

Abstract
The mechanisms by which immunologically activated mast cells stimulate the production of proinflammatory cytokines by T helper type 2 (Th2) lymphocytes were investigated in a human cell culture system. Supernatants collected from cord blood-derived mast cells after treatment with immunoglobulin E (IgE)/anti-IgE contained an activity that stimulated the production of interleukin (IL)-4, IL-5 and IL-13 (both mRNA and protein) by Th2 lymphocytes. This activity was not detected in supernatants from unactivated mast cells and its production was inhibited by treatment of activated mast cells with the cyclo-oxygenase inhibitor diclofenac. The concentration of diclofenac used inhibited completely the production of prostaglandin D2 (PGD2) but did not inhibit the release of histamine or leukotriene C4. The effect of supernatants from activated mast cells was mimicked by exogenous PGD2 at concentrations similar to those detected in the cultures of activated mast cells, and addition of exogenous PGD2 to supernatants from diclofenac-treated mast cells restored their ability to stimulate Th2 cytokine production. The ability of the mast cell supernatants to stimulate production of Th2 cytokines was not affected by addition of diclofenac to the Th2 cells directly, indicating that the production, but not the action, of the factor was sensitive to diclofenac treatment. Inhibition of chemoattractant receptor-homologous molecule expressed on Th2 cells (CRTH2) abolished the effect of the mast cell supernatants on Th2 cytokine production. These data indicate that mast cells have the ability to stimulate Th2 cells to elaborate cytokines independently of T cell receptor activation or co-stimulation and this response is mediated by PGD2 acting upon CRTH2 expressed by Th2 cells.
Keywords: chemoattractant receptor-homologous molecule expressed on Th2 cells, cytokine, mast cells, prostaglandin D2
Introduction
T helper type 2 (Th2) lymphocytes are found in high numbers at sites of allergic inflammation, including the lungs of asthmatic patients, and there is increasing evidence that these cells contribute to disease pathology through the elaboration of cytokines such as interleukin (IL)-4, IL-5 and IL-13 [1–5]. IL-5 has a well-recognized role in promoting blood and tissue eosinophilia [6–9], while both IL-4 and IL-13 have the ability to induce isotype-switching leading to the production of immunoglobulin E (IgE) [10,11]. IL-13 also acts directly on bronchial tissues to promote mucus production and airway hyperresponsiveness [12]. The central role played by Th2 cytokines in allergic disease is illustrated by the findings that transgenic mice that overproduce Th2 cytokines have elevated levels of IgE with associated pathologies that resemble aspects of atopic dermatitis and asthma [13], and that blockade of IL-4 and IL-13 signalling reduces the late-phase airway response to allergen in asthmatic subjects [14].
Mast cells also play a central role in the allergic response [15,16], and the interaction between mast cells and Th2 cells may provide an essential link between the early- and late-phase allergic responses. When allergic individuals are exposed to allergen, cross-linking of IgE on the surface of mast cells leads to the rapid release of preformed mediators such as histamine and newly synthesized mediators such as leukotriene C4 (LTC4) and prostaglandin D2 (PGD2), causing features of the classical early allergic response. In addition to causing the characteristic early-phase responses, mast cell activation may also promote the recruitment and activation of Th2 lymphocytes within allergic tissues, a process which can occur independently of antigen-specific T cell activation [17]. The importance of such interaction between mast cells and Th2 cells may underlie the classic observation that a late-phase lymphocytic allergic response can be induced in non-atopic human subjects by passive transfer of IgE [18], and is consistent with the ability of the anti-IgE antibody omalizumab to inhibit the late-phase airway response to allergen in asthmatic patients [19]. In a chronic model of allergic asthma in mice, mast cells are essential for accumulation of lymphocytes in bronchoalveolar fluid in addition to the development of a number of other features of airway inflammation [20]. Accumulation of various lymphocyte subtypes in inflamed tissues has been shown to be mediated by the release of distinct chemoattractant molecules from mast cells [21–24]. The role of mast cell-derived eicosanoids in promoting activation of T cells has been reviewed comprehensively by Kim and Luster [25]. Supernatants from immunologically activated mast cells display potent chemotactic activity for Th2 lymphocytes, an activity that is mediated by PGD2 acting on the chemotactic receptor chemoattractant receptor-homologous molecule expressed on Th2 cells (CRTH2) [26]. In addition to mediating chemotaxis of Th2 cells in response to PGD2[27], activation of CRTH2 also leads to the production of Th2 cytokines [28], an effect that can occur in the absence of antigen or any other form of co-stimulation [29]. However, a wide variety of inflammatory mediators are released by mast cells upon activation which may have the potential to modulate Th2 cell function, and to date there has been no study investigating whether such mediators promote proinflammatory cytokine production by Th2 cells and, among these diverse mediators, what role PGD2 and CRTH2 might play in such a response. We have therefore investigated the mechanisms by which immunologically activated mast cells stimulate the production of Th2 cytokines in a human in vitro culture system. Supernatants from mast cells activated with IgE/anti-IgE, but not unactivated mast cells, stimulated the production of cytokines by Th2 lymphocytes. Production of the stimulatory activity was inhibited by treatment of mast cells with the cyclo-oxygenase inhibitor diclofenac, and the effect of the supernatant was inhibited by the CRTH2 antagonists ramatroban and TM300089. These data suggest that the interaction of Th2 lymphocytes with activated mast cells leading to the production of cytokines is mediated through a CRTH2-dependent mechanism.
