

Abstract
Immunoproteasome up-regulation enhances the processing of nuclear factor-κB (NF-κB) and degradation of IκBα, which correlates with increased amounts of NF-κB in the various cells. Aberrant activation of NF-κB is involved in the pathogenesis of inflammatory bowel disease (IBD). The aim of this study was to elucidate the effect of proteasome inhibitor MG132 on experimental IBD. We investigated the effects of MG132 on intestinal inflammation and epithelial regeneration in both interleukin-10-deficient (IL-10−/−) mice and mice with dextran sulphate sodium (DSS)-induced colitis. Body weight, histological findings and tumour necrosis factor (TNF)-α mRNA expression, epithelial cell proliferation and NF-κB p65 activity in colonic tissues were examined. The effects of MG132 on cell proliferation, migration and multiple drug resistance 1 (MDR1) gene expression were determined in vitro. MG132 ameliorated intestinal inflammation of IL-10−/− mice by decreasing TNF-α mRNA expression in the colonic tissues, which was associated with suppression of NF-κB activation, and reduced significantly the number of Ki-67-positive intestinal epithelial cells. On the other hand, MG132 did not reduce intestinal inflammation in mice with DSS-induced colitis, and delayed significantly the recovery of body weight and epithelial regeneration. MG132 also suppressed significantly epithelial cell proliferation, cell migration and MDR1 gene expression in vitro. Proteasome inhibition reduces T cell-mediated intestinal inflammation, but may interrupt both epithelial regeneration and barrier function of colonic mucosa. Optimal use of proteasome inhibitor should be kept in mind when we consider its clinical application for patients with IBD.
Keywords: epithelial regeneration, IBD, MDR1, NF-κB, proteasome inhibitor
Introduction
Inflammatory bowel disease (IBD) is characterized by a dysregulated immune response of unknown aetiology. Enteric flora or bacterial products appear to contribute to the recruitment and activation of lamina propria macrophages and T cells [1,2]. Proinflammatory cytokines produced by these cells, such as interleukin (IL)-1β and tumour necrosis factor (TNF)-α, have important roles in the initiation and perpetuation of the intestinal inflammation of IBD [3–6]. Moreover, such inflammatory responses within the intestinal mucosa cause epithelial damage, which might result in impaired epithelial barrier function.
Nuclear factor-κB (NF-κB) is a transcription factor that consists of a homodimer or a heterodimer of two subunits of the NF-κB family; p65 (RelA), RelB, c-Rel, NF-κB1 (p50 and its precursor p105) or NF-κB2 (p52 and its precursor p100) [7]. NF-κB-dependent gene expression is regulated predominantly by a shuttling system that controls the cytoplasmic versus nuclear localization of this transcription factor.
The most abundant form of NF-κB is the p50/p65 heterodimer which remains in an inactive state in the cytoplasm, forming a ternary complex with the inhibitory protein inhibitor of NF-κB, IκBα. Upon stimulation, IκBα is phosphorylated rapidly, ubiquitinated, and subsequently degradated by proteasomes, allowing translocation of p50/ p65 heterodimers into the nucleus [8–13]. During gut inflammation, the excessive immune stimulation by inflammatory cytokines induces NF-κB activation in the intestinal mucosa. For example, the p65 subunit of NF-κB is upregulated in the mucosa of patients with active IBD [14]. Inhibition of NF-κB with specific p65 anti-sense oligonucleotides not only down-regulates cytokine production by intestinal macrophages from patients with Crohn's disease; it is also effective for preventing or treating an experimental model of IBD [15]. Therefore, the control of NF-κB activation has been focused upon as one of the promising therapies for patients with IBD.
The 26S proteasome is a multi-catalytic adenosine triphosphate (ATP)-dependent protease complex that is responsible for the post-translational control of all short-lived and many long-lived proteins [16]. Treatment with proteasome inhibitor results in protein stability and increases markedly a large number of cellular proteins, such as cell regulators, transcription factors and apoptotic regulators [17]. Moreover, proteasomes are involved in two essential steps of NF-κB p50/p65 activation: generation of p50 from its precursor p105 and degradation of IκBα[18–20]. On the basis of these findings, proteasome inhibition is considered to be an emerging strategy to attenuate the inflammatory response associated with NF-κB activation [21]. Recently, Visekruna et al. reported high expression of the immunoproteasome subunits β1i and β2i in the inflamed mucosa of patients with Crohn's disease. Moreover, they showed that processing of NF-κB precursor p105 and degradation of IκBα is enhanced by these immunoproteasomes which they isolated from intestinal tissues of patients with IBD [22]. Taken together, proteasome inhibitor could be useful for reducing intestinal inflammation of patients with IBD. However, little is known about the effect of proteasome inhibition on intestinal inflammation.
In the present study, we examined the effect of proteasome inhibitor MG132 on colitis using two different experimental murine colitis models: IL-10-deficient (IL-10−/−) mice and dextran sulphate sodium (DSS)-induced colitis, and investigated the mechanisms of actions of MG132 on intestinal inflammation.
Materials and methods
Mice
Female C57BL/6 mice (12–15 weeks old, weighing 20–24 g; Japan SLC, Inc., Shizuoka, Japan) and female IL-10−/− mice on a C57BL/6 background (4 weeks old; Jackson Laboratory, Bar Harbor, ME, USA) were used. All mice were housed in specific pathogen-free conditions in the animal facility of Kyoto University. The studies were approved by the Animal Protection Committee of our institution.
