

Abstract
The present study aimed to investigate the role of gastric mucosa for the secretion of interleukin (IL)-23 in chronic gastritis. One hundred and one patients were enrolled; 47 with duodenal ulcer, 33 with gastric ulcer and 31 with chronic gastritis. Biopsies were incubated in the absence/presence of endotoxins. Supernatants were collected and IL-23 and IL-1β were measured by enzyme-linked immunosorbent assay. Scoring of gastritis was performed according to the updated Sydney score. Patients with duodenal and gastric ulcer and those with chronic gastritis had similar scores of gastritis. IL-23 was higher in supernatants of tissue samples of Helicobacter pylori-positive than of H. pylori-negative patients. No differences were recorded in concentrations of IL-23 and IL-1β between patients with duodenal ulcer, gastric ulcer and chronic gastritis. Positive correlations were found between IL-23 of patients with both duodenal and gastric ulcer and chronic gastritis and the degree of infiltration of neutrophils and monocytes. Similar correlations were observed between IL-23 and IL-1β. IL-23 secreted by the gastric mucosa could be implicated in the pathogenesis of chronic gastritis. IL-23 was released in the presence of H. pylori from the inflamed gastric mucosa and followed the kinetics of IL-1β.
Keywords: chronic gastritis, gastric mucosa, IL-23, proinflammatory cytokines
Introduction
Chronic gastritis occurs in the setting of infection by Helicobacter pylori. H. pylori-associated gastritis is characterized by severe infiltration of neutrophils and mononuclear cells in the gastric mucosa [1]. Accumulation and activation of these cells is induced by the local production of cytokines, such as interleukin (IL)-1β. Evidence suggests that H. pylori lipopolysaccharide (LPS) mediates release of cytokines from human monocytes [2,3]. The biological effects of these cytokines may result in the recruitment, influx and activation of neutrophils in the gastric mucosa in the event of infection by H. pylori[4]. IL-1β is a potent inflammatory cytokine that is released as a component of the host response against bacterial infection. It is expressed primarily by activated macrophages [5].
The heterodimeric cytokine IL-12 plays a key role in host defence by differentiating the cells of T helper 1 (Th1) response. Recently, IL-23 was identified as a member of the IL-12 cytokine family secreted by neutrophils and monocytes. Recent data revealed the involvement of IL-23 in inflammatory procedures of the lower gastrointestinal tract, mainly Crohn's disease [6]. Whether IL-23 contributes to the inflammatory reaction taking place in the gastric mucosa in the event of gastritis is not defined. In the present study, biopsies of gastric mucosa taken from patients with peptic ulcer disease and chronic gastritis were cultured to test the release of IL-23.
Patients and methods
Study group
The study was approved by the Medical and Ethics Committee (6th/11·30·05/26962 and 4th/07·16·06/11573) of General Hospital ‘Sismanoglion’, Athens, Greece. A total of 111 patients were enrolled; 47 patients with duodenal ulcer, 33 with gastric ulcer and 31 with chronic gastritis without peptic ulcer disease. Clinical and endoscopic data for 72 of these patients have already been published [7].
Informed consent was obtained from all participants. Indications for endoscopy in these patients were abdominal pain or discomfort, epigastric pain with nausea and vomiting and dyspepsia. All endoscopies were performed by the same endoscopist. Peptic ulcer was defined as a circumscribed break in the mucosa in the duodenum (DU) or in the stomach (GU) with apparent depth covered by an exudate, as described previously [8]. All patients with peptic ulcer disease belonged to the Forrest III score [9]. H. pylori infection was defined by the presence of the bacterium both in the histopathological findings of each biopsy and after a gastric biopsy culture with the proper growth medium [10]. Exclusion criteria for the study were: recent upper gastrointestinal (GI) bleeding, gastric carcinoma, diabetes mellitus, liver cirrhosis, acute or chronic renal failure and the ingestion of any anti-microbial or anti-secretory medication for at least 15 days prior to endoscopy.