Materials and methods
Reagents
The PGD2 was purchased from Biomol (Plymouth Meeting, PA, USA); ramatroban (BAY u3405), SQ29548, PGD2–MOX enzyme immunoassay kits and LTC4 enzyme immunoassay kits were purchased from Cayman Chemical (Ann Arbor, MI, USA); TM30089 was supplied by ChemieTek (Indianapolis, IN, USA); human CD4+ T cell isolation kit II, anti-human CRTH2 MicroBead Kit and T cell activation/expansion kits were from Miltenyi Biotec Ltd (Bergisch Gladbach, Germany); human recombinant stem cell factor, human recombinant IL-6 and human IL-4/5/13 immunoassay kits were purchased from R&D Systems (Minneapolis, MN, USA); Iscove's modified Dulbecco's medium and X-VIVO 15 medium were purchased from Lonza (Walkersville, MD, USA); human myeloma IgE, antibodies against human tryptase and chymase were purchased from Chemicon International (Chandlers Ford, UK); Ficoll–Hypaque was purchased from Amersham Biosciences (Little Chalfont, UK); histamine enzyme immunoassay kit was from SPI-BIO (Montigny le Bretonneux, France); RNeasy Mini kit and Omniscript reverse transcription (RT) kit were supplied from Qiagen (West Sussex, UK); and human recombinant IL-2, human recombinant IL-4, goat anti-human IgE, diclofenac and other chemicals were from Sigma-Aldrich (Dorset, UK).
Human mast cell culture and activation
Human mast cells were cultured from CD34+ progenitor cells, as described in our previous report [26]. Briefly, CD34+ progenitor cells from human cord blood (Lonza) were cultured at a density of 1 × 105 cells/ml with Iscove's modified Dulbecco's medium containing 10% human serum, 0·55 µM 2-mercaptoethanol, penicillin/streptomycin, human recombinant stem cell factor (100 ng/ml) and human recombinant IL-6 (50 ng/ml) in 5% CO2 at 37°C for 8–10 weeks. Half the culture medium was replaced twice weekly with fresh medium containing the same concentration of cytokines. The expression of tryptase and chymase of the cells was tested by immunostaining using the method described by Craig and Schwartz [30]. The cytospin smears were first air-dried for 2 h at room temperature and then fixed with Carnoy's solution (ethanol : chloroform : glacial acetic acid, 6:3:1) for 1 min. The fixed smears were stained using monoclonal antibodies against human mast cell tryptase and human mast cell chymase. The mast cells used in this study were tryptase-positive (> 80%) and chymase-negative (< 1%). The cells were pretreated with 5 µg/ml purified human myeloma IgE and human recombinant IL-4 (10 ng/ml) for 4 days, washed and then sensitized passively with fresh IgE (5 µg/ml) for 2 h. The cells were washed with medium for 20 min and then continued to be incubated with medium or challenged with goat anti-human IgE (1 µg/ml) in the presence or absence of diclofenac (10 µM). The supernatants of the cells were collected 1 h after challenge. The supernatants were assayed for PGD2 using a PGD2– MOX enzyme immunoassay kit and histamine using an enzyme immunoassay kit according to the manufacturer's instructions.
Human CRTH2+CD4+ Th2 cell culture
Human CRTH2+CD4+ Th2 cells were prepared using a modified method described previously [29]. Briefly, peripheral blood mononuclear cells were isolated from buffy coats (National Blood Service, Bristol, UK) by Ficoll Hypaque density gradient centrifugation, followed by CD4+ cell purification using a magnetic affinity cell sorting (MACS) CD4+ T cell isolation kit II. After 7 days’ culture in X-VIVO 15 medium containing 10% human serum, 50 U/ml IL-2 and 100 ng/ml IL-4, CRTH2-positive cells were isolated from the CD4+ cultures by positive selection using an anti-human CRTH2 MicroBead Kit. The harvested CD4+ CRTH2+ cells were treated as Th2 cells and were amplified further by stimulation with the T cell activation/expansion kit and grown in X-VIVO 15 medium containing 10% human serum and 50 U/ml IL-2 before use.
Cytokine release assays
The Th2 cells were treated with X-VIVO 15 culture medium or various mast cell supernatants in the presence or absence of PGD2 or other compounds, as indicated in the results at 37°C and 5% CO2 for 5 h. The supernatants of the treatments were collected. The concentrations of IL-4, IL-5 and IL-13 in the supernatants were assayed using enzyme-linked immunoassay kits, according to the manufacturer's instructions. The results were measured in a Victor2 V-1420 multi-label HTS Counter (PerkinElmer Life Sciences Wellesley, MA, USA).
Reverse transcription–polymerase chain reaction
Reverse transcription-polymerase chain reaction (RT–PCR) was conducted as described previously [29]. Total RNA of Th2 cells after different treatments for 3 h were extracted using an RNeasy Mini kit and quantitated using a GeneQuant Pro (Biochrom, Cambridge, UK). cDNA of the samples was prepared from the same starting amount of RNA using a Omniscript RT kit. PCR products were separated on an agarose gel and detected with a Fluor-S MAX2 Multimager (Bio-Rad, Hercules, CA, USA). The intensity of ethidium bromide-stained bands was quantified using Quantity One software (Bio-Rad). mRNA level of cytokines was normalized with the level of glyceraldehyde-3-phosphate-dehydrogenase (GAPDH). Primers used were as follows – IL-4: 5′-GCTGCCTCCAAGAACACAAC-3′ and 5′-CTCTGGTTGGCTTCCTTCAC-3′ generating a 221-base pairs (bp) fragment; IL-5: 5′-CTGCCTACGTGTATGCCATC-3′ and 5′-CTTTCCACAGTACCCCCTTG-3′ generating a 217-bp fragment; IL-13: 5′-CCTCAATCCTCTCCTGTTGG-3′ and 5′-GTCAGGTTGATGCTCCATACC-3′ generating a 206-bp fragment; and GAPDH: 5′-GCCACTCAGAAGACTGTGGATGGCC-3′ and 5′-GCAATGCCAGCCCCAGCATCAAAGG-3′ generating a 350-bp fragment.