The IL-10−/− mice
Treatments
To investigate the effects of proteasome inhibitor on experimental murine colitis, we used MG132, a proteasome inhibitor (Biomol International LP Inc., Plymouth Meeting, PA, USA), which inhibits inhibitor κB degradation [23–25]. MG132 was dissolved in dimethyl sulphoxide (DMSO) and then diluted in 500 µl sterile phosphate-buffered saline (PBS) for injection. Female IL-10−/− mice at 4 weeks of age were divided into four groups and treated with intraperitoneal injection three times a week as follows: group A, 0·01% DMSO as the control; group B, 0·6 µmol/kg MG132; group C, 3·0 µmol/kg MG132; and group D, 15·0 µmol/kg MG132. All mice were killed after 4 weeks of treatment by cervical dislocation under ether anaesthesia. The colonic tissue was removed from each mouse and examined as described below.
Histological analysis
The entire colon was removed and washed with PBS. The caecum, proximal colon, transverse colon and rectum were dissected transversely and fixed in 10% formalin. The fixed tissue was embedded in paraffin, stained with haematoxylin and eosin (H&E) and analysed histologically in a blind manner. Histological damage was quantified by the histological scoring system described by Sellon et al.[1]. In brief, the sections were graded on a scale of 0–4 based on the degree of lamina propria and submucosal mononuclear cellular infiltration, crypt abscess, crypt hyperplasia, goblet cell depletion and architectural distortion. The mean histological score for each mouse was determined by adding the scores for each sections of the colon examined, and dividing the total by the number of sections examined.
The DSS-induced colitis model
Induction of colitis and treatments
Female C57BL/6 mice were given 3% DSS (molecular weight 36–50 kDa; MP Biomedicals, Inc., Solon, OH, USA) in their drinking water for 5 days, and then switched to regular drinking water. Mice were injected intraperitoneally with 15·0 µmol/kg MG132 (prepared in the same way as for IL-10−/− mice) or 0·01% DMSO as the control three times a week from day 0 to the end of the experiment. On day 10, the mice were killed.
Body weight was measured daily throughout the experiment. After the mice were killed, the colonic tissue was removed from each mouse and examined as described below.
Histological analysis
The rectum was evaluated histologically because this segment is affected most severely in DSS-induced colitis [26]. The rectum (1·5 cm from the anal verge) was removed and dissected transversely every 3 mm, and the pieces were fixed in 10% formalin. The fixed tissues were embedded in paraffin, stained with H&E and analysed histologically in a blind manner. Histological damage was quantified by the histological scoring system described by Williams et al.[27]. In brief, the sections were graded for inflammation severity, inflammation extent and crypt damage. The inflammation severity grading index was as follows: 0, none; 1, mild; 2, moderate; 3, severe. The inflammation extent grading index was as follows: 0, none; 1, mucosa; 2, mucosa and submucosa; 3, transmural. The crypt damage grading index was as follows: 0, none; 1, basal one-third damaged; 2, basal two-thirds damaged; 3, crypts lost, but surface epithelium present; 4, crypts and surface epithelium lost. Each of these grades was also scored as to the percentage of involvement as follows: 0, 0%; 1, 1–25%; 2, 26–50%; 3, 51–75%; 4, 76–100%. Each subscore (inflammation severity, inflammation extent and crypt damage score) was the product of the grade multiplied by the score of the percentage of involvement. The total colitis score was the sum of the three subscores.
mRNA assessment by semiquantitative reverse transcription–polymerase chain reaction
Total RNA was extracted from colonic tissues of IL-10−/− mice and C57BL/6 mice with DSS-induced colitis treated with MG132 or 0·01% DMSO with TRIzol (Invitrogen, Carlsbad, CA, USA). RNA (3 µg) was reverse-transcribed with SuperScript II Reverse Transcriptase (Invitrogen), and the resulting complementary DNAs were analysed for TNF-α mRNA expression by semiquantitative polymerase chain reaction (PCR). PCR was performed in a total volume of 20 µl containing 1 µl of complementary DNA, 10 µmol/l of each primer and a solution of 1 U of AmpliTaq Gold DNA polymerase using the GeneAmp PCR system (Applied Biosystems, Foster City, CA, USA). The PCR primer sequences used are shown in Table 1. PCR products were separated on 1% agarose gels containing ethidium bromide. After gel electrophoresis, band intensities were measured using an autoanalysing system (Printgraph; ATTO Corporation, Tokyo, Japan). The signal of each product was standardized against the β-actin signal for each sample.
Table 1.
Primer pairs for semiquantitative polymerase chain reaction.
| Target gene | Primer sequences | Major product size (base pairs) |
|---|---|---|
| Mouse TNF-α | Forward: 5′-TTCTGTCTACTGAACTTCGGGGTGATCGGTCC-3′ | 354 |
| Reverse: 5′-GTATGAGATAGCAAATCGGCTGACGGTGTGGG-3′ | ||
| Human MDR1 | Forward: 5′-AGATCAACTCGTAGGAGTGTC-3′ | 795 |
| Reverse: 5′-GTTTCTGTATGGTACCTGCAA-3′ | ||
| Mouse β-actin | Forward: 5′-GTGGGCCGCTCTAGGCACCAA-3′ | 540 |
| Reverse: 5′-CTCTTTGATGTCACGCACGATTTC-3′ | ||
| Human β-actin | Forward: 5′-GAGACCTTCAACACCCCAGCC-3′ | 311 |
| Reverse: 5′-GGCCATCTCTTGCTCGAAGTC-3′ |
MDR, multiple drug resistance; TNF, tumour necrosis factor.