Study design and interventions
All patients were examined by upper GI endoscopy. All patients were endoscoped; biopsies were collected during each endoscopy. At the time of endoscopy, two biopsy specimens were obtained from adjacent areas of the gastric antrum. When each biopsy specimen was taken, the forceps were opened fully and aimed at right-angles to the gastric lumen to the extent possible to obtain uniformly sized biopsies. Biopsies were obtained from endoscopically intact mucosa distant from focal lesions such as ulcers and erosions. Each biopsy was used for culture.
In brief, gastric antral mucosal biopsy tissues were weighed and cultured on a culture insert over wells containing RPMI-1640 medium with 10% heat-inactivated fetal bovine serum in a 5% CO2 incubator for 18 h [11]. Biopsies were positioned on the insert with mucosal surfaces uppermost. The first biopsy tissue was left unstimulated and served as control, and the second was stimulated with 10 ng/ml of LPS of Escherichia coli O144 : H4. Stimulation with LPS was performed to mimic in vivo conditions where gastric and dudodenal mucosa are exposed to LPS of H. pylori. At the end of the incubation, the plates were centrifuged for 10 min at 1400 g; supernatants were then collected from the wells and stored at −70°C until assayed for estimation of IL-23 and IL-1β. Results were correlated with histopathological findings.
Estimation of IL-1β and IL-23
The IL-1β was estimated by a developmental enzyme immunoabsorbent assay using capture and detection antibodies and IL-1β standards (R&D Inc., Minneapolis, MN, USA). The lowest limit of detection was 15·6 pg/ml. IL-23 was measured in supernatants with a commercially available enzyme immunoabsorbent assay (eBioscience, San Diego, CA, USA). The lowest limit of detection was 15 pg/ml. All measurements were performed in duplicate and cytokine concentrations were expressed as pg/g of tissue.
Histopathology
Formalin-fixed, paraffin-embedded tissue samples were cut routinely at 3–4 µm and stained with haematoxylin and eosin alcian blue periodic acid Schiff (pH: 2·5) and Giemsa. Immunochemistry was performed for H. pylori detection with 1 : 100 dilution of Renoir Red (Biocare Med., Concord, CA, USA).
The presence of gastritis was evaluated in each biopsy sample after separate scoring for each of the following parameters: (i) disease activity as mucosal infiltration by neutrophils; (ii) chronic inflammation expressed as infiltration by monocytes and lymphocytes; (iii) degree of mucosal atrophy; and (iv) intestinal metaplasia. Each parameter was scored from 0 to 3 (0: absent, 1: mild, 2: moderate, 3: marked). In the case of intestinal metaplasia, scores indicated the following findings: 0: absence; 1: complete or type I; 2: incomplete or type II; and 3: incomplete or type III. As a consequence, the total Sydney score of gastritis ranged between 0 and 15, according to previously reported criteria of the updated Sydney System [12]. The extent of gastric inflammation in the antrum, corpus or both was also recorded. The density of H. pylori was evaluated semiquantitatively by the same criteria [13]. Specimens were classified by one pathologist who was unaware of the corresponding laboratory findings.
Statistical analysis
Patients of the three groups were divided further into subgroups according to the absence or presence of H. pylori. Concentrations of IL-23 and IL-1β were expressed by their mean and standard error for the total number of patients and by their median ±95% confidence intervals or interquartile range when patients were divided further according to the presence or absence of H. pylori infection. Updated Sydney classification scores were given by their means (±standard deviation). Comparison between groups was made by Mann–Whitney U-test with a correction according to Bonferroni; for qualitative data comparisons were performed by χ2 test. Comparisons before and after stimulation with LPS was performed by Wilcoxon's paired test. Correlations between IL-23 and IL-1β and the gastritis score or the density of H. pylori were performed according to Spearman's rank order. Any P-value less than 0·05 was considered significant.
