

Abstract
Allele variants in the L-carnitine (LCAR) transporters OCTN1 (SLC22A4, 1672 C → T) and OCTN2 (SLC22A5, -207 G → C) have been implicated in susceptibility to Crohn's disease (CD). LCAR is consumed in the diet and transported actively from the intestinal lumen via the organic cation transporter OCTN2. While recognized mainly for its role in fatty acid metabolism, several lines of evidence suggest that LCAR may also display immunosuppressive properties. This study sought to investigate the immunomodulatory capacity of LCAR on antigen-presenting cell (APC) and CD4+ T cell function by examining cytokine production and the expression of activation markers in LCAR-supplemented and deficient cell culture systems. The therapeutic efficacy of its systemic administration was then evaluated during the establishment of colonic inflammation in vivo. LCAR treatment significantly inhibited both APC and CD4+ T cell function, as assessed by the expression of classical activation markers, proliferation and cytokine production. Carnitine deficiency resulted in the hyperactivation of CD4+ T cells and enhanced cytokine production. In vivo, protection from trinitrobenzene sulphonic acid colitis was observed in LCAR-treated mice and was attributed to the abrogation of both innate [interleukin (IL)-1β and IL-6 production] and adaptive (T cell proliferation in draining lymph nodes) immune responses. LCAR therapy may therefore represent a novel alternative therapeutic strategy and highlights the role of diet in CD.
Keywords: antigen-presenting cells, carnitine, Crohn's disease, T lymphocytes, trinitrobenzene sulphonic acid colitis
Introduction
Crohn's disease (CD) is a chronic, relapsing inflammatory disease of the gastrointestinal tract thought to result from the aberrant recognition of enteric microbial flora, leading to inappropriate immune responses and chronic intestinal inflammation [1–3]. Although exposure to triggers such as cigarette smoke [4], non-steroidal anti-inflammatory drugs [5] and stress [6] appear to play a role in the relapsing/remitting phases of inflammatory bowel disease, diet is also suspected to influence the behaviour of the disease, either by influencing the microbial flora or by directly modulating the mucosal immune response of the host [7,8].
L-carnitine (LCAR) is consumed in the diet and is absorbed mainly from the lumen of the digestive tract via an active mechanism requiring the organic cation transporter OCTN2 [9,10]. It plays a key role in cell metabolism by regulating the mitochondrial transport of long-chain free fatty acids (LCFAs) and the generation of adenosine triphosphate (ATP) by β-oxidation [11–13]. The role of LCAR in the gastrointestinal tract has recently become a topic of interest, as mutations in the LCAR transporter genes, OCTN1 (SLC22A4, 1672 C → T) and OCTN2 (SLC22A5, -207 G → C), resulted in functional impairments in LCAR uptake and an increased risk of developing CD [14,15]. While these observations have not been replicated worldwide [16], several functional studies have given credence to the hypothesis that LCAR participates in intestinal homeostasis. For instance, OCTN2−/− mice develop colonic atrophy and inflammation spontaneously, a phenotype attributed to the abnormal structure and morphology of intestinal epithelial cells [17]. LCAR has also been shown to be a rate-limiting factor for the maintenance of physiological butyrate β-oxidation in colonocytes, and a protective effect of intrarectal administration of carnitine-loaded liposomes was observed in experimental colitis [18]. However, in addition to its local role in colonocyte function, systemic LCAR may also display immunosuppressive properties, as illustrated by its ability to suppress lipopolysaccharide (LPS)-induced cytokine production and improve murine survival rates during cachexia and septic shock [19]. LCAR has also been shown to reduce CD4+ and CD8+ T cell numbers and interleukin (IL)-2 production in splenocytes isolated from LCAR-treated mice [20] and reduce tumour necrosis factor (TNF)-α production in Staphylococcus aureus-stimulated human polymorphonuclear cells [21]. Interestingly, previous reports have demonstrated that high doses of LCAR can activate glucocorticoid receptor alpha (GR-α) and may share some biological and therapeutic effects with glucocoticoids [22].
While the above data suggest an anti-inflammatory role for LCAR in immune function, other studies have been reported with contradictory results, in part because of the complexity of the immune response and variation between experimental conditions [23–25]. In this study, we present evidence to clarify and directly examine the impact of LCAR on antigen-presenting cell (APC) and T cell function with respect to the expression of key activation markers and cytokines. Our in vitro observations are then validated by investigating the therapeutic efficacy of systemic LCAR supplementation in murine trinitrobenzene sulphonic acid (TNBS) colitis, a model exhibiting many of the same clinical and histological features as human CD.
Materials and methods
Reagents and antibodies
The TNBS and LCAR were purchased from Sigma Chemical Co. (St Louis, MO, USA). Biotinylated anti-CD11c (HL3), anti-CD11b-fluorescein isothiocyanate (FITC) (M1/70), anti-CD4-peridinin chlorophyll (PerCP) (L3T4) and anti-annexin-V-APC were obtained from BD Pharmingen (Mississauga, ON, USA). Anti-major histocompatibility complex class II (MHC II)-FITC (NIMR-4), anti-B220-phycoerythrin (PE) (RA3-6B2), anti-CD86-PE (GL1), anti-CD80-PE (16-10A1), anti-CD3-PE (145-2C11), anti-CD69-FITC (H1.2F3) and anti-CD25-APC (PC61) were obtained from eBioscience (San Diego, CA, USA).
Animals
Male Balb/c and Balb/cByJ mice, 6–8 weeks old, were obtained from Jackson Laboratories, maintained under conventional housing conditions and given free access to standard food and water. All mice were handled according to institutionally recommended animal care guidelines and all experiments were approved by the Animal Studies Ethics Committee of McGill University.
Cell culture conditions
Spleens were harvested from 6–8-week-old male Balb/c or carnitine-deficient mice. Cells were cultured at a concentration of 1 × 106 cells/ml in RPMI-1640 (Invitrogen Life Technologies, Carlsbad, CA, USA), supplemented with 10% fetal calf serum (FCS) (Hyclone, Logan, UT, USA), penicillin (100 U/ml), streptomycin (100 ug/ml), 10 mM Hepes and 50 µM 2-mercaptoethanol (Sigma Chemical Co.). Cells were treated with 0, 10, 100 or 300 mM LCAR. To assess APC function, total splenocytes or purified APCs were stimulated with 1 µg/ml Escherichia coli LPS (Sigma Chemical Co.) for 18 h. To assess T cell function, splenocytes were incubated in the presence of plate-bound hamster anti-mouse CD3 antibody (1 µg/ml) (eBioscience) for 72 h. Pure CD4+ T cells were stimulated with plate-bound anti-CD3 (1 µg/ml) and soluble anti-CD28 (2 µg/ml) for 72 h.