Statistics
Data were analysed using one-way analysis of variance followed by the Newman–Keuls test. Values of P < 0·05 were considered statistically significant.
Results
Supernatants from activated mast cells promote cytokine production by Th2 cells
Supernatants collected from human mast cells 1 h after activation with IgE/anti-IgE contained low levels of IL-4, IL-5 and IL-13 (Table 1). Addition of these supernatants (diluted 1:2) to human Th2 cells (∼5 × 106 cells/ml) for 5 h led to the production of high levels of IL-4, IL-5 and IL-13 (Table 1; Fig. 1). The levels of the cytokines released by these Th2 cells were very similar to those produced by Th2 cells treated with 100 nM PGD2. In contrast, cytokine levels in cultures of Th2 cells treated with supernatants from unactivated mast cells were not increased significantly compared with those treated with media alone.
Table 1.
Cytokine levels in the supernatants from mast cells or T helper type 2 (Th2) cells after various treatments.
| Cytokine levels (pg/ml) |
|||
|---|---|---|---|
| IL-4 | IL-5 | IL-13 | |
| Unactivated mast cell supernatant alone | 4 ± 1 (n = 2) | 35 ± 15 (n = 2) | 18 ± 7 (n = 2) |
| Activated mast cell supernatant alone | 4 ± 2 (n = 2) | 44 ± 21 (n = 2) | 69 ± 24 (n = 2) |
| Th2 cells (∼5 × 106) + medium | 4 ± 2 (n = 8) | 228 ± 40 (n = 8) | 608 ± 95 (n = 8) |
| Th2 cells (∼5 × 106) + unactivated mast cell supernatant | 5 ± 1 (n = 4) | 257 ± 45 (n = 5) | 636 ± 90 (n = 8) |
| Th2 cells (∼5 × 106) + activated mast cell supernatant | 20 ± 7 (n = 4) | 780 ± 81 (n = 5) | 2215 ± 423 (n = 8) |
| Th2 cells (∼5 × 106) + 100 nM PGD2 | 24 ± 9 (n = 8) | 695 ± 40 (n = 8) | 2360 ± 379 (n = 8) |
IL, interleukin; PGD2, prostaglandin D2.
Fig. 1.
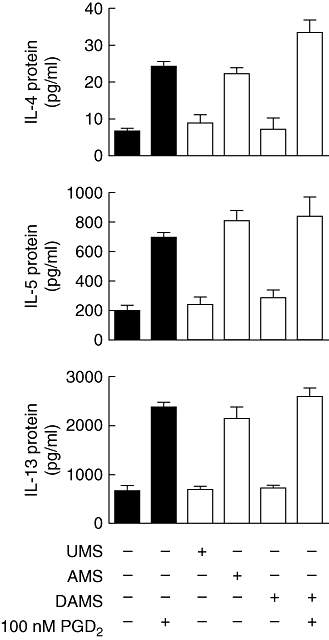
Effect of mast cell supernatants on T helper type 2 (Th2) cytokine production. Th2 cells were incubated with mast cell supernatants (1:2 dilution) (open bars) from unactivated mast cells (UMS), mast cells activated with immunoglobulin E (IgE)/anti-IgE (AMS) or diclofenac-treated mast cells activated with IgE/anti-IgE (DAMS) in the presence or absence of prostaglandin D2 (PGD2) as indicated for 5 h. Responses of Th2 cells to X-VIVO 15 medium or PGD2 alone are also shown as controls (black bars). The concentrations of interleukin (IL)-4, IL-5 and IL-13 in the incubation media were measured by enzyme immunoassay. Results are presented mean ± standard error of the mean (n = 4–6). P < 0·0001 by analysis of variance. P > 0·05 by Newman–Keuls test for (IL-4, IL-5 and IL-13) no additive versus UMS/DAMS, UMS versus DAMS, 100 nM PGD2versus AMS; (IL-5 and IL-13) 100 nM PGD2/AMS versus DAMS + 100 nM PGD2; n = 4–6.
Inhibition of PGD2 production abolishes the release of Th2 cytokine stimulatory activity from mast cells
Co-treatment of IgE/anti-IgE-activated mast cells with diclofenac (10 µM) during the period of anti-IgE stimulation abolished completely production of both Th2 cytokine stimulatory activity (Fig. 1) and PGD2 (Fig. 2a). While unactivated mast cells produced only low levels of PGD2 (< 1 ng/106 cells/ml), high levels of PGD2 (48 ± 8 ng/106 cells/ml) were detected in the supernatants from activated mast cells after 1-h challenge, which was reduced to < 1 ng/106 cells/ml by co-treatment with diclofenac (10 µM). In contrast, diclofenac did not inhibit the release of histamine (Fig. 2b) and enhanced release of LTC4 (Fig. 2c) from mast cells in response to treatment with IgE/anti-IgE. Because the mast cell supernatants were diluted 1 : 2 prior to the treatment of Th2 cells, the Th2 cells were exposed to concentrations of PGD2 in the range of 20–30 ng/ml. Addition of 100 nM PGD2 (equivalent to ∼35 ng/ml) to supernatants from activated mast cells co-treated with diclofenac restored their ability to promote the production of cytokines by Th2 cells (Fig. 1).
Fig. 2.