Immunohistochemistry
Colonic tissue sections of IL-10−/− mice, C57BL/6 mice with DSS-induced colitis and C57BL/6 mice treated with 15·0 µmol/kg MG132 or 0·01% DMSO were stained for phospho-NF-κB p65 and Ki-67 antigen. Paraffin-embedded sections (4 µm thick) were deparaffinized and reacted with 0·3% H2O2 in methanol for 30 min to inhibit endogenous peroxidase activity. The sections were placed in 0·01 mol/l citrate buffer (pH 6·0) and pretreated with microwave heating for antigen retrieval. After blocking with 3% bovine serum albumin (BSA) in PBS, the sections were incubated with each diluted primary antibody overnight at 4°C. The dilutions in PBS with 1% BSA were prepared with anti-mouse phospho-NF-κB p65 polyclonal antibody (1:50; Cell Signaling Technology, Inc., Danvers, MA, USA) and anti-mouse Ki-67 monoclonal antibody (1:50; Dako, Copenhagen, Denmark). After washing with PBS, the sections were incubated for 30 min with biotinylated anti-rabbit immunoglobulin G (IgG) antibody diluted 1:200 in PBS with 1% BSA for anti-mouse phospho-NF-κB p65 antibody, and biotinylated anti-rat IgG antibody diluted 1:200 in PBS with 1% BSA for anti-mouse Ki-67 antibody. The sections were reacted with a peroxidase-linked avidin–biotin complex (Vector Laboratories, Burlingame, CA, USA) for 30 min. Localization of the phospho-NF-κB p65 antigen or the Ki-67 antigen was visualized by incubation with 3,3′-diaminobenzidine tetrahydrochloride in 0·01% H2O2. Haematoxylin was used for nuclear counterstaining. To assess the proliferation activity, the number of Ki-67-positive cells per total crypt epithelial cells was counted. The Ki-67 labelling index was defined as the percentage of Ki-67-positive cells per crypt [28].
Bromodeoxyuridine cell proliferation assay
We examined cell proliferation activity using the bromodeoxyuridine (BrdU) Cell Proliferation Assay kit (Calbiochem, Darmstadt, Germany) [29,30]. The human colon cancer cell line Caco-2 cells were maintained with Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% heat-inactivated fetal bovine serum (FBS) and antibiotics (100 U/ml penicillin and 100 µg/ml streptomycin). The cells were seeded into 96-well plates (1 × 104 cells/well) and grown for 48 h. After confirming adhesion, the cells were incubated in 100 µl DMEM with 0·1% heat-inactivated FBS for 24 h, and then pretreated for 1 h with 0·12 µmol/l, 0·60 µmol/l and 3·0 µmol/l MG132 or 0·006% DMSO as a control (n = 12). After washing with PBS, the cells were stimulated with 100 ng/ml recombinant human epidermal growth factor (EGF) (R&D Systems, Inc., Minneapolis, MN, USA). After an additional incubation for 1 h, 20 µl of BrdU solution was added to each well. After another 12 h incubation, 100 µl of anti-BrdU antibody dilution was added and incubated for 1 h. After washing with wash buffer, 100 µl of peroxidase goat anti-mouse IgG was added and incubated for 30 min. After washing with wash buffer, 100 µl of tetramethylbenzidine solution was added, incubated for 15 min, and 100 µl of 2·5 N sulphuric acid was added. The absorbance was measured at 450 nm and 540 nm as a control using the Ultra Microplate Reader (Bio-Tek Instruments, Inc., Winooski, VT, USA).
Assessment of intestinal epithelial cell migration
To investigate the effects of MG132 on cell migration, we used an in vitro model of a scrape-wounded intestinal epithelial monolayer with the rat intestinal epithelial cell line IEC-6 cells, which are optimal for this assay [31,32]. The cells were maintained with DMEM supplemented with 10% heat-inactivated FBS and antibiotics (as mentioned above). IEC-6 cells were seeded into six-well plates (5 × 105 cells/well) and grown to confluence in basal medium. Cells were incubated in DMEM with 0·1% heat-inactivated FBS for 24 h, and then pretreated for 1 h with 0·024 µmol/l, 0·12 µmol/l and 0·60 µmol/l MG132 or 0·001% DMSO as a control. Cell monolayers were wounded by scraping off with a razor blade, and after 24 h the number of cells crossing 1 mm of the wound was counted at two points in each well. This experiment was performed in triplicate.
Effects of MG132 on multiple drug resistance 1 gene expression
Caco-2 cells were seeded into six-well plates (1 × 105 cells/well) and grown to confluence in DMEM supplemented with 10% heat-inactivated FBS and antibiotics (as mentioned above). Cells were incubated in DMEM with 0·1% heat-inactivated FBS for 24 h, and then pretreated for 1 h with 0·6 µmol/l, 3·0 µmol/l and 15·0 µmol/l MG132 or 0·03% DMSO as a control. Cells were stimulated with 10 ng/ml human recombinant IL-1β (R&D Systems, Inc.), and after another 6 h incubation, total RNA was extracted from the cells using TRIzol. RNA (1·5µg) was reverse-transcribed, and multiple drug resistance 1 (MDR1) gene expression was evaluated by semiquantitative PCR as described above.
Statistical analysis
All numerical data are expressed as the mean ± standard error. A P-value of less than 0·05 was considered statistically significant. The differences in the data between groups were analysed by Student's t-test or the Mann–Whitney U-test.