Flow cytometry
To assess the effect of LCAR on APC activation, splenocytes were stained with anti-MHC II-FITC and CD86-PE or anti-MHC II-FITC and anti-CD80-PE. Mean fluorescence intensity (MFI) was calculated as a measure of surface co-stimulatory molecule expression. Upon 18-h exposure of splenocytes to LCAR, toxicity was evaluated by staining with annexin-V-APC. T cell activation was assessed by staining splenocytes, activated in the presence of anti-CD3, with anti-CD4-PerCP, anti-CD3-PE, anti-CD69-FITC and anti-CD25-APC. All flow cytometric analysis was performed using FlowJo software (version 5.7.2).
Cell sorting
Spleens (for APCs) or mesenteric lymph nodes (mLNs) (for CD4+ T cells) were harvested from Balb/c mice. Splenocytes were stained with anti-CD11b-FITC, anti-B220-PE and anti-CD11c-APC. Dendritic cells (DCs) were selected as CD11c+ cells, macrophages as CD11c−CD11b+ and B cells as CD11c−B220+. Cells isolated from mLNs were stained with anti-CD4-PerCP and anti-CD3-PE (BD Pharmingen, San Diego, CA, USA), and double-positive cells were sorted and cultured. The cell suspensions were sorted by a BD FACSAria cell sorting system (BD Biosciences). Cell purity was > 99%.
Bromodeoxyuridine and [H3]-thymidine incorporation assays
Splenocytes or pure CD4+ T cells cultured in a 96-well microplate were incubated with bromodeoxyuridine (BrdU) for 6 h (Roche Applied Science, Laval, Qc, Canada). The labelled cells were fixed with ethanol and partially digested with nucleases to allow an anti-BrdU antibody [labelled with peroxidase (POD)] to access and bind to BrdU. POD catalysed the cleavage of ammonium 2,2′-azino-bis(3-ethylbenzothiazoline-6-sulphonate) (ABTS), producing a coloured reaction product. The absorbance of the samples (at 405 nm) was determined with a standard microplate reader and represents the number of actively dividing cells during the 6-h incubation period.
[H3]-thymidine incorporation was used to assess proliferation after the ex vivo culture of colon-draining sacral lymph nodes (sLNs). sLNs were isolated after reinduction of TNBS colitis and incubated in the presence of 0·3 mg/ml TNBS for 15 min at room temperature [26]. Cells were then washed extensively and cultured for 4 days in complete RPMI-1640. [H3]-thymidine (0·5 µCi/well) was added for the last 18 h of culture. The amount of [H3]-thymidine incorporated was measured by scintillation counting.
Induction of TNBS colitis
The TNBS (100 mg/kg) dissolved in 50% ethanol was introduced into the colon via a 3.5 F catheter, fitted to a 1 ml syringe, in isofluorane-anaesthetized mice. Control mice received intrarectal saline using the same technique. LCAR (100 or 150 mg/kg dissolved in saline) or vehicle (saline alone) was administered intraperitoneally once daily during the entire duration of colitis, with the first dose administered 30 min prior to induction of colitis. To assess T cell responses, colitis was reinduced 7 days after the first injection and the mice were killed on day 10.
Assessment of colonic damage
The macroscopic severity of colon damage was assessed according to the Wallace criteria, as described previously [27]. For histological assessment, 2 µM-thick sections were stained with haematoxylin and eosin [28] and histological changes were graded semi-quantitatively based on a set of previously established criteria [29]. The grading scale ranged from 0 to 13, and was calculated as the sum of scores for: expansion of submucosa (0–4), expansion of lamina propria (0–4), loss of goblet cells (0–4) and neutrophil infiltration (0–1). All macroscopic and microscopic scoring was performed in a blinded fashion.
Quantitative real-time polymerase chain reaction for inflammatory cytokines
Colonic RNA was extracted following the TRIzol protocol (Invitrogen, Burlington, ON, Canada). Total RNA was reverse-transcribed using the cDNA reverse transcription kit (Applied Biosystems, Foster City, CA, USA). Quantitative real-time polymerase chain reaction (PCR) was performed using an ABI Prism 7900HT Sequence Detection System (Applied Biosystems) (1 PCR cycle, 95°C, 10 min; 40 PCR cycles, 60°C, 1 min, 95°C, 15 s). cDNA was amplified in a 10 µl final reaction mix containing TaqMan Universal PCR Master Mix (Applied Biosystems) and corresponding TaqMan® Gene Expression Assays [Mm00446190_m1 (IL-6), Mm00434228_m1 (IL-1β), Hs99999901_s1 (Eukaryotic 18 s rRNA), Applied Biosystems]. Signals were analysed by the ABI Prism Sequence Detection System software version 2.2 (Applied Biosystems). The comparative Ct method for relative quantification was used, where all threshold cycles (Ct) are first normalized to the expression of 18 s rRNA. Here, the cytokine expression is represented as a fold-change relative to control mice.
Cytokine quantification/enzyme-linked immunosorbent assay
Whole blood was withdrawn immediately post-mortem and sera were frozen at −20°C until use. Serum (IL-6 and IL-1β) and culture supernatant cytokines [IL-1β, IL-6, interferon (IFN)-γ, IL-4 and IL-5] were quantified by Quantikine enzyme-linked immunosorbent assay kits (R&D Systems, Minneapolis, MN, USA).
Statistical analysis
All values are expressed as mean ± standard error of the mean. Changes in body weight were compared by Kruskal–Wallis anova. The macroscopic and histological scores between TNBS and TNBS plus LCAR groups and in vitro data were analysed by two-tailed Student's t-test for unpaired samples. Quantitive reverse transcription–PCR cytokine mRNA expression data were analysed with a Mann–Whitney U-test.