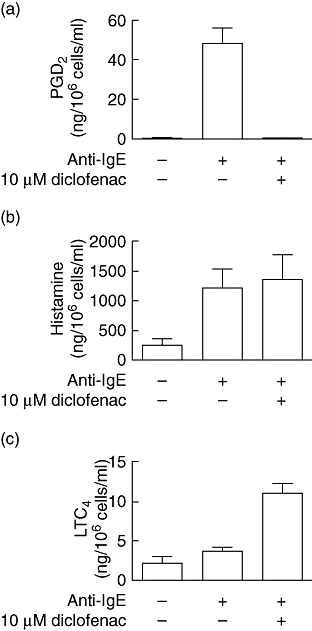
Production of prostaglandin D2 (PGD2), histamine and leukotriene C4 (LTC4) by mast cells. Mast cells were preincubated with immunoglobulin E (IgE) followed by treatment with culture medium or anti-IgE antibody in the presence or absence of diclofenac, as described in Materials and methods. One h after the treatments the supernatants were collected and assayed for PGD2 (a), histamine (b) and LTC4 (c) by enzyme immunoassay. Results are expressed as the mean ± standard error of the mean (n = 3–4). P < 0·0001 for (a) or P < 0·05 for (b,c) by analysis of variance. P > 0·05 by Newman–Keuls test for (a) no additive versus anti-IgE + 10 µM diclofenac, n = 4; (b) anti-IgE versus anti-IgE + 10 µM diclofenac, n = 3; (c) no additive versus anti-IgE, n = 3.
The Th2 cytokine production in response to activated mast cell supernatants is mediated by CRTH2
The IL-4, IL-5 and IL-13 production induced by activated mast cell supernatants was inhibited completely by the dual CRTH2/thromboxane A2 receptor (TP) antagonist ramatroban (1 µM) and selective CRTH2 antagonist TM30089 (1 µM), while the selective TP antagonist SQ29548 (1 µM) was without effect (Fig. 3).
Fig. 3.
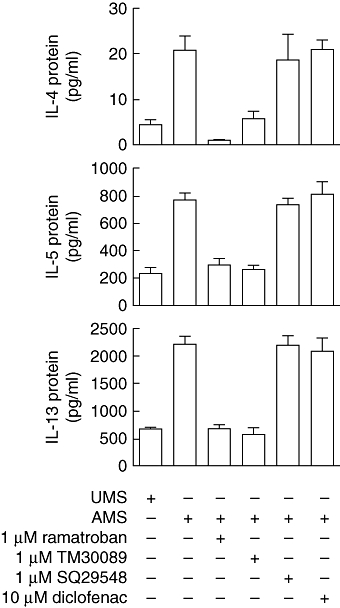
Effect of ramatroban, TM30089, SQ29548 and diclofenac on T helper type 2 (Th2) cytokine production in response to activated mast cell supernatants. Th2 cells were treated with mast cell supernatants (1:2 dilution) in the presence or absence ramatroban, TM30089, SQ29548 or diclofenac as indicated for 5 h. The concentrations of interleukin (IL-4), IL-5 and IL-13 in the incubation media were measured by enzyme immunoassay. Results are expressed as the mean ± standard error of the mean (n = 2–8). P < 0·001 for IL-4 or P < 0·0001 for IL-5 and IL-13 by analysis of variance. P > 0·05 by Newman–Keuls test for unactivated mast cells (UMS) versus mast cells activated with IgE/anti-IgE (AMS) + 1 µM ramatroban/AMS + 1 µM TM30089, AMS + 1 µM ramatroban versus AMS + 1 µM TM30089, AMS versus AMS + 1 µM SQ29548/AMS + 10 µM diclofenac, AMS + 1 µM SQ29548 versus AMS + 10 µM diclofenac, n = 2–8.
To rule out the possibility of a direct effect of diclofenac on Th2 cytokine production, we also stimulated Th2 cells with activated mast cell supernatants in the presence of diclofenac (Fig. 3). In this situation, diclofenac did not affect significantly the response of Th2 cells to the supernatants.
Regulation of Th2 cytokine production by activated mast cell supernatant is at gene transcriptional level
To elucidate further the mechanism of Th2 cytokine production in response to the mast cell supernatants, the levels of mRNA encoding IL-4, IL-5 and IL-13 in Th2 cells were measured after different treatments for 3 h (Fig. 4). Addition of activated mast cell supernatants to Th2 cells up-regulated mRNA for the cytokines to levels similar to those observed after treatment with 100 nM PGD2. The supernatants from unactivated mast cells or from diclofenac-treated activated mast cells did not increase the levels of cytokine mRNA significantly. The elevation of Th2 cytokine mRNA levels induced by activated mast cell supernatants was inhibited almost completely by ramatroban but not by SQ29548.
Fig. 4.
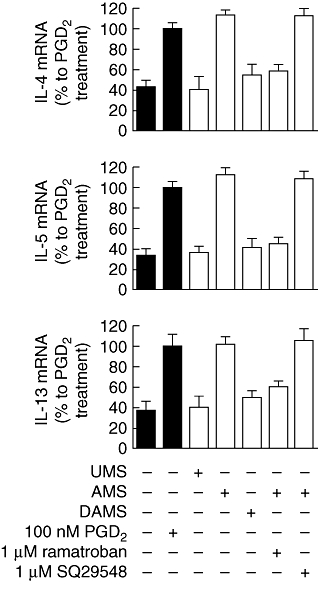
Effect of mast cell supernatants and selective antagonists on cytokine mRNA levels in T helper type 2 (Th2) cells. Th2 cells were incubated with X-VIVO 15 medium alone or medium containing prostaglandin D2 (PGD2) (black bars) or mast cell supernatants (1:2 dilution) (open bars) in the presence or absence of ramatroban or SQ29548 as indicated for 3 h. The total RNA from the cell pellets was extracted. Semi-quantitative reverse transcription–polymerase chain reaction was conducted to measure the change on gene expression of interleukin (IL)-4, IL-5 and IL-13. Results are presented mean ± standard error of the mean of % mean response induced by PGD2 alone (n = 6). P < 0·0001 by analysis of variance. P > 0·05 by Newman–Keuls test for no additive versus unactivated mast cells (UMS)/diclofenac-treated mast cells activated with IgE/anti-IgE (DAMS)/mast cells activated with IgE/anti-IgE (AMS) + 1 µM ramatroban, UMS versus DAMS/AMS + 1 µM ramatroban, DAMS versus AMS + 1 µM ramatroban, 100 nM PGD2versus AMS/AMS + 1 µM SQ29548, AMS versus AMS + 1 µM SQ29548, n = 6.
