Holt AC, Takeda K, Akira S, Medzhitov R. Toll-like receptors control activation of adaptive immune responses. Nat Immunol. 2001;2:947–50. doi: 10.1038/ni712. [DOI] [PubMed] [Google Scholar]
- 54.Yarovinsky F, Kanzler H, Hieny S, Coffman RL, Sher A. Toll-like receptor recognition regulates immunodominance in an antimicrobial CD4+ T cell response. Immunity. 2006;25:655–64. doi: 10.1016/j.immuni.2006.07.015. [DOI] [PubMed] [Google Scholar]
- 55.Larosa DF, Stumhofer JS, Gelman AE, et al. T cell expression of MyD88 is required for resistance to Toxoplasma gondii. Proc Natl Acad Sci USA. 2008;105:3855–60. doi: 10.1073/pnas.0706663105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bolz DD, Sundsbak RS, Ma Y, et al. MyD88 plays a unique role in host defense but not arthritis development in Lyme disease. J Immunol. 2004;173:2003–10. doi: 10.4049/jimmunol.173.3.2003. [DOI] [PubMed] [Google Scholar]
- 57.Kursar M, Mittrucker HW, Koch M, Kohler A, Herma M, Kaufmann SH. Protective T cell response against intracellular pathogens in the absence of Toll-like receptor signaling via myeloid differentiation factor 88. Int Immunol. 2004;16:415–21. doi: 10.1093/intimm/dxh047. [DOI] [PubMed] [Google Scholar]
- 58.von Bernuth H, Picard C, Jin Z, et al. Pyogenic bacterial infections in humans with MyD88 deficiency. Science. 2008;321:691–6. doi: 10.1126/science.1158298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Way SS, Kollmann TR, Hajjar AM, Wilson CB. Cutting edge: protective cell-mediated immunity to Listeria monocytogenes in the absence of myeloid differentiation factor 88. J Immunol. 2003;171:533–7. doi: 10.4049/jimmunol.171.2.533. [DOI] [PubMed] [Google Scholar]
- 60.Kim L, Butcher BA, Lee CW, Uematsu S, Akira S, Denkers EY. Toxoplasma gondii genotype determines MyD88-dependent signaling in infected macrophages. J Immunol. 2006;177:2584–91. doi: 10.4049/jimmunol.177.4.2584. [DOI] [PubMed] [Google Scholar]
- 61.Liesenfeld O, Kosek J, Remington JS, Suzuki Y. Association of CD4+ T cell-dependent, IFN-γ-mediated necrosis of the small intestine with genetic susceptibility of mice to peroral infection with Toxoplasma gondii. J Exp Med. 1996;184:597–607. doi: 10.1084/jem.184.2.597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Jankovic D, Kullberg MC, Feng CG, et al. Conventional T-bet+Foxp3- Th1 cells are the major source of host-protective regulatory IL-10 during intracellular protozoan infection. J Exp Med. 2007;204:273–83. doi: 10.1084/jem.20062175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Liesenfeld O. Oral infection of C57BL/6 mice with Toxoplasma gondii: a new model of inflammatory bowel disease? J Infect Dis. 2002;185:S96–101. doi: 10.1086/338006. [DOI] [PubMed] [Google Scholar]
- 64.Xavier RJ, Podolsky DK. Unravelling the pathogenesis of inflammatory bowel disease. Nature. 2007;448:427–34. doi: 10.1038/nature06005. [DOI] [PubMed] [Google Scholar]
- 65.Heimesaat MM, Bereswill S, Fischer A, et al. Gram-negative bacteria aggravate murine small intestinal Th1-type immunopathology following oral infection with Toxoplasma gondii. J Immunol. 2006;177:8785–95. doi: 10.4049/jimmunol.177.12.8785. [DOI] [PubMed] [Google Scholar]
- 66.Heimesaat MM, Fischer A, Jahn HK, et al. Exacerbation of murine ileitis by Toll-like receptor 4 mediated sensing of lipopolysaccharide from commensal Escherichia coli. Gut. 2007;56:941–8. doi: 10.1136/gut.2006.104497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Liesenfeld O, Kang H, Park D, et al. TNF-α, nitric oxide and IFN-γ are all critical for development of necrosis in the small intestine and early mortality in genetically susceptible mice infected perorally with Toxoplasma gondii. Parasite Immunol. 1999;21:365–76. doi: 10.1046/j.1365-3024.1999.00237.x. [DOI] [PubMed] [Google Scholar]