Results
The LCAR displays immunosuppressive properties
To assess the immunosuppressive actions of LCAR, total splenocytes were stimulated with LPS and cultured in the presence of LCAR in vitro. The seemingly high doses of LCAR were selected based on previously published data demonstrating pharmacological activity in the absence of toxicity at these doses [22]. LPS stimulation led to a significant increase in cell proliferation compared with unstimulated cultures (Fig. 1a), and a dose-dependent suppression of proliferation was induced by LCAR treatment of LPS-stimulated cells, reaching statistical significance at 100 mM LCAR (Fig. 1a). Next, the effect of LCAR was assessed on APC function by CD80 (B7-1) and CD86 (B7-2) expression (MFI) on MHC II positive cells, a marker of APCs. CD80 and CD86 are co-stimulatory molecules expressed on APCs that provide the necessary stimuli to prime T cells via CD28 and promote activation and T cell survival [30,31]. At 100 mM LCAR, a significant reduction in both CD80 and CD86 MFI was observed, signifying a reduction in the number of surface co-stimulatory molecules present per APC (Fig. 1b and c). Notably, no changes in proliferation or expression of co-stimulatory molecules were observed in unstimulated cultures treated with LCAR (Fig. 1a–c). The suppressive effect of LCAR on LPS-stimulated cells could not be attributed to the induction of cell death, as the percentages of live cells [DiOC6(3)+] (data not shown) and apoptotic cells (annexin V+) (Fig. 1d) were not altered by LCAR treatment, corroborating previously published data [22]. The percentage of apoptotic cells was significantly increased at a dose of 300 mM LCAR (data not shown) and this dose was therefore eliminated from further assessment of LCAR function. These data demonstrate that LCAR exerts immunosuppressive effects on APC function without displaying toxicity.
Fig. 1.
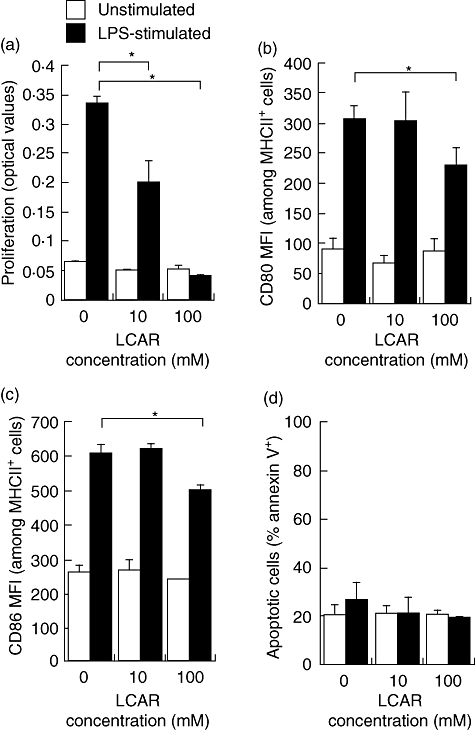
L-carnitine (LCAR) suppresses antigen-presenting cell (APC) function in vitro. Splenocytes were harvested from healthy Balb/c mice and stimulated with lipopolysaccharide (LPS) (1 µg/ml) for 18 h in the presence of LCAR (0, 10, or 100 mM). (a) Cell proliferation was assessed by bromodeoxyuridine (BrdU) incorporation. APC activation was assessed by flow cytometric analysis of CD80 (b) and CD86 (c) surface expression on individual major histocompatibility complex class II (MHC II+) cells, as represented by the mean fluorescence intensity (MFI). (d) Apoptosis was assessed by flow cytometric analysis of annexin-V staining. Data represent the mean ± standard error of the mean (n = 3). *P < 0·05.
The LCAR suppresses DC and macrophage function in vitro
Because MHC II is expressed on all professional APCs, including DCs, macrophages and B cells, the cell type affected by LCAR treatment could not be determined in the previous experiment. We therefore purified CD11c+ DCs, CD11b+ macrophages and B220+ B cells by cell sorting and stimulated them individually in the presence of LPS to assess their responsiveness to LCAR. CD86 expression was significantly reduced in DC and macrophage cultures, but not in B cell cultures (Fig. 2a–c). These data implicate DCs and macrophages specifically in LCAR's immunosuppressive action and indicate that they can be suppressed efficiently in the absence of T cells and other cell types normally present in the spleen. We also assessed cytokine production by splenocytes, pure DCs, pure macrophages and pure B cells. IL-6, IL-1β and TNF-α production were suppressed dose-dependently by LCAR in DC, macrophage and mixed splenocyte cultures (Fig. 2d–f), while B cell cultures were unaffected (data not shown). Therefore, LCAR can directly suppress DC and macrophage activation and cytokine production.
Fig. 2.
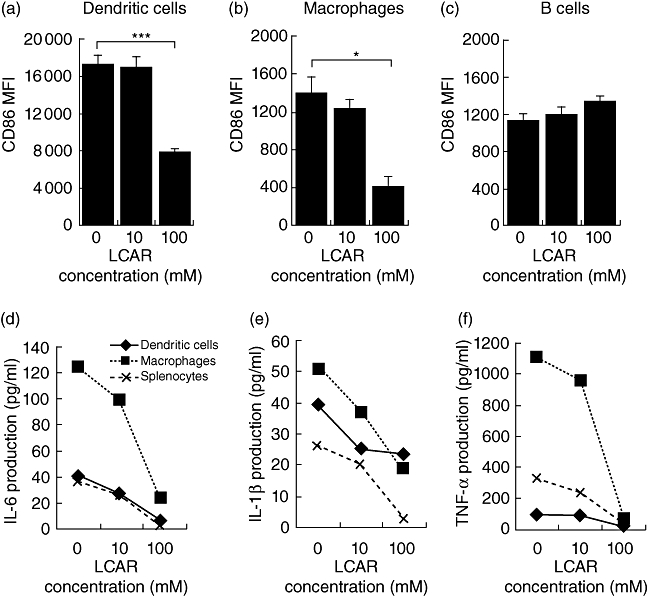
L-carnitine (LCAR) specifically suppresses dendritic cell (DC) and macrophage activation and cytokine production. Total splenocytes were sorted by flow cytometry to obtain pure DCs (CD11c+), macrophages (CD11c−CD11b+) or B cells (CD11c−B220+) and cultured in parallel with unsorted splenocytes. Cells were stimulated with lipopolysaccharide (LPS) (1 µg/ml) and cultured for 18 h in the presence of LCAR (0, 10 or 100 mM). (a–c) CD86 expression was assessed by flow cytometric analysis of mean fluorescence intensity (MFI) among purified DCs (a), macrophages (b) and B cells (c). (d) Interleukin (IL)-6, (e) IL-1β and (f) tumour necrosis factor (TNF)-α production were quantified in culture supernatants of sorted and unsorted splenocytes. Data represent the mean ± standard error of the mean (n = 3). *P < 0·05; ***P < 0·001.