Results
Patients’ characteristics are shown in Table 1. No differences were found between the three groups regarding age, sex and intake of non-steroidal anti-inflammatory drugs. All patients, except one, suffering from duodenal ulcer had presence of H. pylori on tissue biopsy.
Table 1.
Demographic characteristics of patients enrolled in the study.
| Parameters | Duodenal ulcer | Gastric ulcer | Chronic gastritis | |||
|---|---|---|---|---|---|---|
| Patients (n) | 47 | 33 | 31 | |||
| Age (mean ± s.d.) | 66·51 ± 9·85 | 66·27 ± 11·47 | 64·93 ± 11·54* | |||
| Male/female | 29/18 | 20/13 | 19/12* | |||
| Non-smoking/smoking | 22/25 | 16/17 | 14/17* | |||
| History of NSAID use (yes/no) | 29/18 | 20/13 | 19/12* | |||
| Helicobacter pylori positive/negative | 46/1 | 21/12 | 21/10† | |||
| Site of gastric inflammation | ||||||
| Antrum (no. of patients) | 23 | 16 | 12* | |||
| Corpus (no. of patients) | 13 | 7 | 9* | |||
| Disseminated (no of patients) | 11 | 10 | 10* | |||
| H. pylori (+) | H. pylori (−) | H. pylori (+) | H. pylori (−) | H. pylori (+) | H. pylori (−) | |
| Total updated Sydney score (mean ± s.d.) | 5·47 ± 2·09 | n.a. | 6·33 ± 1·87 | 5·25 ± 2·56 | 5·09 ± 1·41 | 4·50 ± 1·70 |
| Degree of neutrophil infiltration (mean ± s.d.) | 1·91 ± 0·73 | n.a. | 2·52 ± 0·60 | 1·50 ± 0·79‡ | 2·14 ± 0·79 | 1·20 ± 0·92‡ |
| Degree of monocyte infiltration (mean ± s.d.) | 1·96 ± 0·82 | n.a. | 1·90 ± 0·77 | 1·83 ± 0·72 | 1·86 ± 0·73 | 1·30 ± 0·82 |
| Degree of lymphocyte infiltration (mean ± s.d.) | 0·78 ± 0·89 | n.a. | 0·76 ± 0·76 | 1·08 ± 1·16 | 0·67 ± 0·86 | 1·00 ± 067 |
| Degree of mucosal atrophy (mean ± s.d.) | 0·57 ± 0·83 | n.a. | 0·76 ± 0·94 | 0·42 ± 0·67 | 0·33 ± 0·66 | 0·70 ± 0·82 |
| Degree of intestinal metaplasia (mean ± s.d.) | 0·26 ± 0·55 | n.a. | 0·38 ± 0·74 | 0·42 ± 0·67 | 0·10 ± 0·30 | 0·30 ± 0·68 |
| Density of Helicobacter pylori (mean ± s.d.) | 1·98 ± 0·91 | 1·90 ± 0·83 | 2·10 ± 0·83 | |||
P = n.s. (non-significant) between the three groups.
P < 0·0001 between the three groups.
P < 0·0001 compared with the respective Helicobacter pylori-positive patients. Updated Sydney scores are given. NSAID, non-steroidal anti-inflammatory drugs; s.d., standard deviation; n.a., not applicable.
The degree of infiltration of the gastric mucosa by neutrophils was greater among H. pylori-positive patients compared with H. pylori-negative patients. No differences were recorded between them regarding the other histological parameters of gastritis (Table 1).
Concentrations of IL-23 and of IL-1β in supernatants of samples of gastric mucosa taken from patients with duodenal ulcer disease with gastric ulcer disease and with chronic gastritis are shown in Table 2. They were higher among the population of H. pylori-positive patients compared with H. pylori-negative patients.
Table 2.