Discussion
The IgE-dependent interaction of mast cells and Th2 cells leading to the elaboration of cytokines such as IL-4, IL-5 and IL-13 is postulated to play a central role in asthma and related allergic disorders. Treatment with anti-IgE antibody omalizumab reduces both the early- and late-phase airway response to bronchial challenge with allergen in allergic subjects [19] and reduces airway inflammation (including Th2 cell accumulation) in chronic asthma [31]. While the effects of anti-IgE may, in part, be mediated by inhibition of IgE-facilitated antigen presentation [32], there is increasing evidence that activated mast cells may also contribute to Th2 cell recruitment and activation within allergic tissues [17,33]. Supernatants collected from activated mast cells promoted the migration of Th2 cells in vitro and the factor(s) responsible for this activity is most likely to be PGD2 or a closely related arachidonic acid metabolite that activates Th2 cells through high-affinity interaction with CRTH2 [26], which is expressed abundantly on the surface of Th2 cells [27,34]. In addition to promoting migration of Th2 cells, PGD2 is also known to stimulate production of IL-4, IL-5 and IL-13 by these cells [29]. However, the capacity of supernatants from activated mast cells to induce Th2 cytokine production has not been tested directly. As mast cells produce high levels of PGD2in vitro[35] and in vivo upon challenge with allergen [36–38], it is of interest to determine the contribution of mast cell-derived PGD2 to the production of cytokines by Th2 cells. Supernatants collected from human mast cells after activation with IgE/anti-IgE contained high levels of histamine and PGD2, but LTC4, IL-4, IL-5 and IL-13 were detected only at low levels. This was expected, as although activated mast cells can be a rich source of both IL-4 [39] and IL-13 [40], release of these cytokines is delayed compared with performed mediators and rapidly synthesized eicosanoids [41,42], and are therefore not likely to be present in significant quantities by 1 h after stimulation in absence of priming with stem cell factor [43]. However, addition of these supernatants to Th2 cells stimulated increased production of IL-4, IL-5 and IL-13, while supernatants from unactivated mast cells were without effect. It is generally thought that activation of the T cell receptor or co-stimulation, as occurs during antigen presentation, is required to activate T cells [44]. However, these data indicate that a soluble factor produced from activated mast cells is sufficient to drive production of cytokines by Th2 cells. Production of this activity was inhibited by diclofenac, suggesting that a cyclo-oxygenase product of arachidonic metabolism was responsible for this activity. This cyclo-oxygenase product is likely to be PGD2 based on the following observations:
The effect of the mast cell supernatants was mimicked by PGD2.
PGD2 was detected in the mast cell supernatants at concentrations sufficient to stimulate cytokine production by Th2 cells.
Diclofenac abolished both the production of Th2 cytokine stimulatory activity and PGD2 without affecting the release of histamine.
Addition of exogenous PGD2 to supernatants from diclofenac-treated mast cells restored their ability to stimulate Th2 cytokine production.
Although PGD2 was detected in the mast cell supernatants at sufficient concentrations to stimulate Th2 cytokine production, it is possible that biologically active metabolites of PGD2 may also make a contribution in vivo. In particular, both Δ12PGD2 and Δ12PGJ2 are formed rapidly in biological fluids such as plasma [45] and are potent CRTH2 agonists [46,47].
The IgE-mediated activation of mast cells orchestrates an inflammatory cascade through secretion of variety of biologically active mediators. These mediators may be categorized into three groups: preformed secretory granule-associated mediators, lipid-derived mediators and cytokines. The well-known mediators in the first group include histamine, which we measured and showed was not inhibited by diclofenac, so although it has been reported that histamine is able to enhance the production of Th2 cytokines [48,49], this mediator is unlikely to play a major role in this response. The most important second-group mediators are the cyclo-oxygenase and lipoxygenase metabolites of arachidonic acid. We also measured the levels of LTC4, a downstream product of lipoxygenase, in the mast cell supernatants. Treatment of mast cells with diclofenac did not suppress but rather enhanced LTC4 secretion from activated mast cells. This result suggested that blockade of the cyclo-oxygenase pathway diverts arachidonic acid to lipoxygenase products. Because these diclofenac-treated mast cells lack Th2 cytokine stimulatory activity, this effectively rules out leukotrienes as stimulators of cytokine production by Th2 cells. Therefore, the high correlation between inhibition of PGD2 secretion from mast cells and cytokine production from Th2 cells by diclofenac suggested strongly that PGD2 is the dominant mast cell mediator driving cytokine production by Th2 cells.
The effect of the activated mast cell supernatant on the production of Th2 cytokines was inhibited by ramatroban, a dual CRTH2/TP antagonist. The selective TP antagonist SQ29548 was without effect, implicating CRTH2 in mediating Th2 cytokine production in response to mast cell supernatants. This conclusion is supported by findings that the selective CRTH2 antagonist TM300089 also inhibited Th2 cytokine in response to mast cell supernatants. These data extend our earlier observation that mast cell supernatants promote chemotaxis of Th2 cells through a CRTH2-dependent mechanism, and suggest that CRTH2 may play a central role in both the recruitment of Th2 cells and their subsequent activation to elaborate proinflammatory cytokines. This mechanism may underlie the observed ability of selective CRTH2 antagonists to inhibit allergen-induced airway inflammation in the mouse [50] and the guinea pig [51]. Furthermore, these effects are relevant to the effects observed in mice genetically deficient in CRTH2. In a mouse model of skin inflammation, genetic ablation of CRTH2 was associated with diminished dermal infiltration of various leucocyte populations, including lymphocytes and eosinophils, reduced tissue swelling and a reduction in the levels of serum IgE [52]. It is plausible that the reduction in IgE observed in CRTH2−/− mice is related to the effects on Th2 cytokine production. Production of IgE was also reduced in CRTH2-deficient mice exposed to Japanese cedar pollen intranasally, an effect associated with reduced inflammation of the nasal mucosa and signs of rhinitis [53]. In Japanese cedar pollen-induced dermatitis, skin inflammation is dependent upon both mast cell activation and the presence of CRTH2 [54]. Taken together, these studies support the view that mast cell-derived PGD2 plays a critical role in the development of allergic responses via activation of CRTH2.