The LCAR suppresses CD4+ T cell function in vitro
In addition to aberrant innate immune responses, CD involves inappropriate T cell responses to harmless antigens [32]. We therefore sought to examine the effect of LCAR on CD4+ T cell function. Splenocytes were stimulated with plate-bound anti-CD3 to activate T cells and cultured for 72 h in the presence of LCAR. LCAR significantly suppressed anti-CD3-induced CD4+ T cell activation, with a greater than 50% reduction in the number of double-positive (CD69+CD25+) cells (Fig. 3a).
Fig. 3.
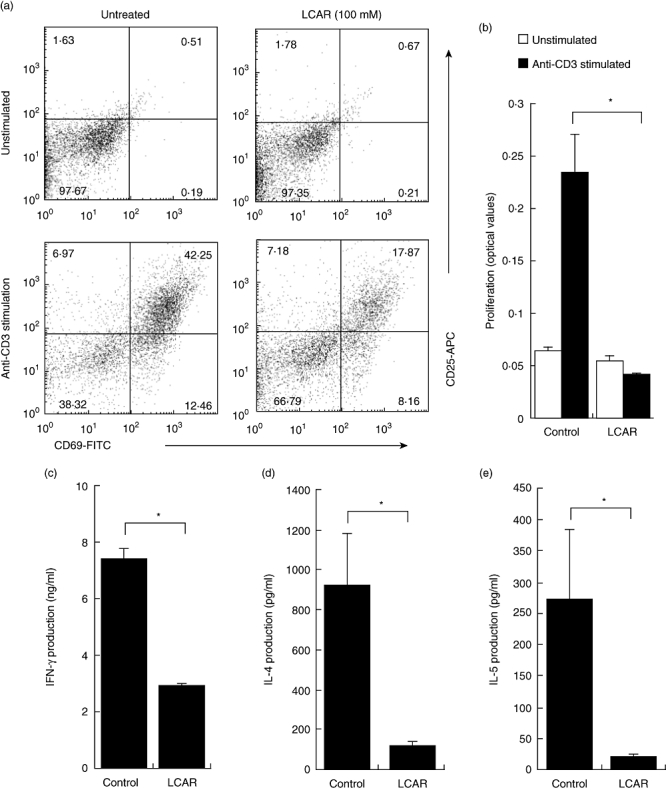
L-carnitine (LCAR) suppresses CD4+ T cell activation, proliferation and cytokine production. (a) Splenocytes were stimulated with plate-bound anti-CD3 (1 µg/ml) in the presence of LCAR (0 or 100 mM) for 72 h. CD4+ T cell activation was assessed by gating on CD3+CD4+ cells and determining the percentage of CD69+, CD25+ or double-positive cells after overnight culture. (b) Purified CD4+ T cells were cultured in the presence of LCAR (0 or 100 mM) and stimulated with plate-bound anti-CD3 for 72 h to assess proliferation by bromodeoxyuridine (BrdU) incorporation. (c–e) Purified CD4+ T cells were cultured in the presence of LCAR (0 or 100 mM) and stimulated with plate-bound anti-CD3 and soluble anti-CD28 for 72 h. (c) Interferon (IFN)-γ, (d) interleukin (IL)-4 and (e) IL-5 production were assessed by enzyme-linked immunosorbent assay. Data represent mean ± standard error of the mean (n = 3). *P < 0·05.
Given that splenocytes contain a mixture of cell types, assessments of T cell proliferation and cytokine production were performed on purified CD4+ T cells. This experiment also addressed whether LCAR could suppress T cell responses independently of the presence of APCs. mLNs were selected as the source of T cells as they contain a greater percentage of T cells than spleens, and represent mucosal immune responses more accurately. After 72 h of plate-bound anti-CD3 stimulation, LCAR completely abolished CD4+ T cell proliferation, as assessed by BrdU incorporation (Fig. 3b), while no effect was observed in unstimulated cultures. LCAR-mediated suppression of purified CD4+ T cell proliferation was also observed when T cells were stimulated with soluble anti-CD3 in the presence of soluble anti-CD28 or mitomycin C-treated APCs, as assessed by [H3]-thymidine incorporation (data not shown). In response to soluble anti-CD3 plus anti-CD28 stimulation, the production of the classical T helper type 1 (Th1) cytokine, IFN-γ, as well as two Th2-associated cytokines, IL-4 and IL-5, were significantly diminished by treatment with LCAR (P < 0·05) (Fig. 3c–e). At this time-point, IL-2 concentration was too low to quantify. Therefore, in vitro treatment with LCAR appears to exhibit immunosuppressive properties at the level of both APCs and CD4+ T cells.
Carnitine-deficient CD4+ T cells become hyperactivated upon stimulation
Because the addition of LCAR to APC and T cell cultures resulted in immunosuppression, we sought to ascertain whether a carnitine deficiency might affect the sensitivity of CD4+ T cells to stimulation. Balb/cByJ mice are SCAD (short-chain Acyl-CoA dehydrogenase)-deficient and display a defect in the conversion of short chain fatty acids such as butyrate into acetyl-CoA [33]. Butyrate therefore accumulates inside the mitochondria and is converted to butyrylcarnitine by carnitine acetyltransferase. During this conversion, carnitine stores are used up, resulting in a secondary carnitine deficiency [33].
In this study, splenocytes were obtained from unmanipulated Balb/c and carnitine-deficient mice and stimulated for 72 h in the presence of plate-bound anti-CD3. CD4+ T cells from carnitine-deficient mice displayed a hyperactivated phenotype characterized by the expression of CD69 and CD25 and a significant enhancement of IFN-γ production (Fig. 4a and b). CD4+ T cell hyperactivation and IFN-γ production were both reversed by supplementation with LCAR (100 mM) in the culture medium (Fig. 4a and b). We therefore conclude that carnitine supplementation can restore a normal immune response in otherwise hyperactivated carnitine-deficient CD4+ T cells.
Fig. 4.
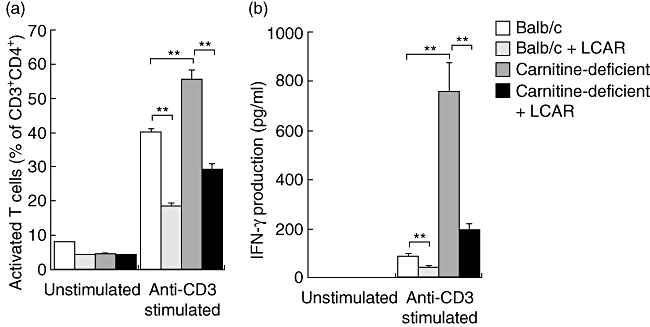
Hyperactivation of carnitine-deficient cells is reversed by treatment with L-carnitine (LCAR). Splenocytes were isolated from healthy Balb/c and carnitine-deficient mice and stimulated with anti-CD3 overnight in the presence or absence of LCAR (0 or 100 mM). (a) CD4+ T cell activation was assessed by gating on CD3+CD4+ cells and determining the percentage of CD69+CD25+ cells after overnight culture. (b) Interferon (IFN)-γ production in the supernatants of cultures was assessed by enzyme-linked immunosorbent assay. Data represent mean ± standard error of the mean (n = 3). *P < 0·05; **P < 0·01.
