

Abstract
The most prevalent primary immunodeficiency is common variable immunodeficiency (CVID). Mutations have been described in four genes, ICOS, CD19, BAFF-R and TNFRSF13B (encoding TACI), together associated with 10–15% of CVID cases. We investigated a family with CVID and identified the heterozygous C104R TNFRSF13B mutation in two of the three index-children with CVID, a mother with selective immunoglobulin A deficiency, a mother with recurrent infections and a healthy grandfather. Remarkably, we did not find the TNFRSF13B mutation in the third index-child with CVID, despite his hypogammaglobulinaemia and decreased response to unconjugated pneumococcal vaccine. This family illustrates that TNFRSF13B mutations induce disease susceptibility rather than cause disease directly. Apparently, other genetic or environmental factors, still to be identified, contributed to the development of CVID in this family. Consequently, TNFRSF13B mutations must be interpreted with caution in the clinical setting.
Keywords: common variable immunodeficiency, primary immunodeficiency, TNFRSF13B gene, transmembrane activator and CAML interactor protein
Introduction
The most frequent primary immunodeficiency is common variable immunodeficiency disorder (CVID). It is a heterogeneous group of disorders characterized by late-onset hypogammaglobulinaemia that presents often in childhood or adolescence. Clinical manifestations include recurrent respiratory tract infections with the development of bronchiectasis and intermittent or chronic diarrhoea. Polyclonal lymphoproliferation with splenomegaly, lymphadenopathy or nodular lymphoid hyperplasia of the small bowel occurs in approximately one-third of patients, autoimmune phenomena such as idiopathic thrombocytopenic purpura and autoimmune haemolytic anaemia in up to 25% and granulomatous disease in 8–22%. CVID is associated with an increased risk of gastrointestinal and lymphoid malignancies [1–4]. Together, the European Society for Immunodeficiencies (ESID) and Pan-American Group for Immunodeficiencies (PAGID) developed diagnostic criteria for CVID, which are published on the ESID website (http://www.esid.org). Since 2003 mutations have been described in four genes: ICOS, CD19, BAFF-R and TNFRSF13B[encoding transmembrane activator and CAML interactor (TACI)], together associated with 10–15% of CVID cases [5–12]. Inducible co-stimulator of activated T cells (ICOS) deficiency has been found in only four families with the same mutation, who most probably descend from a common founder [7,8]. Three families with CD19 deficiency were identified, all having a different mutation [9,11]. Finally, so far a single B cell-activating factor-receptor (BAFF-R) deficiency has been found in one family [10]. All three deficiencies show an autosomal recessive pattern of inheritance and are extremely rare. TACI deficiency has been found in as many as 10% of CVID patients, with either one or two mutated alleles [5,6]. TACI is one of the tumour necrosis factor receptor family members expressed on peripheral B lymphocytes which are engaged in their final maturation [13,14]. TACI helps B lymphocytes to switch their production from immunoglobulin (Ig)M to IgG, IgA and IgE. It also enhances the generation and maintenance of isotype-switched memory B lymphocytes and plasma cells, and increases the affinity of the produced antibody by the process of somatic hypermutation of the Ig gene. The association of TNFRSF13B mutations with CVID is highly significant, but healthy family members and unrelated controls have also been reported to carry mutations [15]. Inversely, we describe a CVID family with the C104R TNFRSF13B mutation in several family members, but an index-child with a complete CVID phenotype lacked the TNFRSF13B mutation. Consequently, the interpretation of TNFRSF13B mutations in CVID is subject to discussion. Because of this, it is not advocated to screen CVID patients routinely for this mutation.
Materials and methods
Patients and family members
Clinical and laboratory data were obtained from the medical records of three index patients diagnosed with CVID according to the ESID/PAGID criteria who were all related to each other. Family members were interviewed. Blood samples were taken after informed consent, when there was no infection. In the index patients, blood samples were obtained immediately prior to their Ig infusion. The study was approved by the local Medical Ethics Committee.
Immunochemistry and immunoserology
The IgG, IgA, IgM and IgG subclasses were determined by nephelometry. Pneumococcal antibodies were assayed by enzyme-linked immunosorbent assay, diphtheria and tetanus anti-toxin antibodies with a toxin-binding inhibition assay [16,17].