Concentrations of interleukin (IL)-23) and IL-1β of supernatants of tissue samples taken from patients with either duodenal or gastric ulcer disease or chronic gastritis.
| Duodenal ulcer |
Gastric ulcer |
Chronic gastritis |
||||
|---|---|---|---|---|---|---|
| H. pylori (+) | H. pylori (−) | H. pylori (+) | H. pylori (−) | H. pylori (+) | H. pylori (−) | |
| No. of patients | 46 | 1 | 21 | 12 | 21 | 10 |
| IL-23 [median (IQR), pg/g of tissue] | ||||||
| −LPS | 223·8 (137·8) | – | 1748·3 (332·2)* | 138·0 (260·8) | 323·3 (60·89)† | 204·1 (86·91) |
| +LPS | 355·4 (369·4) | – | 289·7 (567·3)* | 249·3 (456·1) | 534·1 (119·16)‡ | 377·2 (91·26) |
| IL-1β[median (IQR), pg/g of tissue] | ||||||
| −LPS | 723·1 (799·8) | – | 617·3 (945·9)† | 532·3 (441·1) | 782·3 (666·8)† | 704·3 (720·4) |
| +LPS | 1593·1 (1663·2) | – | 1486·9 (1001·4)* | 856·4 (963·2) | 1213·9 (1489·7)† | 1453·6 (1759·9) |
P of comparisons with Helicobacter pylori-negative patients:
P < 0·05,
P = non-significant,
P < 0·01. Patients were divided as either H. pylori-positive or H. pylori-negative. Lipopolysaccahride (LPS): endotoxin of Escherichia coli O144 : H4. IQR, interquartile range.
Comparisons of concentrations of IL-23 and IL-1β in supernatants before and after stimulation with LPS are shown in Fig. 1. Stimulation with LPS resulted in a significant increase of the production of both IL-23 and IL-1β.
Fig. 1.
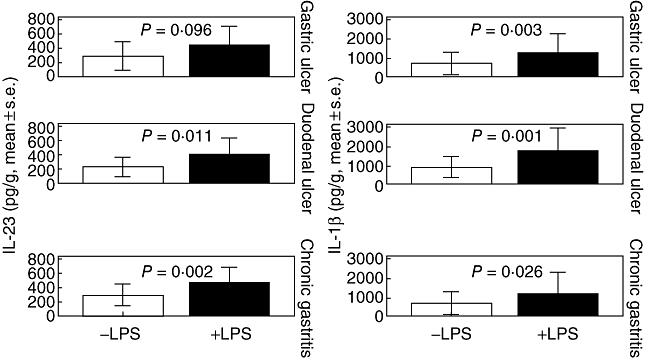
Concentrations of interleukin (IL)-23 and IL-1β in supernatants of gastric mucosa in patients with duodenal and gastric ulcer, and chronic gastritis before and after stimulation with lipopolysaccharide (LPS). P-values refer to comparisons before and after stimulation with LPS.
Concentrations of IL-23 in tissue supernatants after stimulation with LPS were correlated positively with the degree of infiltration by monocytes (P = 0·013, r2 = +0·498) and by neutrophils (P = 0·001, rs = +0·631). Similar correlations were found between concentrations of IL-1β after stimulation with LPS with the degree of infiltration by monocytes (P < 0·001, r2 = +0·509) and by neutrophils (P < 0·001, rs = +0·553).
Discussion
Chronic active gastritis is the leading inflammatory disorder of the upper gastrointestinal tract caused by H. pylori infection. H. pylori-related gastric inflammation may also evolve into peptic ulcer disease [14]. The latter process is mediated by various mechanisms implicating several proinflammatory cytokines.