The LCAR therapy impairs the expression of intestinal proinflammatory cytokines and abrogates TNBS colitis
The TNBS colitis is driven by the interplay between innate and adaptive immune responses. Given LCAR's immunosuppressive properties on both arms of the immune system, we sought to investigate the therapeutic efficacy of systemic LCAR administration in the TNBS colitis model. Colitis was induced in Balb/c mice and LCAR (100 or 150 mg/kg) was administered intraperitoneally once daily. The intraperitoneal route was selected to minimize the trauma associated with daily intravenous injections or oral gavage. As feeding behaviour is reduced and highly variable between mice after the induction of TNBS colitis, LCAR supplementation of the food or water was also not a feasible option.
Within 1 day of intrarectal instillation of TNBS, severe wasting of body weight and diarrhoea were observed in both the LCAR-treated (high dose: 150 mg/kg; low dose 100 mg/kg) and untreated groups, while control mice maintained their original body weights (Fig. 5a). However, LCAR treatment resulted in significant improvements in the body weights of mice with colitis by day 2 for the high-dose group and by day 3 for the low-dose group (Fig. 5a).
Fig. 5.
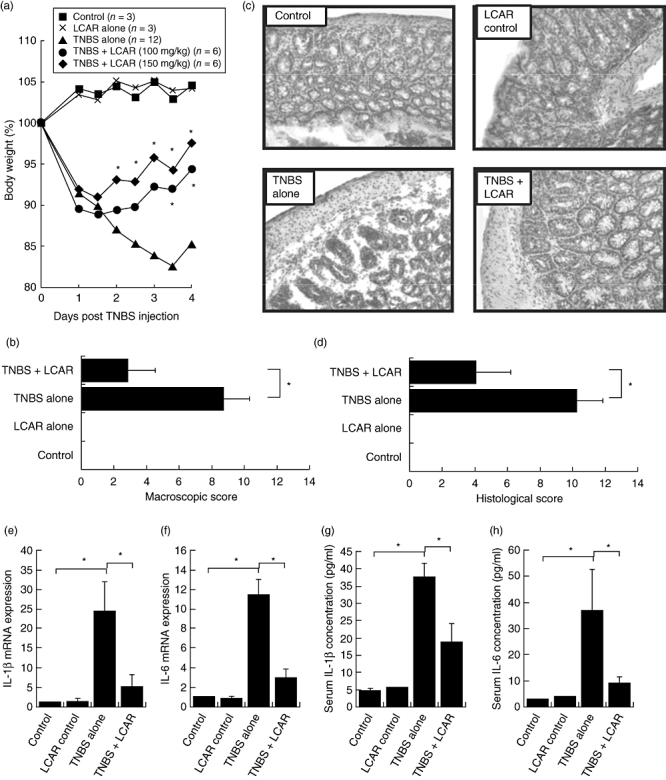
L-carnitine (LCAR) therapy is effective in treating trinitrobenzene sulphonic acid (TNBS) colitis. TNBS colitis was induced by intrarectal administration of 100 mg/kg TNBS dissolved in 50% ethanol and mice were treated every 24 h with LCAR (100 or 150 mg/kg), starting 30 min prior to the induction of colitis. (a) Body weight was recorded twice daily and is expressed as a percentage of body weight on day 0. Each data point represents the mean ± standard error of the mean (s.e.m.) of three control mice and six mice with TNBS colitis per group. *P < 0·05 compared with untreated TNBS. (b) The macroscopic score of colon inflammation was determined by the Wallace criteria. (c) Haematoxylin and eosin-stained sections of distal colon (original magnification, 400 ×). (d) The histological score of colon damage. (e) mRNA expression of interleukin (IL)-1β and (f) IL-6 was determined in whole colon homogenates by quantitative real-time polymerase chain reaction. Data are expressed as mean fold-change relative to control ± s.e.m. (g) Serum cytokine levels of IL-1β and (h) IL-6 were quantified by enzyme-linked immunosorbent assay. Data are expressed as mean ± s.e.m. (n = 3 for control groups, n = 6 for TNBS groups). *P < 0·05.
Because high-dose LCAR proved most effective in ameliorating body weight loss, an in-depth analysis of markers of inflammation was performed on colon tissues from these mice. Upon visual inspection, the macroscopic severity of colitis was rated by the Wallace criteria, where LCAR-treated mice displayed an approximately 70% reduction in inflammation (Fig. 5b). Control mice showed no macroscopic signs of inflammation (score = 0). Histological grading of frozen sections also showed no inflammatory infiltrates in non-colitic mice (score = 0). Importantly, the administration of LCAR in healthy mice did not result in any noticeable effects on body weight, the appearance and histology of the colon or any other criteria examined. In mice with TNBS colitis, the area of most severe inflammation was the distal half of the colon, where a loss of goblet cells, distortion of the crypts and infiltration of mononuclear cells were evident. Such histological changes were significantly reduced by treatment with LCAR (Fig. 5c and d). Therefore, LCAR was effective in suppressing the development of intestinal inflammation and associated body weight loss in mice with TNBS colitis.
The inflammatory cytokines IL-6 and IL-1β are instrumental in the initiation and maintenance of the inflammation characteristic of both human CD and TNBS colitis [32,34]. Here, the mRNA expression of these key proinflammatory cytokines was assessed in colonic tissues of mice, and treatment with LCAR resulted in an approximately fivefold reduction in the colonic expression of both IL-1β and IL-6 mRNA when compared with untreated mice with TNBS colitis (Fig. 5e and f). In addition to its local anti-inflammatory effects, LCAR therapy also significantly reduced the serum levels of IL-1β and IL-6, underscoring the beneficial systemic outcome of LCAR's local anti-inflammatory effects (Fig. 5g and h). Therefore, LCAR's therapeutic efficacy in treating TNBS colitis may be attributed to its ability to suppress the expression of proinflammatory cytokines, corroborating our in vitro data.