Immunophenotyping of lymphocyte subpopulations
Flow cytometric analysis of peripheral blood (PB) was performed to determine the absolute counts of B and T lymphocytes and natural killer cells using a fluorescence activated cell sorter (FACS)Calibur (BD Biosciences, San Jose, CA, USA). PB B lymphocytes were analysed further using antibodies against IgM [mouse polyclonal antibody – phycoerythrin (PE)], IgD [mouse polyclonal antibody – fluorescein isothiocyanate), CD19 (SJ25C1 – peridinin chlorophyll), CD21 (LB21 – PE), CD27 (L128 – allophycocyanin), CD24 (1B5-FITC), CD38 (HB7-PE) and TACI (biotin-conjugated mouse polyclonal antibody in combination with PE-conjugated streptavidin).
Genetic analysis
Polymerase chain reaction was performed to amplify the coding exons of the TNFRSF13B gene (MIM 604907; NCBI NM_012452). Primer sequences are available upon request. All sequencing was performed on an ABI Prism 3100 fluorescent sequencer (Applied Biosystems, Foster City, CA, USA).
Results
The pedigree of the family is shown in Fig. 1. Clinical and laboratory parameters of the tested family members are shown in Tables 1–3. The proband patient III:2 was the first in the family to be diagnosed with ‘possible CVID’ at the age of 3 years, with recurrent upper respiratory tract infections and low IgG. She started intravenous immunoglobulin substitution (IVIG) at the age of 4, when her IgG decreased further, and infections continued to recur despite trimethoprim–sulphamethoxazole prophylaxis. Later, when her pneumococcal antibodies disappeared and her IgA and IgM decreased below normal, she could be classified as ‘probable CVID’. Her brother (III:1) suffered increasingly from recurrent respiratory infections and was started on IVIG 6 years after his sister, at the age of 10, with a diagnosis of ‘probable CVID’. Shortly after that, their mother (II:2) started to suffer from recurrent respiratory infections, rheumatic complaints and chronic fatigue, but she had normal Ig levels and normal responses to vaccines. Their cousin (III:3) was diagnosed with ‘probable CVID’ 6 years after the first patient (III:2), at the age of 2. His mother (II:4) had suffered from recurrent respiratory infections and chronic fatigue since early childhood and was diagnosed with selective IgA-deficiency. (Grand)mother I:2 died of renal carcinoma at the age of 47. The 70-year-old (grand)father I:1 is healthy. Aunt II:1 could not be tested. The heterozygous TNFRSF13B mutation C104R was found in III:2, III:3, II:2, II:4 and I:1, but not in III:1.
Fig. 1.
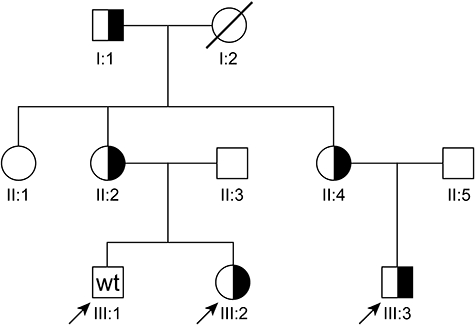
Pedigree of the family. Circles represent females; squares, males. Half-solid symbols are heterozygous carriers of the TNFRSF13B mutation C104R and wt family members who are known not to be carriers. The other family members were not tested. Slashed symbols represent deceased family members. Index cases are indicated by an arrow.
Table 1.
Clinical parameters of the tested family members.
| III:1 | III:2 | III:3 | II:2 | II:4 | I:1 | |
|---|---|---|---|---|---|---|
| Onset of symptoms (years) | 6 | 1 | 2 | 41 | Early childhood | No symptoms |
| Age at start IVIG (years) | 10·3 | 4 | 2·3 | No IVIG | No IVIG | No IVIG |
| Age when TACI mutation was found (years) | No mutation | 10 | 3 | 43 | 40 | 70 |
| Recurrent respiratory infections | + | + | + | + | + | − |
| Autoimmune disease | − | − | − | − | − | − |
| Lymphoproliferation | − | − | − | − | − | − |
| Granulomatous disease | − | − | − | − | − | − |
| Rheumatic complaints | − | − | − | + | − | − |
| TNFRSF13B mutation | No mutation | C104R heterozygous | C104R heterozygous | C104R heterozygous | C104R heterozygous | C104R heterozygous |
| Diagnosis according to ESID/PAGID criteria | Probable CVID | Possible CVID; later probable CVID | Probable CVID | No PID | Definitive IgA-deficiency | No PID |
CVID, common variable immunodeficiency disorder; ESID/PAGID, European Society for Immunodeficiencies/Pan-American Group for Immunodeficiencies; IVIG, intravenous immunoglobulin; IgA, immunoglobulin A; PID, primary immunodeficiency.