Former results of our group revealed that the inflamed gastric mucosa was potent for the release of soluble triggering receptor expressed on myeloid cells (sTREM-1) in peptic ulcer disease. Proinflammatory cytokines such as tumour necrosis factor-α were released from the inflamed gastric mucosa independently from the inflammatory status [7]. Other authors have stated that cell loss and apoptosis of gastric mucous cells was enhanced by H. pylori LPS. The latter is less potent compared with E. coli LPS for the induction of release of proinflammatory mediators. The low immunological potency of H. pylori LPS may contribute to low-grade gastritis [15]. In an attempt to simulate the latter process, cultured biopsies were stimulated with LPS. The applied LPS derived from E. coli O144 : H4 acts as a Toll-like receptor-4 agonist. It was used for this study because of the lack of commercially available of H. pylori. It should be emphasized that this may also be a limitation of our study, as LPS of H. pylori has also been described to behave in a different manner [16].
Results revealed that the inflamed gastric mucosa of H. pylori-positive patients could secrete IL-23 (Fig. 1). Gastric mucosa of patients with both duodenal and gastric ulcers was equally potent for secretion of IL-23 compared with patients with chronic active gastritis with no signs of peptic ulcer disease. The release of IL-23 was greater by H. pylori-infected gastric mucosa than by gastric mucosa not infected by H. pylori mainly for patients with chronic gastritis and only after stimulation with LPS. Similar findings have been published elsewhere [17,18].
Release of IL-23 after stimulation of gastric mucosa of patients with gastric ulcer and H. pylori infection was reduced. This probably implies a different effect of E. coli LPS and H. pylori on the gastric mucosa regarding the release of IL-23 in the event of gastric ulcer.
Similar kinetics was also observed for IL-1β. IL-1β was increased in strict correlation with the degree of mucosal inflammation independently from the underlying pathogenetic status.
The release of IL-23 in supernatants of gastric mucosa in peptic ulcer disease and chronic gastritis creates the hypothesis that IL-23 is a cytokine implicated in the pathogenesis of chronic gastric inflammation. Both IL-23 and IL-1β were correlated with the degree of acute and chronic inflammation. IL-23 belongs to the IL-12 superfamily, which is implicated in the Th1 immune response. On that basis, it might be proposed that IL-23 participated in the orchestration of chronic inflammation of the gastric mucosa.
The present study has reported for the first time in the literature two separate findings for the capacity of inflamed gastric mucosa for the release of proinflammatory cytokines. First, gastric mucosa secretes IL-23 at a rate dependent on the degree of infiltration by inflammatory cells; and secondly, the kinetics of release of IL-23 followed those of IL-1β. Whether IL-23 is an independent mediator in the pathogenesis of peptic ulcer disease or not cannot be excluded with safety from the presented findings. It is anticipated that IL-23 participated in the process of inflammation irrespective of the presence or not of ulcer. Further investigation is necessary to elucidate fully the exact role of IL-23 in the pathogenesis of chronic gastritis.
References
- 1.Yuan Y, Padol IT, Hunt RH. Peptic ulcer disease today. Nat Clin Pract Gastroenterol Hepatol. 2006;3:80–9. doi: 10.1038/ncpgasthep0393. [DOI] [PubMed] [Google Scholar]