The LCAR therapy inhibits T cell responses in TNBS colitis
Our in vitro data demonstrated that LCAR could inhibit both the innate and adaptive arms of the immune response. Because one injection of TNBS in Balb/c mice results typically in an acute, T cell-independent inflammatory response, a second injection of TNBS is required to produce a chronic, T cell-driven form of intestinal inflammation. We therefore reinduced colitis 7 days after the initial TNBS injection and isolated colon-draining sLNs. Total sLN cells were restimulated ex vivo with TNBS, and antigen-specific T cell responses were assessed (Fig. 6). In vivo treatment with LCAR resulted in a significant reduction of ex vivo cell proliferation, indicating that in addition to its immunosuppressive effect on the acute inflammatory response, LCAR administration also suppressed adaptive immune responses, an important consideration for the potential of this therapy in CD.
Fig. 6.
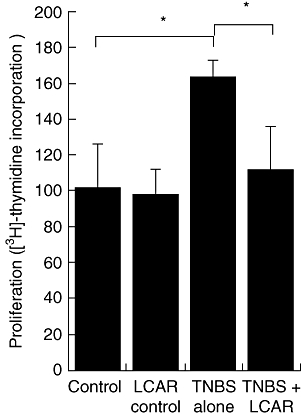
L-carnitine (LCAR) suppresses T cell responses in vivo. Chronic trinitrobenzene sulphonic acid (TNBS) colitis was induced by two intrarectal injections (day 0 and day 7) of 100 mg/kg TNBS dissolved in 50% ethanol. Mice were treated every 24 h with LCAR (150 mg/kg), starting 30 min prior to the induction of colitis. Colon-draining lymph nodes were isolated 3 days after the final TNBS injection and restimulated ex vivo with TNBS. Antigen-specific proliferation was assessed by [H3]-thymidine incorporation after 4 days of culture. Data are expressed as mean ± standard error of the mean (n = 3 for control groups, n = 5 for TNBS groups). *P < 0·05.
Discussion
The interplay between both innate and adaptive immune responses is crucial to perpetuate inflammation in human CD. In this study, we confirm that LCAR can suppress DC and macrophage co-stimulatory molecule expression dose-dependently, and to our knowledge are the first to describe its effects on purified CD4+ T cell activation and cytokine production. Furthermore, we demonstrate that carnitine deficiency results in T cell hyperactivation, which can be reversed by LCAR supplementation. Finally, we demonstrate the therapeutic potential of LCAR in treating the acute and chronic aspects of intestinal inflammation.
The TNBS colitis mimics human CD in that it generates mucosal inflammation which is dependent upon the presence of bacteria in the gut lumen and results in the transmural infiltration of mononuclear cells [35]. Although once thought to be driven primarily by adaptive immune responses, innate cells are now recognized as playing a key role in the initiation phase of TNBS colitis [36]. T cells, on the other hand, are probably implicated in the amplification and perpetuation of inflammation [37]. In support of our in vitro data, systemic administration of free LCAR was effective in treating TNBS colitis. This protection was characterized by an improvement in all clinical and histological criteria in mice treated with daily injections of LCAR and was associated with a suppressive effect on the colonic mRNA expression and serum levels of IL-1β and IL-6. Importantly, LCAR was also effective in dampening antigen-specific T cell responses in sLNs of mice with chronic TNBS colitis.
A recent study investigating the role of carnitine transporters in butyrate metabolism in colonocytes demonstrated a protective role of the local administration of carnitine-loaded liposomes in TNBS colitis [18]. However, in contrast with the current study, the direct effect of carnitine administration on the immune response was not examined. Additionally, we provide evidence that systemic administration of pure carnitine, as opposed to local administration of carnitine-loaded liposomes, is also protective in the development of TNBS colitis. This observation may therefore have implications for the clinical translation of LCAR therapy, both in terms of cost and route of administration. Taken together, LCAR's therapeutic efficacy in treating TNBS colitis may be due to the combination of its protective effects on colonocyte structure and metabolism as well as its immunosuppressive action during the generation of immune responses.
We had initially aimed to assess whether mice with a carnitine deficiency were predisposed to developing intestinal inflammation induced by TNBS. However, upon induction of colitis, carnitine-deficient mice were significantly more susceptible to TNBS-induced mortality compared with wild-type mice, with up to 75% mortality per experiment. The sudden deaths of carnitine-deficient mice upon exposure to TNBS may have resulted from metabolic disturbances, as carnitine-deficient mice develop hypoglycaemia after 18 h of fasting [33]. Alternatively, as mice deficient in the carnitine transporter, OCTN2, develop spontaneous atrophy of intestinal epithelial cells and colonic inflammation [17], a disturbance in the intestinal barrier function of carnitine-deficient mice may also have resulted in a similar defect and warrants further investigation. Because of the as-yet unexplained high mortality rate of carnitine-deficient mice upon exposure to TNBS, the immune response of these mice could be observed only in vitro. Here, we have shown that carnitine deficiency promotes the hyperactivation of CD4+ T cells and the production of the classical Th1 cytokine, IFN-γ.
In this study, the precise immunosuppressive mechanism of action of LCAR on APC and T cell activation remains elusive. However, there are a number of potential mechanisms which may participate in this effect. First, anti-oxidants have recognized protective roles on the intestinal mucosa by preventing reactive oxygen species (ROS) production [38,39] and play a critical role in preventing inflammation and cancer [40,41]. Recent studies have demonstrated that LCAR can act as an anti-oxidant and protect from ROS-induced tissue damage [42,43]. In fact, LCAR is more effective at inhibiting lipid peroxidation than both trolox and alpha-tocopherol (vitamin E), two widely recognized anti-oxidants [44]. Thus, LCAR's anti-oxidant properties may therefore participate in its immunosuppressive capacity. Alternatively, LCAR has been shown to directly enhance the nuclear translocation and transcriptional activity of GR-α[22]. The optimal LCAR concentration to induce GR-α translocation was 100 mM, the same concentration used in this study. The physiological relevance of this similarity is underscored by the fact that tissue LCAR concentrations as high as 100 mM have been described [22]. Therefore, among other potential mechanisms, LCAR may suppress immune responses by either quenching ROS, and thereby inhibiting the third signal for T cell activation, or by activating GR-α translocation directly and mimicking the known immunosuppressive properties of glucocortocoids.