- 68.Furuta T, Kikuchi T, Akira S, Watanabe N, Yoshikawa Y. Roles of the small intestine for induction of Toll-like receptor 4-mediated innate resistance in naturally acquired murine toxoplasmosis. Int Immunol. 2006;18:1655–62. doi: 10.1093/intimm/dxl099. [DOI] [PubMed] [Google Scholar]
- 69.Minns LA, Menard LC, Foureau DM, et al. TLR9 is required for the gut-associated lymphoid tissue response following oral infection of Toxoplasma gondii. J Immunol. 2006;176:7589–97. doi: 10.4049/jimmunol.176.12.7589. [DOI] [PubMed] [Google Scholar]
- 70.Denkers EY, Kim L, Butcher BA. In the belly of the beast: subversion of macrophage proinflammatory signaling cascades during Toxoplasma gondii infection. Cell Microbiol. 2003;5:75–83. doi: 10.1046/j.1462-5822.2003.00258.x. [DOI] [PubMed] [Google Scholar]
- 71.Shapira S, Harb OS, Caamano J, Hunter CA. The NF-kappaB signaling pathway: immune evasion and immunoregulation during toxoplasmosis. Int J Parasitol. 2004;34:393–400. doi: 10.1016/j.ijpara.2003.12.005. [DOI] [PubMed] [Google Scholar]
- 72.Butcher BA, Denkers EY. Mechanism of entry determines ability of Toxoplasma gondii to inhibit macrophage proinflammatory cytokine production. Infect Immun. 2002;70:5216–24. doi: 10.1128/IAI.70.9.5216-5224.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Butcher BA, Kim L, Johnson PF, Denkers EY. Toxoplasma gondii tachyzoites inhibit proinflammatory cytokine induction in infected macrophages by preventing nuclear translocation of the transcription factor NFκB. J Immunol. 2001;167:2193–201. doi: 10.4049/jimmunol.167.4.2193. [DOI] [PubMed] [Google Scholar]
- 74.Shapira SS, Speirs K, Gerstein A, Caamano J, Hunter CA. Suppression of NF-κB activation by infection with Toxoplasma gondii. J Infect Dis. 2002;185:S66–72. doi: 10.1086/338000. [DOI] [PubMed] [Google Scholar]
- 75.Shapira S, Harb O, Margarit J, et al. Initiation and termination of NFκB signaling by the intracellular protozoan parasite Toxoplasma gondii. J Cell Sci. 2005;118:3501–8. doi: 10.1242/jcs.02428. [DOI] [PubMed] [Google Scholar]
- 76.Kim L, Butcher BA, Denkers EY. Toxoplasma gondii interferes with lipopolysaccharide-induced mitogen-activated protein kinase activation by mechanisms distinct from endotoxin tolerance. J Immunol. 2004;172:3003–10. doi: 10.4049/jimmunol.172.5.3003. [DOI] [PubMed] [Google Scholar]
- 77.Lee CW, Bennouna S, Denkers EY. Screening for Toxoplasma gondii regulated transcriptional responses in LPS-activated macrophages. Infect Immun. 2006;74:1916–23. doi: 10.1128/IAI.74.3.1916-1923.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Leng J, Butcher BA, Egan CE, Abi Abdallah DS, Denkers EY. Toxoplasma gondii prevents chromatin remodeling initiated by Toll-like receptor-triggered macrophages activation. J Immunol. 2009;182:489–97. doi: 10.4049/jimmunol.182.1.489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Saeij JP, Coller S, Boyle JP, Jerome ME, White MW, Boothroyd JC. Toxoplasma co-opts host gene expression by injection of a polymorphic kinase homologue. Nature. 2007;445:324–7. doi: 10.1038/nature05395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Pepper M, Dzierszinski F, Wilson E, et al. Plasmacytoid dendritic cells are activated by Toxoplasma gondii to present antigen and produce cytokines. J Immunol. 2008;180:6229–36. doi: 10.4049/jimmunol.180.9.6229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Bierly AL, Shufesky WJ, Sukhumavasi W, Morelli A, Denkers EY. Dendritic cells expressing plasmacytoid marker PDCA-1 are Trojan horses during Toxoplasma gondii infection. J Immunol. 2008;181:8445–91. doi: 10.4049/jimmunol.181.12.8485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Aliberti J, Reis e Sousa C, Schito M, et al. CCR5 provides a signal for microbial induced production of IL-12 by CD8α+ dendritic cells. Nat Immunol. 2000;1:83–7. doi: 10.1038/76957. [DOI] [PubMed] [Google Scholar]