In conclusion, activated mast cells promote the production of cytokines by Th2 cells. This effect is mediated by the production of PGD2 from mast cells which stimulates Th2 cells by an action on the cell surface receptor CRTH2. These findings, combined with previous studies, suggest that PGD2 is the dominant mast cell mediator that activates Th2 cells. In addition to promoting the recruitment of Th2 cells during the initiation of an allergic response, activation of CRTH2 may contribute to Th2 cytokine production at sites of allergic inflammation.
References
- 1.Robinson DS, Hamid Q, Ying S, et al. Predominant Th2-like bronchoalveolar T-lymphocyte population in atopic asthma. N Engl J Med. 1992;326:298–304. doi: 10.1056/NEJM199201303260504. [DOI] [PubMed] [Google Scholar]
- 2.Bentley AM, Meng Q, Robinson DS, Hamid Q, Kay AB, Durham SR. Increases in activated T lymphocytes, eosinophils, and cytokine mRNA expression for interleukin-5 and granulocyte/macrophage colony-stimulating factor in bronchial biopsies after allergen inhalation challenge in atopic asthmatics. Am J Respir Cell Mol Biol. 1993;8:35–42. doi: 10.1165/ajrcmb/8.1.35. [DOI] [PubMed] [Google Scholar]
- 3.Robinson DS. Th-2 cytokines in allergic disease. Br Med Bull. 2000;56:956–68. doi: 10.1258/0007142001903625. [DOI] [PubMed] [Google Scholar]
- 4.Larche M, Robinson DS, Kay AB. The role of T lymphocytes in the pathogenesis of asthma. J Allergy Clin Immunol. 2003;111:450–63. doi: 10.1067/mai.2003.169. [DOI] [PubMed] [Google Scholar]
- 5.Medoff BD, Thomas SY, Luster AD. T cell trafficking in allergic asthma: the ins and outs. Annu Rev Immunol. 2008;26:205–32. doi: 10.1146/annurev.immunol.26.021607.090312. [DOI] [PubMed] [Google Scholar]
- 6.Campbell HD, Tucker WQ, Hort Y, et al. Molecular cloning, nucleotide sequence, and expression of the gene encoding human eosinophil differentiation factor (interleukin 5) Proc Natl Acad Sci USA. 1987;84:6629–33. doi: 10.1073/pnas.84.19.6629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lopez AF, Sanderson CJ, Gamble JR, Campbell HD, Young IG, Vadas MA. Recombinant human interleukin 5 is a selective activator of human eosinophil function. J Exp Med. 1988;167:219–24. doi: 10.1084/jem.167.1.219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Leckie MJ, ten Brinke A, Khan J, et al. Effects of an interleukin-5 blocking monoclonal antibody on eosinophils, airway hyper-responsiveness, and the late asthmatic response. Lancet. 2000;356:2144–8. doi: 10.1016/s0140-6736(00)03496-6. [DOI] [PubMed] [Google Scholar]
- 9.Menzies-Gow A, Flood-Page P, Sehmi R, et al. Anti-IL-5 (mepolizumab) therapy induces bone marrow eosinophil maturational arrest and decreases eosinophil progenitors in the bronchial mucosa of atopic asthmatics. J Allergy Clin Immunol. 2003;111:714–9. doi: 10.1067/mai.2003.1382. [DOI] [PubMed] [Google Scholar]
- 10.Lebman DA, Coffman RL. Interleukin 4 causes isotype switching to IgE in T cell-stimulated clonal B cell cultures. J Exp Med. 1988;168:853–62. doi: 10.1084/jem.168.3.853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Punnonen J, Aversa G, Cocks BG, et al. Interleukin 13 induces interleukin 4-independent IgG4 and IgE synthesis and CD23 expression by human B cells. Proc Natl Acad Sci USA. 1993;90:3730–4. doi: 10.1073/pnas.90.8.3730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kuperman DA, Huang X, Koth LL, et al. Direct effects of interleukin-13 on epithelial cells cause airway hyperreactivity and mucus overproduction in asthma. Nat Med. 2002;8:885–9. doi: 10.1038/nm734. [DOI] [PubMed] [Google Scholar]
- 13.Lee GR, Flavell RA. Transgenic mice which overproduce Th2 cytokines develop spontaneous atopic dermatitis and asthma. Int Immunol. 2004;16:1155–60. doi: 10.1093/intimm/dxh117. [DOI] [PubMed] [Google Scholar]
- 14.Wenzel S, Wilbraham D, Fuller R, Getz EB, Longphre M. Effect of an interleukin-4 variant on late phase asthmatic response to allergen challenge in asthmatic patients: results of two phase 2a studies. Lancet. 2007;370:1422–31. doi: 10.1016/S0140-6736(07)61600-6. [DOI] [PubMed] [Google Scholar]
- 15.Brightling CE, Bradding P, Pavord ID, Wardlaw AJ. New insights into the role of the mast cell in asthma. Clin Exp Allergy. 2003;33:550–6. doi: 10.1046/j.1365-2222.2003.01636.x. [DOI] [PubMed] [Google Scholar]
- 16.Boyce JA. The role of mast cells in asthma. Prostaglandins Leukot Essent Fatty Acids. 2003;69:195–205. doi: 10.1016/s0952-3278(03)00081-4. [DOI] [PubMed] [Google Scholar]