Results
Effects of MG132 on IL-10−/− mice
Histological findings and TNF-α gene expression in colonic tissues
In our animal facility, IL-10−/− mice develop intestinal inflammation spontaneously at approximately 5 weeks of age. In 8-week-old IL-10−/− mice (after 4 weeks’ treatment with vehicle), the histological findings of the colon revealed epithelial hyperplasia, inflammatory cell infiltration, goblet cell depletion and crypt abscesses. In contrast, in IL-10−/− mice treated with 15·0 µmol/kg MG132 for 4 weeks, goblet cells remained and inflammatory cell infiltration was milder than that in the vehicle-treated IL-10−/− mice (Fig. 1a). MG132 administration reduced the histological score, and the histological score in IL-10−/− mice treated with 15·0 µmol/kg MG132 was significantly lower than that of vehicle-treated IL-10−/− mice (P < 0·05; Fig. 1b).
Fig. 1.
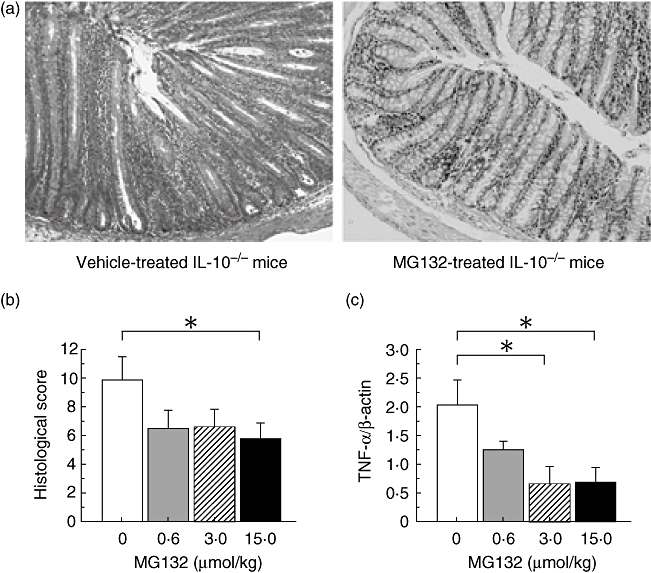
Effects of MG132 on interleukin-10-deficient (IL-10−/−) mice. (a) Histological findings in IL-10−/− mice. Sections of the cecum, proximal colon, transverse colon and rectum were collected at necropsy (on day 28) and stained with haematoxylin and eosin. Left panel, vehicle-treated IL-10−/− mice; right panel, MG132-treated (15·0 µmol/kg) IL-10−/− mice (original magnification 100×). (b) Histological score in IL-10−/− mice treated with the indicated doses of MG132. Histological damage was quantified by the scoring system described by Sellon et al. (n = 8 in each group). *P < 0·05 compared with vehicle-treated IL-10−/− mice. (c) Tumour necrosis factor-α gene expression in the colonic tissues of IL-10−/− mice treated with MG132. *P < 0·05 compared with vehicle-treated IL-10−/− mice (n = 8 in each group).
MG132 inhibited TNF-α gene expression in the colonic tissues of IL-10−/− mice (Fig. 1c), reaching significantly different levels from vehicle-treated mice with doses of 3·0 and 15·0 µmol/kg (P < 0·05).
Immunohistochemical staining with anti-phospho-NF-κB p65 antibody and anti-Ki-67 antibody
In the vehicle-treated IL-10−/− mice, phospho-NF-κB p65 immunohistochemical staining was strongly positive in the nucleus of the bottom of the crypt epithelial cells. In contrast, in IL-10−/− mice treated with MG132, nuclear translocation of phospho-NF-κB p65 in the epithelial cells of the crypts was not observed (Fig. 2a).
Fig. 2.
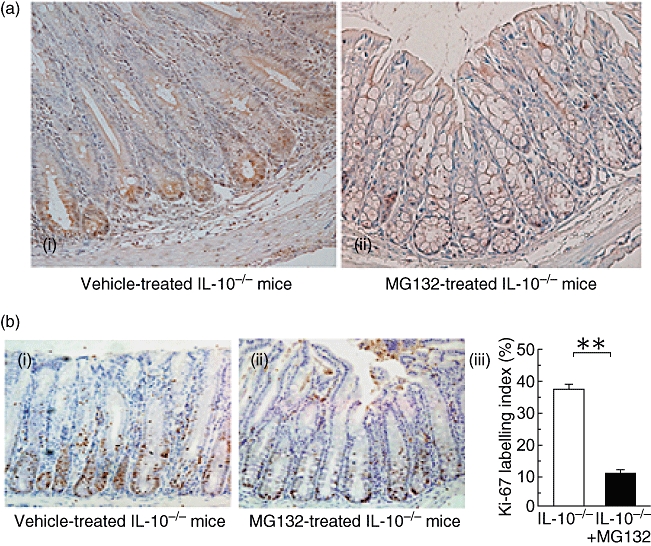
Immunohistochemical staining of phospho-nuclear factor-kappa B (NF-κB) p65 and Ki-67 in interleukin-10-deficient (IL-10−/−) mice treated with MG132. (a) Immunohistochemical staining with anti-phospho-NF-κB p65 antibody. (i) Vehicle-treated IL-10−/− mice; (ii) MG132-treated (15·0 µmol/kg) IL-10−/− mice (original magnification 200×). (b) Immunohistochemical staining with anti-Ki-67 antibody (i, ii) and the Ki-67 labelling index (iii). (i) Vehicle-treated IL-10−/− mice; (ii) MG132-treated (15·0 µmol/kg) IL-10−/− mice (original magnification 200×). The Ki-67 labelling index was defined as the percentage of Ki-67-positive cells per crypt. Six crypts per mouse were evaluated (n = 3 in each group). **P< 0·01 compared with vehicle-treated IL-10−/− mice.