- 2.Calam J, Baron JH. ABC of the upper gastrointestinal tract. Pathophysiology of duodenal and gastric ulcer and gastric cancer. BMJ. 2001;323:980–2. doi: 10.1136/bmj.323.7319.980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Eckmann L. Sensor molecules in intestinal innate immunity against bacterial infections. Curr Opin Gastroenterol. 2006;22:95–101. doi: 10.1097/01.mog.0000208458.38772.2a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Polenghi A, Bossi F, Fischetti F, et al. The neutrophil-activating protein of Helicobacter pylori crosses endothelia to promote neutrophil adhesion in vivo. J Immunol. 2007;178:1312–20. doi: 10.4049/jimmunol.178.3.1312. [DOI] [PubMed] [Google Scholar]
- 5.Rydstrom A, Wick MJ. Monocyte recruitment, activation, and function in the gut-associated lymphoid tissue during oral Salmonella infection. J Immunol. 2007;178:5789–801. doi: 10.4049/jimmunol.178.9.5789. [DOI] [PubMed] [Google Scholar]
- 6.Hue S, Ahern P, Buonocore S, et al. Interleukin-23 drives innate and T cell-mediated intestinal inflammation. J Exp Med. 2006;203:2473–83. doi: 10.1084/jem.20061099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Koussoulas V, Vassiliou S, Spyridaki E, et al. Evidence for the role of gastric mucosa at the secretion of Soluble Triggering Receptor expressed on myeloid cells (sTREM-1) in peptic ulcer disease. World J Gastroenterol. 2007;13:4610–14. doi: 10.3748/wjg.v13.i34.4610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Peek RM, Miller GG, Tham KT, et al. Heightened inflammatory response and cytokine expression in vivo to cagA Helicobacter pylori strains. Lab Invest. 1995;71:760–70. [PubMed] [Google Scholar]
- 9.Kohler B, Maier M, Benz C. Acute ulcer bleeding. A prospective randomized trial to compare Doppler and Forrest classifications in endoscopic diagnosis and therapy. Dig Dis Sci. 1997;42:1370–4. doi: 10.1023/a:1018877602113. [DOI] [PubMed] [Google Scholar]
- 10.Peterson WL, Fendrick M, Cave DR, et al. Helicobacter pylori-related disease: guidelines for testing and treatment. Arch Intern Med. 2000;160:1285–91. doi: 10.1001/archinte.160.9.1285. [DOI] [PubMed] [Google Scholar]
- 11.Kusugami K, Fukatsu A, Tanimoto M, et al. Elevation of interleukin-6 in inflammatory bowel disease is macrophage and epithelial cell-dependent. Dig Dis Sci. 1995;40:949–59. doi: 10.1007/BF02064182. [DOI] [PubMed] [Google Scholar]
- 12.Dixon MF, Genta RM, Yardley JH, Correa P. Classification and grading of gastritis. The updated Sydney System. Am J Surg Pathol. 1996;20:1161–81. doi: 10.1097/00000478-199610000-00001. [DOI] [PubMed] [Google Scholar]
- 13.Zhang C, Yamada N, Wu YL, et al. Comparison of Helicobacter pylori infection and gastric mucosal histological features of gastric ulcer patients with chronic gastritis patients. World J Gastroenterol. 2005;11:976–81. doi: 10.3748/wjg.v11.i7.976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lindholm C, Quiding- Jarbrink M, Lonroth H, et al. Local cytokine response in Helicobacter pylori-infected subjects. Infect Immun. 1998;66:5964–71. doi: 10.1128/iai.66.12.5964-5971.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Durkin E, Moran AP, Hanson P. Apoptosis induction in gastric mucous cells in vitro: lesser potency of Helicobacter pylori than Escherichia coli lipopolysaccharide, but positive interaction with ibuprofen. J Endotoxin Res. 2006;12:47–56. doi: 10.1179/096805106X89080. [DOI] [PubMed] [Google Scholar]
- 16.Lepper PM, Triantafilou M, Schumann C, Schneider EM, Triantafilou K. Lipopolysaccharides from Helicobacter pylori can act as antagonists for Toll-like receptor 4. Cell Microbiol. 2005;7:519–28. doi: 10.1111/j.1462-5822.2005.00482.x. [DOI] [PubMed] [Google Scholar]
- 17.Vivas JR, Regnault B, Michel V, et al. Interferon gamma-signature transcript profiling and IL-23 upregulation in response to Helicobacter pylori infection. Int J Immunopathol Pharmacol. 2008;21:515–26. doi: 10.1177/039463200802100305. [DOI] [PubMed] [Google Scholar]
- 18.Caruso R, Fina D, Paoluzi OA, et al. IL-23-mediated regulation of IL-17 production in Helicobacter pylori-infected gastric mucosa. Eur J Immunol. 2008;38:470–8. doi: 10.1002/eji.200737635. [DOI] [PubMed] [Google Scholar]