Table 3.
Flow cytometric results of the tested family members.
| III:1 | III:2 | III:3 | II:2 | II:4 | I:1 | |
|---|---|---|---|---|---|---|
| T lymphocytes (×109/l)* | 1·47 (0·8–3·5) | 1·64 (0·8–3·5) | 2·26 (0·9–4·5) | 1·6 (0·7–2·1) | 1·65 (0·7–2·1) | 1·2 (0·7–2·1) |
| B lymphocytes (% of PBL) | 10·2 | 8·9 | 18·1 | 13·8 | 10·7 | 8·1 |
| B lymphocytes (×109/l) | 0·22 (0·2–0·6) | 0·20 (0·2–0·6) | 0·64 (0·2–2·1) | 0·3 (0·1–0·5) | 0·33 (0·1–0·5) | 0·2 (0·1–0·5) |
| NK cells (×109/l) | 0·45 (0·07–1·2) | 0·26 (0·07–1·2) | 0·09 (0·1–1) | 0·3 (0·09–0·6) | 0·36 (0·09–0·6) | 0·4 (0·09–0·6) |
| Switched memory B cells (% of B lymphocytes) (CD19+/IgD−/CD27+) | 4·5 | 7·0 | 1·3 | 20·5 ↑ (5–10) | 6·7 ↑ (5–10) | 29·6 ↑ (5–10) |
| Transitional B cells (% of B lymphocytes) (CD19+/IgM+/CD38+) | 6·3 | 6·2 | 13·1 | n.d. | 3·9 (3·5–5·6) | n.d. |
| CD21low (% of B lymphocytes) | 5·3 | 9·2 | 9·2 | 16·8 ↑ (8·7–13·5) | 4·5 ↓ (8·7–13·5) | 33·0 ↑ (8·7–13·5) |
| TNFRSF13B mutation | No mutation | C104R heterozygous | C104R heterozygous | C104R heterozygous | C104R heterozygous | C104R heterozygous |
Age-matched reference values between parentheses.
n.d., not determined; PBL, peripheral blood lymphocytes; ↓, below the reference value for age; NK, natural killer.
Table 2.
Laboratory parameters of the tested family members.
| III:1 | III:2 | III:3 | II:2 | II:4 | I:1 | |||
|---|---|---|---|---|---|---|---|---|
| IgG (g/l)† | 4·2 ↓ (6–15·4) | 3 ↓ (4–11) | 10·4‡ (IVIG) | 2·5 ↓ (4–11) | 8·7 (7–16) | 7·8 (7–16) | 7·8 (7–16) | |
| IgG1 (g/l) | 3·3 ↓ (3·8–10) | 2·6 ↓ (3·5–10) | 1·9 ↓ (3·5–10) | 4·6 (3·8–10) | 4·2 (3·8–10) | n.d. | ||
| IgG2 (g/l) | 0·7 ↓ (0·9–5) | 0·5 ↓ (0·6–3·5) | 0·17 ↓ (0·6–3·5) | 2·97 (0·9–5) | 1·49 (0·9–5) | n.d. | ||
| IgG3 (g/l) | 0·14 ↓ (0·15–1·5) | 0·16 (0·14–1·3) | 0·09 ↓ (0·14–1·3) | 0·31 (0·15–1·5) | 0·12 ↓ (0·15–1·5) | n.d. | ||
| IgG4 (g/l) | 0·12 (< 0·03–2·1) | < 0·04 (< 0·03–1·2) | < 0·01 (< 0·03–1·2) | 0·75 (< 0·03–2·1) | 0·59 (< 0·03–2·1) | n.d. | ||
| IgA (g/l)† | 0·70 (0·3–2) | 0·27 (0·1–1·6) | 0·25‡↓ (0·3–2) | < 0·25 ↓ (0·1–1·6) | 1·31 (0·7–4) | 0·60 ↓ (0·7–4) | 1·12 (0·7–4) | |
| IgM (g/l)† | 0·41 ↓ (0·5–2) | 0·5 (0·5–1·8) | 0·33‡↓ (0·5–2) | 0·24 ↓ (0·5–1·8) | 0·51 (0·4–2·3) | 0·74 (0·4–2·3) | 0·30 ↓ (0·4–2·3) | |
| Response to unconjugated pneumococcal vaccine§ | (6 years) | (10 years) | (3 years) | (4 years) | (3 years) | (43 years) | (39 years) | n.d. |
| Serotype 3: before/after (U/ml) | 7/32 | 12/33 ↓ | 7/52 | 7 | < 1/1 ↓ | 4/29 | 4/77 | |
| Serotype 4: before/after (U/ml) | 11/23 ↓ | 11/46 | 3/22 | 5 | < 1/< 1 ↓ | 2/70 | 2/> 100 | |
| Serotype 9: before/after (U/ml) | 6/17 ↓ | 6/13 ↓ | 1/16 ↓ | 3 | < 1/< 1 ↓ | 7/38 | 1/35 | |
| Response to diphtheria toxoid (IU/ml)¶ | 0·7/3·08* | 0·02/3·06 | 0·05/0·55 | 0·1/5·52 | 3·84/n.d. | n.d. | ||
| Response to tetanus toxoid (IU/ml)¶ | 4·03/> 16* | 0·13/2·24 | 0·07/3·60 | 1·0/> 16 | 6·29/n.d. | n.d. | ||