In the vehicle-treated IL-10−/− mice, Ki-67-positive cells were located up to the lower half of the crypts in which phospho-NF-κB p65 was detected in the nucleus. On the other hand, in MG132 (15·0 µmol/kg)-treated IL-10−/− mice, the presence of Ki-67-positive cells was limited to the bottom of the crypts (Fig. 2b). The Ki-67 labelling index of IL-10−/− mice treated with 15·0 µmol/kg MG132 was significantly lower than that of vehicle-treated IL-10−/− mice (P < 0·01).
Effects of MG132 on mice with DSS-induced colitis
Body weight change
We then investigated the effects of MG132 on mucosal regeneration in DSS-induced colitis. In both MG132-treated and vehicle-treated mice with DSS-induced colitis, body weight decreased from day 5 to day 7. In the vehicle-treated mice, body weight began to recover rapidly beginning on day 8, but the recovery was impaired in MG132-treated mice. There was a significant difference in the percentage of change in body weight on day 9 and 10 between MG132-treated and vehicle-treated mice administered DSS (day 9, P < 0·05; day 10, P < 0·01; Fig. 3a).
Fig. 3.
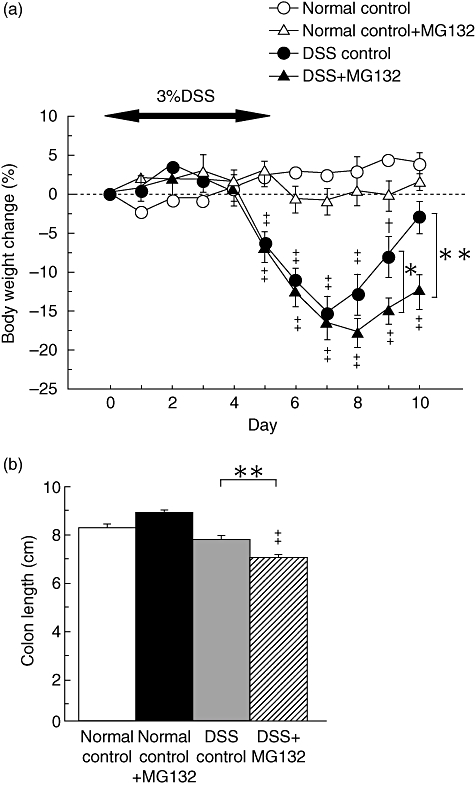
The effects of MG132 on body weight change and colon length of mice with dextran sulphate sodium (DSS)-induced colitis. (a) Serial changes in body weight throughout the experiment in vehicle-treated mice not administered DSS (normal control; open circles), MG132-treated (15·0 µmol/kg) mice not administered DSS (normal control + MG132; open triangles), vehicle-treated mice with DSS-induced colitis (DSS control; closed circles) and MG132-treated (15·0 µmol/kg) mice with DSS-induced colitis (DSS + MG132; closed triangles) (n = 8 in each group). *P < 0·05 and **P < 0·01 between vehicle-treated and MG132-treated mice with DSS-induced colitis. †P < 0·05 and ‡P < 0·01 compared with respective normal mice not administered DSS. (b) Effects of MG132 (15·0 µmol/kg) on colon length in mice with DSS-induced colitis (n = 8 in each group). Colon length was measured from the ileocecal junction to the anal verge at necropsy (on day 10). **P < 0·01 between vehicle-treated and MG132-treated mice with DSS-induced colitis. ‡P < 0·01 compared with respective normal mice not administered DSS.
Colon length
The shortening of the colon and oedema of the rectum in the vehicle-treated mice administered DSS improved on day 10 (data not shown) and colon length on day 10 was not different from that of normal mice (Fig. 3b). In contrast, in the MG132-treated mice administered DSS, the colon was significantly shorter than that of the vehicle-treated mice administered DSS (P < 0·01; Fig. 3b), and oedema of the rectum remained.
Histological findings and TNF-α gene expression in colonic tissues
The treatment of intraperitoneal injection of 15·0 µmol/kg MG132 in C57BL/6 mice not administered DSS did not affect the histological findings, such as crypt height or cell infiltration (Fig. 4a, i,ii).
Fig. 4.