- 83.Aliberti J, Valenzuela JG, Carruthers VB, et al. Molecular mimicry of a CCR5 binding-domain in the microbial activation of dendritic cells. Nat Immunol. 2003;4:485–90. doi: 10.1038/ni915. [DOI] [PubMed] [Google Scholar]
- 84.Kim L, Denkers EY. Toxoplasma gondii triggers Gi-dependent phosphatidylinositol 3-kinase signaling required for inhibition of host cell apoptosis. J Cell Sci. 2006;119:2119–26. doi: 10.1242/jcs.02934. [DOI] [PubMed] [Google Scholar]
- 85.Luder CGK, Gross U. Apoptosis and its modulation during infection with Toxoplasma gondii: molecular mechanisms and role in pathogenesis. Curr Top Microbiol Immunol. 2005;289:219–38. doi: 10.1007/3-540-27320-4_10. [DOI] [PubMed] [Google Scholar]
- 86.Lee CW, Sukhumavasi W, Denkers EY. Phosphoinositide-3-kinase-dependent, MyD88-independent induction of CC-type chemokines characterizes the macrophage response to Toxoplasma gondii strains with high virulence. Infect Immun. 2007;75:5788–97. doi: 10.1128/IAI.00821-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Butcher BA, Kim L, Panopoulos A, Watowich SS, Murray PJ, Denkers EY. Cutting edge: IL-10-independent STAT3 activation by Toxoplasma gondii mediates suppression of IL-12 and TNF-α in host macrophages. J Immunol. 2005;174:3148–52. doi: 10.4049/jimmunol.174.6.3148. [DOI] [PubMed] [Google Scholar]
- 88.Pashine A, Valiante NM, Ulmer JB. Targeting the innate immune response with improved vaccine adjuvants. Nat Med. 2005;11:S63–8. doi: 10.1038/nm1210. [DOI] [PubMed] [Google Scholar]
- 89.Schofield L, Hewitt MC, Evans K, Siomos A, Seeberger PH. Synthetic GPI as a candidate anti-toxic vaccine in a model of malaria. Nature. 2002;418:785–9. doi: 10.1038/nature00937. [DOI] [PubMed] [Google Scholar]
- 90.Chaudhuri N, Whyte MK, Sabroe I. Reducing the toll of inflammatory lung disease. Chest. 2007;131:1550–6. doi: 10.1378/chest.06-2869. [DOI] [PubMed] [Google Scholar]
- 91.Goldman M. Translational mini-review series on Toll-like receptors: Toll-like receptor ligands as novel pharmaceuticals for allergic disorders. Clin Exp Immunol. 2007;147:208–16. doi: 10.1111/j.1365-2249.2006.03296.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Eder W, Klimecki W, Yu L, et al. Toll-like receptor 2 as a major gene for asthma in children of European farmers. J Allergy Clin Immunol. 2004;113:482–8. doi: 10.1016/j.jaci.2003.12.374. [DOI] [PubMed] [Google Scholar]
- 93.Lazarus R, Raby BA, Lange C, et al. Toll-like receptor 10 genetic variation is associated with asthma in two independent samples. Am J Respir Crit Care Med. 2004;170:594–600. doi: 10.1164/rccm.200404-491OC. [DOI] [PubMed] [Google Scholar]
- 94.Creticos PS, Schroeder JT, Hamilton RG, et al. Immunotherapy with a ragweed-Toll-like receptor 9 agonist vaccine for allergic rhinitis. N Engl J Med. 2006;355:1445–55. doi: 10.1056/NEJMoa052916. [DOI] [PubMed] [Google Scholar]
- 95.Ehlers M, Ravetch JV. Opposing effects of Toll-like receptor stimulation induce autoimmunity or tolerance. Trends Immunol. 2007;28:74–9. doi: 10.1016/j.it.2006.12.006. [DOI] [PubMed] [Google Scholar]