| TNFRSF13B mutation | No mutation | C104R heterozygous | C104R heterozygous | C104R heterozygous | C104R heterozygous | C104R heterozygous | ||
Measured at the age of 9 years.
Immunoglobulin (Ig) levels at time of diagnosis; age-matched reference values between parentheses.
Immunoglobulin levels at 8 years.
Measured before and 3–4 weeks after vaccination; impaired antibody response was defined as failure to increase the pre-immunization titre by fourfold.
Measured before and 3–4 weeks after vaccination with a combined diphtheria toxoid–tetanus toxoid–inactivated polio vaccine.
n.d., not determined; ↓, below the reference value for age.
Discussion
We identified the heterozygous C104R TNFRSF13B mutation (encoding TACI) in two of three related CVID patients on IVIG. It is remarkable that we did not find this TNFRSF13B mutation in patient III:1, despite his hypogammaglobulinaemia and decreased response to unconjugated pneumococcal vaccine, which is a clinical presentation that fits perfectly with the diagnosis of ‘probable CVID’. His clinical phenotype is indistinguishable from the other two index patients, his sister (III:2) and cousin (III:3). This has not been described previously, and provides new evidence that the C104R TNFRSF13B mutation is not a straightforward disease-determining mutation. Pan-Hammarström et al.[15] described that 0·3% of the normal population carries heterozygous TNFRSF13B mutations, but the frequency of the C104R, A181E and ins204A mutations was increased significantly in CVID patients compared with controls, suggesting that these variants, even in heterozygous forms, contribute to the development of CVID. We found the C104R TNFRSF13B mutation in three additional family members: one with selective IgA-deficiency and recurrent infections, one with recurrent infections only and one healthy 70-year-old grandfather. This study shows that mutations in the TNFRSF13B gene must be interpreted with caution: they should be considered as disease-susceptibility polymorphisms, rather than disease-determining mutations. So far no association of TNFRSF13B mutations with clinical complications, such as severity or frequency of infections, lymphoproliferation, autoimmunity or granulomatous disease, has been established within the CVID population. Other genetic or environmental factors that contribute to the development of (TACI-related) CVID need to be identified. If such factors appear to have predictive value for the occurrence and progression of clinical complications, this will greatly support the care for these patients and their families.
Acknowledgments
We are grateful to the family for their co-operation. We would like to thank Tom de Vries Lentsch for help in preparing Fig. 1. This study was funded by a grant from the JBZ Research Fund.