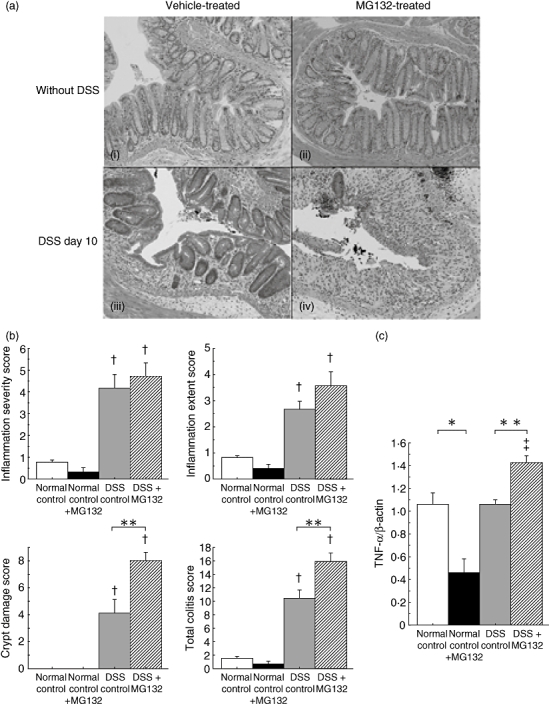
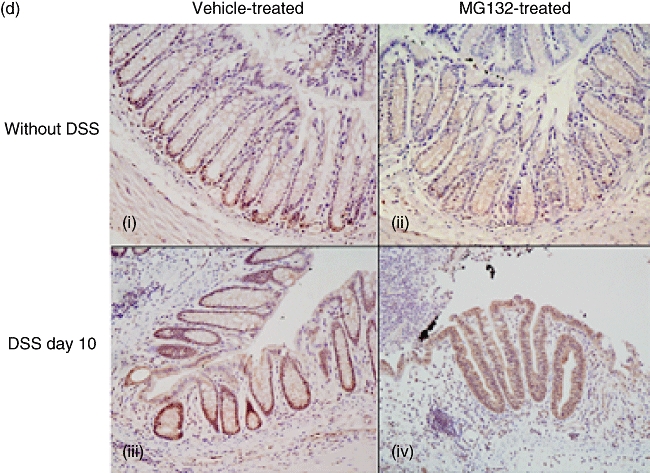
The effects of MG132 on histological changes, tumour necrosis factor (TNF)-α gene expression and immunohistochemical staining of phospho-nuclear factor-kappa B (NF-κB) p65 in the colonic tissues of mice with dextran sulphate sodium (DSS)-induced colitis. (a) Histological findings of the rectum at necropsy (on day 10) stained with haematoxylin and eosin. (i) Vehicle-treated mice not administered DSS, (ii) MG132-treated (15·0 µmol/kg) mice not administered DSS, (iii) vehicle-treated mice with DSS-induced colitis and (iv) MG132-treated (15·0 µmol/kg) mice with DSS-induced colitis (original magnification 100×). (b) Histological scores of MG132-treated (15·0 µmol/kg) mice with DSS-induced colitis. The total colitis score is the sum of the three subscores (inflammation severity, inflammation extent, crypt damage score), which was quantified by the scoring system described by Williams et al. (n = 8 in each group). **P < 0·01 between vehicle-treated and MG132-treated mice with DSS-induced colitis. †P < 0·05 compared with respective normal mice not administered DSS. (c) TNF-α gene expression in the colonic tissues of MG132-treated (15·0 µmol/kg) mice with DSS-induced colitis (n = 8 in each group). *P < 0·05 between vehicle-treated and MG132-treated mice not administered DSS. **P < 0·01 between vehicle-treated and MG132-treated mice with DSS-induced colitis. (d) Immunohistochemical staining with anti-phospho-NF-κB p65 antibody. (i) vehicle-treated mice not administered DSS, (ii) MG132-treated (15·0 µmol/kg) mice not administered DSS, (iii) vehicle-treated mice with DSS-induced colitis and (iv) MG132-treated (15·0 µmol/kg) mice with DSS-induced colitis (original magnification 200×).
On day 5, in both the vehicle-treated and MG132-treated mice administered DSS, the histological findings of the colon revealed epithelial destruction, severe inflammatory cell infiltration and submucosal oedema (data not shown). There was no significant difference in the histological score and TNF-α gene expression in the colonic tissues between the two groups (data not shown).
On day 10, however, in the vehicle-treated mice administered DSS, the colonic mucosa was covered by a monolayer of epithelial cells and crypt regeneration was observed (Fig. 4a, iii). In contrast, in the MG132-treated mice administered DSS, little crypt regeneration was observed, and remarkable inflammatory cell infiltration and submucosal oedema remained (Fig. 4a, iv).
The histological score indicated that DSS administration induced severe colitis in both vehicle-treated and MG132-treated mice and there was no significant difference in the inflammatory severity or inflammatory extent scores between vehicle-treated and MG132-treated mice. On the other hand, both the crypt damage and total colitis scores were significantly higher in MG132-treated mice than in vehicle-treated mice administered DSS (P < 0·01) (Fig. 4b). Although MG132 reduced TNF-α gene expression significantly in the colonic tissues of normal mice (P < 0·05, Fig. 4c), it enhanced TNF-α gene expression significantly in DSS-treated mice (P < 0·01, Fig. 4c) on day 10.
Immunohistochemical staining with anti-phospho-NF-κB p65 antibody
In the vehicle-treated mice not administered DSS, phospho-NF-κB p65 immunohistochemical staining was positive in the nucleus of the epithelial cells of the lower third of the crypts (Fig. 4d, i). On the other hand, in the MG132-treated mice not administered DSS, nuclear translocation of phospho-NF-κB p65 in the epithelial cells was suppressed through the crypts (Fig. 4d, ii).
In the vehicle-treated mice administered DSS, phospho-NF-κB p65 staining was strongly positive in the nucleus of the epithelial cells of the almost entire regenerating crypts (Fig. 4d, iii). In contrast, only a few crypts were observed in the MG132-treated mice administered DSS, and nuclear translocation of phospho-NF-κB p65 in the epithelial cells was not observed at all (Fig. 4d, iv).
Effects of MG132 on intestinal epithelial cells
Immunohistochemical staining with anti-Ki-67 monoclonal antibody
To investigate the effects of MG132 on intestinal epithelial cell proliferation activity in vivo, we performed immunohistochemical staining with the anti-Ki-67 antibody. In C57BL/6 mice, Ki-67-positive cells were located mainly in the lower third of the crypts (Fig. 5a, i). In contrast, in C57BL/6 mice treated with 15·0 µmol/kg MG132, Ki-67-positive cells were located only at the bottom of the crypts (Fig. 5a, ii). The Ki-67 labelling index of MG132-treated mice was significantly lower than that of vehicle-treated mice (P < 0·01, Fig. 5a, iii).
Fig. 5.