- 17.Maezawa Y, Nakajima H, Seto Y, et al. IgE-dependent enhancement of Th2 cell-mediated allergic inflammation in the airways. Clin Exp Immunol. 2004;135:12–8. doi: 10.1111/j.1365-2249.2004.02337.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Solley GO, Gleich GJ, Jordon RE, Schroeter AL. The late phase of the immediate wheal and flare skin reaction. Its dependence upon IgE antibodies. J Clin Invest. 1976;58:408–20. doi: 10.1172/JCI108485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fahy JV, Fleming HE, Wong HH, et al. The effect of an anti-IgE monoclonal antibody on the early- and late-phase responses to allergen inhalation in asthmatic subjects. Am J Respir Crit Care Med. 1997;155:1828–34. doi: 10.1164/ajrccm.155.6.9196082. [DOI] [PubMed] [Google Scholar]
- 20.Yu M, Tsai M, Tam SY, Jones C, Zehnder J, Galli SJ. Mast cells can promote the development of multiple features of chronic asthma in mice. J Clin Invest. 2006;116:1633–41. doi: 10.1172/JCI25702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ott VL, Cambier JC, Kappler J, Marrack P, Swanson BJ. Mast cell-dependent migration of effector CD8+ T cells through production of leukotriene B4. Nat Immunol. 2003;4:974–81. doi: 10.1038/ni971. [DOI] [PubMed] [Google Scholar]
- 22.Taube C, Miyahara N, Ott V, et al. The leukotriene B4 receptor (BLT1) is required for effector CD8+ T cell-mediated, mast cell-dependent airway hyperresponsiveness. J Immunol. 2006;176:3157–64. doi: 10.4049/jimmunol.176.5.3157. [DOI] [PubMed] [Google Scholar]
- 23.Gonzalo JA, Qiu Y, Lora JM, et al. Coordinated involvement of mast cells and T cells in allergic mucosal inflammation: critical role of the CC chemokine ligand 1:CCR8 axis. J Immunol. 2007;179:1740–50. doi: 10.4049/jimmunol.179.3.1740. [DOI] [PubMed] [Google Scholar]
- 24.Nakae S, Ho LH, Yu M, et al. Mast cell-derived TNF contributes to airway hyperreactivity, inflammation, and TH2 cytokine production in an asthma model in mice. J Allergy Clin Immunol. 2007;120:48–55. doi: 10.1016/j.jaci.2007.02.046. [DOI] [PubMed] [Google Scholar]
- 25.Kim N, Luster AD. Regulation of immune cells by eicosanoid receptors. Sci World J. 2007;7:1307–28. doi: 10.1100/tsw.2007.181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gyles SL, Xue L, Townsend ER, Wettey F, Pettipher R. A dominant role for chemoattractant receptor-homologous molecule expressed on T helper type 2 (Th2) cells (CRTH2) in mediating chemotaxis of CRTH2+ CD4+ Th2 lymphocytes in response to mast cell supernatants. Immunology. 2006;119:362–8. doi: 10.1111/j.1365-2567.2006.02440.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hirai H, Tanaka K, Yoshie O, et al. Prostaglandin D2 selectively induces chemotaxis in T helper type 2 cells, eosinophils, and basophils via seven-transmembrane receptor CRTH2. J Exp Med. 2001;193:255–61. doi: 10.1084/jem.193.2.255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tanaka K, Hirai H, Takano S, Nakamura M, Nagata K. Effects of prostaglandin D2 on helper T cell functions. Biochem Biophys Res Commun. 2004;316:1009–14. doi: 10.1016/j.bbrc.2004.02.151. [DOI] [PubMed] [Google Scholar]
- 29.Xue L, Gyles SL, Wettey FR, et al. Prostaglandin D2 causes preferential induction of proinflammatory Th2 cytokine production through an action on chemoattractant receptor-like molecule expressed on Th2 cells. J Immunol. 2005;175:6531–6. doi: 10.4049/jimmunol.175.10.6531. [DOI] [PubMed] [Google Scholar]
- 30.Craig SS, Schwartz LB. Tryptase and chymase, markers of distinct types of human mast cells. Immunol Res. 1989;8:130–48. doi: 10.1007/BF02919075. [DOI] [PubMed] [Google Scholar]
- 31.Djukanovic R, Wilson SJ, Kraft M, et al. Effects of treatment with anti-immunoglobulin E antibody omalizumab on airway inflammation in allergic asthma. Am J Respir Crit Care Med. 2004;170:583–93. doi: 10.1164/rccm.200312-1651OC. [DOI] [PubMed] [Google Scholar]
- 32.van Neerven RJ, Knol EF, Ejrnaes A, Wurtzen PA. IgE-mediated allergen presentation and blocking antibodies: regulation of T-cell activation in allergy. Int Arch Allergy Immunol. 2006;141:119–29. doi: 10.1159/000094714. [DOI] [PubMed] [Google Scholar]
- 33.Maezawa Y, Nakajima H, Kumano K, Kubo S, Karasuyama H, Iwamoto I. Role of IgE in Th2 cell-mediated allergic airway inflammation. Int Arch Allergy Immunol. 2003;131(Suppl.)(1):2–6. doi: 10.1159/000070473. [DOI] [PubMed] [Google Scholar]
- 34.Nagata K, Tanaka K, Ogawa K, et al. Selective expression of a novel surface molecule by human Th2 cells in vivo. J Immunol. 1999;162:1278–86. [PubMed] [Google Scholar]
- 35.Lewis RA, Soter NA, Diamond P, Kubo S, Karasuyama H, Iwamoto I. Prostaglandin D2 generation after activation of rat and human mast cells with anti-IgE. J Immunol. 1982;129:1627–31. [PubMed] [Google Scholar]