While the association between mutations in the OCTN genes and CD susceptibility has not been replicated worldwide, our results support the aforementioned candidate gene in predisposing individuals to CD and highlight the potential therapeutic efficacy of LCAR supplementation. Here, we confirm and expand the evidence to support an immunosuppressive role for LCAR on APC and T cell function and demonstrate the therapeutic value of its systemic administration in treating intestinal inflammation.
Acknowledgments
The authors wish to thank Dr Jeremy Jass (Department of Pathology, McGill University, Montreal, Qc) for his assistance in grading histological changes in the colons of mice with TNBS colitis, and Dr Ciriacco Piccirillo (Department of Microbiology and Immunology, McGill University, Montreal, Qc) for insightful discussion and use of his laboratory equipment. This study was funded in part by the Crohn's and Colitis Foundation of Canada, grant number 3801 and the Research Institute of the McGill University Health Center.
References
- 1.Elson CO, Sartor RB, Tennyson GS, Riddell RH. Experimental models of inflammatory bowel disease. Gastroenterology. 1995;109:1344–67. doi: 10.1016/0016-5085(95)90599-5. [DOI] [PubMed] [Google Scholar]
- 2.Fujino S, Andoh A, Bamba S, et al. Increased expression of interleukin 17 in inflammatory bowel disease. Gut. 2003;52:65–70. doi: 10.1136/gut.52.1.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schmidt C, Giese T, Ludwig B, et al. Expression of interleukin-12-related cytokine transcripts in inflammatory bowel disease: elevated interleukin-23p19 and interleukin-27p28 in Crohn's disease but not in ulcerative colitis. Inflamm Bowel Dis. 2005;11:16–23. doi: 10.1097/00054725-200501000-00003. [DOI] [PubMed] [Google Scholar]
- 4.Tobin MV, Logan RF, Langman MJ, McConnell RB, Gilmore IT. Cigarette smoking and inflammatory bowel disease. Gastroenterology. 1987;93:316–21. doi: 10.1016/0016-5085(87)91021-3. [DOI] [PubMed] [Google Scholar]
- 5.Evans JM, McMahon AD, Murray FE, McDevitt DG, MacDonald TM. Non-steroidal anti-inflammatory drugs are associated with emergency admission to hospital for colitis due to inflammatory bowel disease. Gut. 1997;40:619–22. doi: 10.1136/gut.40.5.619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Duffy LC, Zielezny MA, Marshall JR, et al. Relevance of major stress events as an indicator of disease activity prevalence in inflammatory bowel disease. Behav Med. 1991;17:101–10. doi: 10.1080/08964289.1991.9937553. [DOI] [PubMed] [Google Scholar]
- 7.Levi AJ. Diet in the management of Crohn's disease. Gut. 1985;26:985–8. doi: 10.1136/gut.26.10.985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hartman C, Berkowitz D, Weiss B, et al. Nutritional supplementation with polymeric diet enriched with transforming growth factor-beta 2 for children with Crohn's disease. Isr Med Assoc J. 2008;10:503–7. [PubMed] [Google Scholar]
- 9.Rebouche CJ, Seim H. Carnitine metabolism and its regulation in microorganisms and mammals. Annu Rev Nutr. 1998;18:39–61. doi: 10.1146/annurev.nutr.18.1.39. [DOI] [PubMed] [Google Scholar]
- 10.Wu X, Huang W, Prasad PD, et al. Functional characteristics and tissue distribution pattern of organic cation transporter 2 (OCTN2), an organic cation/carnitine transporter. J Pharmacol Exp Ther. 1999;290:1482–92. [PubMed] [Google Scholar]
- 11.Rebouche CJ, Engel AG. Tissue distribution of carnitine biosynthetic enzymes in man. Biochim Biophys Acta. 1980;630:22–9. doi: 10.1016/0304-4165(80)90133-6. [DOI] [PubMed] [Google Scholar]
- 12.Rebouche CJ. Carnitine function and requirements during the life cycle. FASEB J. 1992;6:3379–86. [PubMed] [Google Scholar]
- 13.Fritz IB, Marquis NR. The role of acylcarnitine esters and carnitine palmityltransferase in the transport of fatty acyl groups across mitochondrial membranes. Proc Natl Acad Sci USA. 1965;54:1226–33. doi: 10.1073/pnas.54.4.1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Peltekova VD, Wintle RF, Rubin LA, et al. Functional variants of OCTN cation transporter genes are associated with Crohn disease. Nat Genet. 2004;36:471–5. doi: 10.1038/ng1339. [DOI] [PubMed] [Google Scholar]
- 15.Vermeire S, Pierik M, Hlavaty T, et al. Association of organic cation transporter risk haplotype with perianal penetrating Crohn's disease but not with susceptibility to IBD. Gastroenterology. 2005;129:1845–53. doi: 10.1053/j.gastro.2005.10.006. [DOI] [PubMed] [Google Scholar]
- 16.Van Limbergen J, Russell RK, Nimmo ER, Satsangi J. The genetics of inflammatory bowel disease. Am J Gastroenterol. 2007;102:2820–31. doi: 10.1111/j.1572-0241.2007.01527.x. [DOI] [PubMed] [Google Scholar]
- 17.Shekhawat PS, Srinivas SR, Matern D, et al. Spontaneous development of intestinal and colonic atrophy and inflammation in the carnitine-deficient jvs (OCTN2(−/−)) mice. Mol Genet Metab. 2007;92:315–24. doi: 10.1016/j.ymgme.2007.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.D'Argenio G, Calvani M, Casamassimi A, et al. Experimental colitis: decreased Octn2 and Atb0+ expression in rat colonocytes induces carnitine depletion that is reversible by carnitine-loaded liposomes. FASEB J. 2006;20:2544–6. doi: 10.1096/fj.06-5950fje. [DOI] [PubMed] [Google Scholar]
- 19.Winter BK, Fiskum G, Gallo LL. Effects of L-carnitine on serum triglyceride and cytokine levels in rat models of cachexia and septic shock. Br J Cancer. 1995;72:1173–9. doi: 10.1038/bjc.1995.482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Athanassakis I, Mouratidou M, Sakka P, Evangeliou A, Spilioti M, Vassiliadis S. L-carnitine modifies the humoral immune response in mice after in vitro or in vivo treatment. Int Immunopharmacol. 2001;1:1813–22. doi: 10.1016/s1567-5769(01)00105-9. [DOI] [PubMed] [Google Scholar]