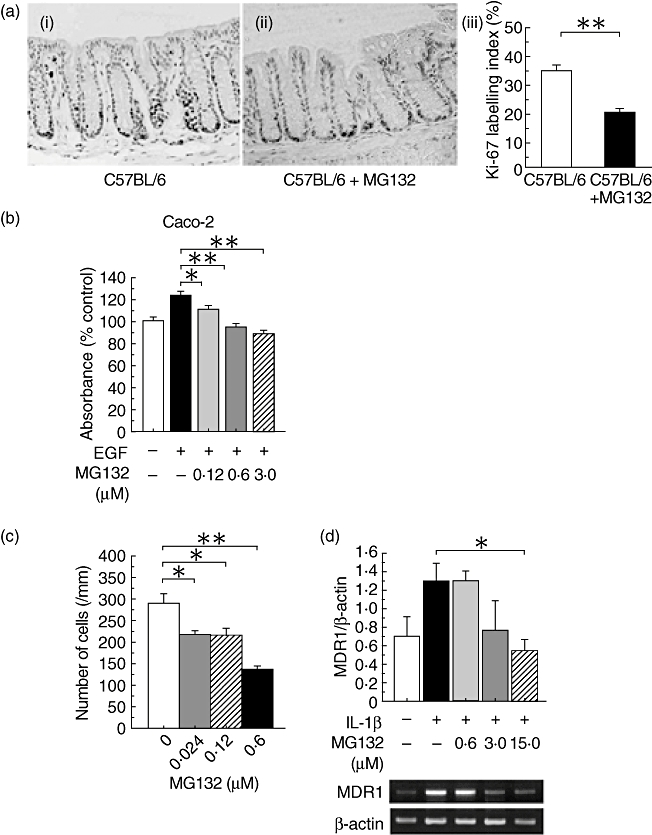
The effects of MG132 on intestinal epithelial cells. (a) Immunohistochemical staining with anti-Ki-67 antibody (i, ii) and the Ki-67 labelling index (iii). (i) Vehicle-treated C57BL/6 mice, (ii) MG132-treated (15·0 µmol/kg) C57BL/6 mice (original magnification 200×). **P < 0·01 between vehicle-treated and MG132-treated C57BL/6 mice (n = 3 in each group). (b) Bromodeoxyuridine (BrdU) cell proliferation assay in Caco-2 cells. *P < 0·05 and **P < 0·01 compared with vehicle-treated cells stimulated with 100 ng/ml human recombinant epidermal growth factor (EGF) (n = 12 in each group). (c) Migration assay using intestinal epithelial cell line (IEC-6) cells. The number of cells crossing 1 mm of the wound 24 h after scraping is shown. *P < 0·05 and **P < 0·01 compared with vehicle-treated cells (n = 6 in each group). (d) Effects of MG132 on multiple drug resistance 1 (MDR1) gene expression in Caco-2 cells. Cells were incubated with MG132 for 1 h and stimulated with 10 ng/ml human recombinant interleukin (IL)-1β for 6 h. Expression of the MDR1 mRNA transcript was determined by semiquantitative polymerase chain reaction. The results are expressed as the relative ratio of MDR1 to β-actin. *P < 0·05 compared with vehicle-treated cells (n = 3 in each group). This experiment was performed three times and similar results were obtained each time.
BrdU cell proliferation assay
We investigated the in vitro effects of MG132 on cell proliferation activity. In Caco-2 cells, MG132 suppressed significantly cell proliferation induced by 100 ng/ml recombinant human EGF in a dose-dependent manner (Fig. 5b).
Migration assay
Migration assay in a scrape-wounded monolayer model with IEC-6 cells revealed that MG132 suppressed cell migration significantly (0·024 and 0·12 µmol/l, P < 0·05; 0·60 µmol/l, P < 0·01; Fig. 5c), which is one of the important factors for epithelial regeneration.
Effect of MG132 on MDR1 gene expression
MG132 suppressed MDR1 gene expression induced by 10 ng/ml human recombinant IL-1β in Caco-2 cells in a dose-dependent manner. MDR1 gene expression in the presence of 15·0 µmol/L MG132 was significantly lower than that in controls (P < 0·05; Fig. 5d).
Discussion
In the pathophysiology of IBD, much attention has been paid to immunological dysregulation of the intestinal mucosa. Indeed, many drugs that suppress immunological responses have been developed for treating IBD [33,34]. NF-κB regulates many genes involved in the pathogenesis of IBD [7] and it is up-regulated markedly in the intestinal mucosa of patients with IBD. Notably, this up-regulation occurs in not only the intestinal epithelial cells but also in lamina propria mononuclear cells in the inflammatory mucosa [14,15,35]. Therefore, the control of NF-κB activation is considered to be one of the promising molecules for treatment of patients with IBD.
Recently, it has been reported that the proteasome-mediated degradation of IκBα and processing of p105, which resulted in NF-κB activation, is involved in the pathophysiology of IBD [22]. In this point, we focused upon treatment with proteasome inhibitor for patients with IBD, which could be one of the promising therapies by controlling NF-κB activation.
Previous reports revealed that NF-κB activation is sustained in inflamed colonic mucosa of IL-10−/− mice [15,36], suggesting that NF-κB activation participates in intestinal inflammation. Interestingly, we observed in the present study that administration of a proteasome inhibitor MG132 clearly ameliorated colonic inflammation of IL-10−/− mice in association with inhibition of TNF-α expression in the colonic mucosa. In addition to its inhibitory effects on colonic inflammation, we also observed that MG132 inhibited the translocation of phospho-NF-κB p65 to the nucleus, and decreased significantly the number of Ki-67 positive cells in the colonic epithelium. We also evaluated the effect of MG 132 on another signalling pathway such as extracellular-signal regulated kinase (ERK), p38 mitogen-activated protein kinase (MAPK) and c-Jun N-terminal kinase (JNK) in colonic epithelial cells. Immunohistochemistry study showed that translocation of phospho-ERK, phospho-p38 MAPK and phospho-JNK into nucleus was not inhibited in colonic epithelial cells of MG132-treated IL-10−/− mice compared with vehicle-treated mice. Taken together, we considered that MG132 treatment is involved in perturbation of NF-κB but is not due to the MAPK and JNK pathways (data not shown). These data suggested that the proteasome-mediated NF-κB activation plays a critical role in intestinal inflammation of IL-10−/− mice.