- 36.Naclerio RM, Meier HL, Kagey-Sobotka A, et al. Mediator release after nasal airway challenge with allergen. Am Rev Respir Dis. 1983;128:597–602. doi: 10.1164/arrd.1983.128.4.597. [DOI] [PubMed] [Google Scholar]
- 37.Murray JJ, Tonnel AB, Brash AR, et al. Release of prostaglandin D2 into human airways during acute antigen challenge. N Engl J Med. 1986;315:800–4. doi: 10.1056/NEJM198609253151304. [DOI] [PubMed] [Google Scholar]
- 38.Charlesworth EN, Kagey-Sobotka A, Schleimer RP, Norman PS, Lichtenstein LM. Prednisone inhibits the appearance of inflammatory mediators and the influx of eosinophils and basophils associated with the cutaneous late-phase response to allergen. J Immunol. 1991;146:671–6. [PubMed] [Google Scholar]
- 39.Bradding P, Feather IH, Howarth PH, et al. Interleukin 4 is localized to and released by human mast cells. J Exp Med. 1992;176:1381–6. doi: 10.1084/jem.176.5.1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Burd PR, Thompson WC, Max EE, Mills FC. Activated mast cells produce interleukin 13. J Exp Med. 1995;181:1373–80. doi: 10.1084/jem.181.4.1373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Toru H, Pawankar R, Ra C, Yata J, Nakahata T. Human mast cells produce IL-13 by high-affinity IgE receptor cross-linking: enhanced IL-13 production by IL-4-primed human mast cells. J Allergy Clin Immunol. 1998;102:491–502. doi: 10.1016/s0091-6749(98)70140-x. [DOI] [PubMed] [Google Scholar]
- 42.Pawankar R, Yamagishi S, Yagi T. Revisiting the roles of mast cells in allergic rhinitis and its relation to local IgE synthesis. Am J Rhinol. 2000;14:309–17. doi: 10.2500/105065800781329582. [DOI] [PubMed] [Google Scholar]
- 43.Kanbe N, Kurosawa M, Yamashita T, Kurimoto F, Yanagihara Y, Miyachi Y. Cord-blood-derived human cultured mast cells produce interleukin 13 in the presence of stem cell factor. Int Arch Allergy Immunol. 1999;119:138–42. doi: 10.1159/000024189. [DOI] [PubMed] [Google Scholar]
- 44.Song J, Lei FT, Xiong X, Haque R. Intracellular signals of T cell costimulation. Cell Mol Immunol. 2008;5:239–47. doi: 10.1038/cmi.2008.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Schuligoi R, Schmidt R, Geisslinger G, Kollroser M, Peskar BA, Heinemann A. PGD2 metabolism in plasma: kinetics and relationship with bioactivity on DP1 and CRTH2 receptors. Biochem Pharmacol. 2007;74:107–17. doi: 10.1016/j.bcp.2007.03.023. [DOI] [PubMed] [Google Scholar]
- 46.Heinemann A, Schuligoi R, Sabroe I, Hartnell A, Peskar BA. Delta 12-prostaglandin J2, a plasma metabolite of prostaglandin D2, causes eosinophil mobilization from the bone marrow and primes eosinophils for chemotaxis. J Immunol. 2003;170:4752–8. doi: 10.4049/jimmunol.170.9.4752. [DOI] [PubMed] [Google Scholar]
- 47.Gazi L, Gyles S, Rose J, et al. Delta12-prostaglandin D2 is a potent and selective CRTH2 receptor agonist and causes activation of human eosinophils and Th2 lymphocytes. Prostaglandins Other Lipid Mediat. 2005;75:153–67. doi: 10.1016/j.prostaglandins.2004.11.003. [DOI] [PubMed] [Google Scholar]
- 48.Schmidt J, Fleissner S, Heimann-Weitschat I, Lindstaedt R, Szelenyi I. Histamine increases anti-CD3 induced IL-5 production of TH2-type T cells via histamine H2-receptors. Agents Actions. 1994;42:81–5. doi: 10.1007/BF01983469. [DOI] [PubMed] [Google Scholar]
- 49.Elliott KA, Osna NA, Scofield MA, Khan MM. Regulation of IL-13 production by histamine in cloned murine T helper type 2 cells. Int Immunopharmacol. 2001;1:1923–37. doi: 10.1016/s1567-5769(01)00117-5. [DOI] [PubMed] [Google Scholar]
- 50.Uller L, Mathiesen JM, Alenmyr L, et al. Antagonism of the prostaglandin D2 receptor CRTH2 attenuates asthma pathology in mouse eosinophilic airway inflammation. Respir Res. 2007;8:16. doi: 10.1186/1465-9921-8-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pettipher R. The roles of the prostaglandin D2 receptors DP1 and CRTH2 in promoting allergic responses. Br J Pharmacol. 2008;153(Suppl.)(1):S191–9. doi: 10.1038/sj.bjp.0707488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Satoh T, Moroi R, Aritake K, et al. Prostaglandin D2 plays an essential role in chronic allergic inflammation of the skin via CRTH2 receptor. J Immunol. 2006;177:2621–29. doi: 10.4049/jimmunol.177.4.2621. [DOI] [PubMed] [Google Scholar]
- 53.Nomiya R, Okano M, Fujiwara T, et al. CRTH2 plays an essential role in the pathophysiology of Cry j 1-induced pollinosis in mice. J Immunol. 2008;180:5680–8. doi: 10.4049/jimmunol.180.8.5680. [DOI] [PubMed] [Google Scholar]
- 54.Oiwa M, Satoh T, Watanabe M, et al. CRTH2-dependent, STAT6-independent induction of cedar pollen dermatitis. Clin Exp Allergy. 2008;38:1357–66. doi: 10.1111/j.1365-2222.2008.03007.x. [DOI] [PubMed] [Google Scholar]