- 21.Fattorossi A, Biselli R, Casciaro A, Tzantzoglou S, de Simone C. Regulation of normal human polyrnorphonuclear leucocytes by carnitine. Mediators Inflamm. 1993;2:S37–41. doi: 10.1155/S0962935193000742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Alesci S, De Martino MU, Mirani M, et al. A nutritional modulator of glucocorticoid receptor functions. FASEB J. 2003;17:1553–5. doi: 10.1096/fj.02-1024fje. [DOI] [PubMed] [Google Scholar]
- 23.Kouttab NM, De Simone C. Modulation of cytokine production by carnitine. Mediators Inflamm. 1993;2:S25–8. doi: 10.1155/S0962935193000717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.De Simone C, Ferrari M, Meli D, Midiri G, Sorice F. Reversibility by L-carnitine of immunosuppression induced by an emulsion of soya bean oil, glycerol and egg lecithin. Arzneimittelforschung. 1982;32:1485–8. [PubMed] [Google Scholar]
- 25.De Simone C, Famularo G, Tzantzoglou S, Trinchieri V, Moretti S, Sorice F. Carnitine depletion in peripheral blood mononuclear cells from patients with AIDS: effect of oral L-carnitine. AIDS. 1994;8:655–60. doi: 10.1097/00002030-199405000-00012. [DOI] [PubMed] [Google Scholar]
- 26.Dohi T, Fujihashi K, Rennert PD, Iwatani K, Kiyono H, McGhee JR. Hapten-induced colitis is associated with colonic patch hypertrophy and T helper cell 2-type responses. J Exp Med. 1999;189:1169–80. doi: 10.1084/jem.189.8.1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wallace JL, McKnight W, Asfaha S, Liu DY. Reduction of acute and reactivated colitis in rats by an inhibitor of neutrophil activation. Am J Physiol. 1998;274:G802–8. doi: 10.1152/ajpgi.1998.274.5.G802. [DOI] [PubMed] [Google Scholar]
- 28.Dieleman LA, Elson CO, Tennyson GS, Beagley KW. Kinetics of cytokine expression during healing of acute colitis in mice. Am J Physiol. 1996;271:G130–6. doi: 10.1152/ajpgi.1996.271.1.G130. [DOI] [PubMed] [Google Scholar]
- 29.Ameho CK, Adjei AA, Harrison EK, et al. Prophylactic effect of dietary glutamine supplementation on interleukin 8 and tumour necrosis factor alpha production in trinitrobenzene sulphonic acid induced colitis. Gut. 1997;41:487–93. doi: 10.1136/gut.41.4.487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Freeman GJ, Gribben JG, Boussiotis VA, et al. Cloning of B7-2: a CTLA-4 counter-receptor that costimulates human T cell proliferation. Science. 1993;262:909–11. doi: 10.1126/science.7694363. [DOI] [PubMed] [Google Scholar]
- 31.Hathcock KS, Laszlo G, Pucillo C, Linsley P, Hodes RJ. Comparative analysis of B7-1 and B7-2 costimulatory ligands: expression and function. J Exp Med. 1994;180:631–40. doi: 10.1084/jem.180.2.631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Elson CO, Beagley KW, Sharmanov AT, et al. Hapten-induced model of murine inflammatory bowel disease: mucosa immune responses and protection by tolerance. J Immunol. 1996;157:2174–85. [PubMed] [Google Scholar]
- 33.Wood PA, Amendt BA, Rhead WJ, Millington DS, Inoue F, Armstrong D. Short-chain acyl-coenzyme A dehydrogenase deficiency in mice. Pediatr Res. 1989;25:38–43. doi: 10.1203/00006450-198901000-00010. [DOI] [PubMed] [Google Scholar]
- 34.Fiocchi C. Lymphokines and the intestinal immune response. Role in inflammatory bowel disease. Immunol Invest. 1989;18:91–102. doi: 10.3109/08820138909112230. [DOI] [PubMed] [Google Scholar]
- 35.Neurath MF, Fuss I, Kelsall BL, Stuber E, Strober W. Antibodies to interleukin 12 abrogate established experimental colitis in mice. J Exp Med. 1995;182:1281–90. doi: 10.1084/jem.182.5.1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Santucci L, Agostini M, Bruscoli S, et al. GITR modulates innate and adaptive mucosal immunity during the development of experimental colitis in mice. Gut. 2007;56:52–60. doi: 10.1136/gut.2006.091181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sheibanie AF, Yen JH, Khayrullina T, et al. The proinflammatory effect of prostaglandin E2 in experimental inflammatory bowel disease is mediated through the IL-23–> IL-17 axis. J Immunol. 2007;178:8138–47. doi: 10.4049/jimmunol.178.12.8138. [DOI] [PubMed] [Google Scholar]
- 38.D'Odorico A, Bortolan S, Cardin R, et al. Reduced plasma antioxidant concentrations and increased oxidative DNA damage in inflammatory bowel disease. Scand J Gastroenterol. 2001;36:1289–94. doi: 10.1080/003655201317097146. [DOI] [PubMed] [Google Scholar]
- 39.D'Argenio G, Cosenza V, Delle Cave M, et al. Butyrate enemas in experimental colitis and protection against large bowel cancer in a rat model. Gastroenterology. 1996;110:1727–34. doi: 10.1053/gast.1996.v110.pm8964397. [DOI] [PubMed] [Google Scholar]
- 40.Frenkel K. Carcinogen-mediated oxidant formation and oxidative DNA damage. Pharmacol Ther. 1992;53:127–66. doi: 10.1016/0163-7258(92)90047-4. [DOI] [PubMed] [Google Scholar]
- 41.Halliwell B, Gutteridge JM. Lipid peroxidation, oxygen radicals, cell damage, and antioxidant therapy. Lancet. 1984;1:1396–7. doi: 10.1016/s0140-6736(84)91886-5. [DOI] [PubMed] [Google Scholar]
- 42.Wang C, Sadovova N, Ali HK, et al. L-carnitine protects neurons from 1-methyl-4-phenylpyridinium-induced neuronal apoptosis in rat forebrain culture. Neuroscience. 2007;144:46–55. doi: 10.1016/j.neuroscience.2006.08.083. [DOI] [PubMed] [Google Scholar]
- 43.Rauchova H, Koudelova J, Drahota Z, Mourek J. Hypoxia-induced lipid peroxidation in rat brain and protective effect of carnitine and phosphocreatine. Neurochem Res. 2002;27:899–904. doi: 10.1023/a:1020339530924. [DOI] [PubMed] [Google Scholar]
- 44.Gulcin I. Antioxidant and antiradical activities of L-carnitine. Life Sci. 2006;78:803–11. doi: 10.1016/j.lfs.2005.05.103. [DOI] [PubMed] [Google Scholar]