References
- 1.Wood P, Stanworth S, Burton J, et al. Recognition, clinical diagnosis and management of patients with primary antibody deficiencies: a systematic review. Clin Exp Immunol. 2007;149:410–23. doi: 10.1111/j.1365-2249.2007.03432.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Quinti I, Soresina A, Spadaro G, et al. Long-term follow-up and outcome of a large cohort of patients with common variable immunodeficiency. J Clin Immunol. 2007;27:308–16. doi: 10.1007/s10875-007-9075-1. [DOI] [PubMed] [Google Scholar]
- 3.Goldacker S, Warnatz K. Tackling the heterogeneity of CVID. Curr Opin Allergy Clin Immunol. 2005;5:504–9. doi: 10.1097/01.all.0000191888.97397.b3. [DOI] [PubMed] [Google Scholar]
- 4.Di Renzo M, Pasqui AL, Auteri A. Common variable immunodeficiency: a review. Clin Exp Med. 2004;3:211–7. doi: 10.1007/s10238-004-0027-2. [DOI] [PubMed] [Google Scholar]
- 5.Castigli E, Wilson SA, Garibyan L, et al. TACI is mutant in common variable immunodeficiency and IgA deficiency. Nat Genet. 2005;37:829–34. doi: 10.1038/ng1601. [DOI] [PubMed] [Google Scholar]
- 6.Salzer U, Chapel HM, Webster AD, et al. Mutations in TNFRSF13B encoding TACI are associated with common variable immunodeficiency in humans. Nat Genet. 2005;37:820–8. doi: 10.1038/ng1600. [DOI] [PubMed] [Google Scholar]
- 7.Grimbacher B, Hutloff A, Schlesier M, et al. Homozygous loss of ICOS is associated with adult-onset common variable immunodeficiency. Nat Immunol. 2003;4:261–8. doi: 10.1038/ni902. [DOI] [PubMed] [Google Scholar]
- 8.Salzer U, Maul-Pavicic A, Cunningham-Rundles C, et al. ICOS deficiency in patients with common variable immunodeficiency. Clin Immunol. 2004;113:234–40. doi: 10.1016/j.clim.2004.07.002. [DOI] [PubMed] [Google Scholar]
- 9.van Zelm MC, Reisli I, van der Burg M, et al. An antibody-deficiency syndrome due to mutations in the CD19 gene. N Engl J Med. 2006;354:1901–12. doi: 10.1056/NEJMoa051568. [DOI] [PubMed] [Google Scholar]
- 10.Warnatz K, Salzer U, Gutenberger S, et al. Finally found: human BAFF-R deficiency causes CVID. XIth Meeting of the European Society for Immunodeficiencies; Versailles. 2004. October. [Google Scholar]
- 11.Kanegane H, Agematsu K, Futatani T, et al. Novel mutations in a Japanese patient with CD19 deficiency. Genes Immun. 2007;8:663–70. doi: 10.1038/sj.gene.6364431. [DOI] [PubMed] [Google Scholar]
- 12.Schaffer AA, Salzer U, Hammarstrom L, et al. Deconstructing common variable immunodeficiency by genetic analysis. Curr Opin Genet Dev. 2007;17:201–12. doi: 10.1016/j.gde.2007.04.002. [DOI] [PubMed] [Google Scholar]
- 13.Salzer U, Jennings S, Grimbacher B. To switch or not to switch – the opposing roles of TACI in terminal B cell differentiation. Eur J Immunol. 2007;37:17–20. doi: 10.1002/eji.200636914. [DOI] [PubMed] [Google Scholar]
- 14.Sakurai D, Kanno Y, Hase H, et al. TACI attenuates antibody production costimulated by BAFF-R and CD40. Eur J Immunol. 2007;37:110–8. doi: 10.1002/eji.200636623. [DOI] [PubMed] [Google Scholar]
- 15.Pan-Hammarstrom Q, Salzer U, Du L, et al. Reexamining the role of TACI coding variants in common variable immunodeficiency and selective IgA deficiency. Nat Genet. 2007;39:429–30. doi: 10.1038/ng0407-429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hendriksen CF, van der Gun JW, Kreeftenberg JG. Combined estimation of tetanus and diphtheria antitoxin in human sera by the in vitro toxin-binding inhibition (ToBI) test. J Biol Stand. 1989;17:191–200. doi: 10.1016/0092-1157(89)90009-7. [DOI] [PubMed] [Google Scholar]
- 17.de Melker HE, Berbers GA, Nagelkerke NJ, et al. Diphtheria antitoxin levels in the Netherlands: a population-based study. Emerg Infect Dis. 1999;5:694–700. doi: 10.3201/eid0505.990511. [DOI] [PMC free article] [PubMed] [Google Scholar]