Next, we investigated the effect of proteasome inhibitor on intestinal inflammation of the DSS-induced colitis model. In mice with DSS-induced colitis there was no significant difference in body weight loss, colon length, histological scores or TNF-α expression in the colonic tissues between MG132-treated mice and control mice during the first few days following DSS administration. The recovery of body weight, however, was delayed significantly in MG132-treated mice and moreover, 10 days after starting DSS administration, colon length was significantly shorter and the histological scores were significantly higher in MG132-treated mice than in control mice in association with up-regulated expression of TNF-α. Further, our in vitro study showed that MG132 inhibited epithelial cell migration in a dose-dependent manner, and the results of both in vivo and in vitro studies indicated that MG132 suppressed intestinal epithelial cell proliferation. In general, because the development of DSS-induced colitis is considered to be due primarily to the injurious effects of DSS on intestinal epithelial cells [37,38], our findings that MG132 disrupted the recovery of DSS-induced colitis appear to be due mainly to the suppression of NF-κB activation in epithelial cells. Taken together, these findings suggest strongly that proteasome-mediated NF-κB activation has an important role in epithelial regeneration after mucosal injury in the DSS-induced colitis model.
In IL-10−/− mice having chronic intestinal inflammation, sustained activation of NF-κB occurred in both epithelial cells and immune cells. In immune-mediated colitis, chronic activation of NF-κB in epithelial cells induces epithelial cell proliferation, which could result histologically in epithelial hyperplasia. On the other hand, in the acute DSS-induced colitis model, NF-κB activation in epithelial cells was involved mainly in epithelial regeneration during the recovery phase. Taken together, NF-κB activation in epithelial cells in chronic intestinal inflammation is considered to play a different role from that in acute intestinal inflammation. As a result, blockade of proteasome-mediated NF-κB activation in epithelial cells ameliorated chronic immune-mediated colitis but not DSS-induced colitis.
Another interesting finding of this study is the effect of MG132 on MDR1 expression. The MDR1 gene encodes P-glycoprotein, a 170-kDa transmembrane protein that belongs to a family of ATP-binding cassette transporters [39]. MDR1 has an important role in cell protection by transporting xenobiotics that enter into the cells [40]. Moreover, targeted disruption of the mdr1a gene in rodents results in the spontaneous development of colitis despite an intact immune system [41]. Accordingly, MDR1 has received great attention recently as a molecule that contributes to intestinal barrier function. In vitro study revealed that MG132 decreased MDR1 gene expression of human colon cancer cell lines in a dose-dependent manner. Supporting our finding, the MDR1 gene has an NF-κB binding site [42]. Taken together, these findings suggest that the impaired recovery of DSS-induced colitis observed in MG132-treated mice might involve decreased mdr1 expression because of NF-κB inhibition with MG132. Thus, P-glycoprotein appears to have an important function in the protection of epithelial cells in the inflamed intestinal mucosa.
In summary, our study revealed that proteasome inhibition resulted in amelioration of colonic inflammation of IL-10−/− mice, but impaired recovery from DSS-induced colitis. Amelioration of colonic inflammation appears to involve the inhibition of immune cell activation by NF-κB inhibition with MG132. On the other hand, delayed recovery of colitis by MG132 might be due to not only impaired epithelial cell regeneration, but also to the disruption of epithelial barrier function associated with decreased mdr1 gene expression. Thus, proteasome-mediated NF-κB activation has dual actions during colonic inflammation: aggravation of inflammation and enhancement of recovery of epithelial injury, depending on the inflammatory conditions.
Acknowledgments
This work was supported by Grant-in-aid for Scientific Research 16017240, 15209024, 15659169, and 18209027 from the Ministry of Education, Culture, Sports, Science and Technology of Japan, and Grant-in-Aid for Research on Measures for Intractable Diseases and Research on Advanced Medical Technology from the Ministry of Health, Labor and Welfare, Japan and the Kato Memorial Trust for Nambyo Research.
References
- 1.Sellon RK, Tonkonogy S, Schultz M, et al. Resident enteric bacteria are necessary for development of spontaneous colitis and immune system activation in interleukin-10-deficient mice. Infect Immun. 1998;66:5224–31. doi: 10.1128/iai.66.11.5224-5231.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kim SC, Tonkonogy SL, Albright CA, et al. Variable phenotype of enterocolitis in interleukin 10-deficient mice monoassociated with two different commensal bacteria. Gastroenterology. 2005;128:891–906. doi: 10.1053/j.gastro.2005.02.009. [DOI] [PubMed] [Google Scholar]
- 3.Sartor RB. Cytokines in intestinal inflammation: pathophysiological and clinical considerations. Gastroenterology. 1994;106:533–9. doi: 10.1016/0016-5085(94)90614-9. [DOI] [PubMed] [Google Scholar]
- 4.Mahida YR, Wu K, Jewell DP. Enhanced production of interleukin 1-β by mononuclear cells isolated from mucosa with active ulcerative colitis of Crohn's disease. Gut. 1989;30:835–8. doi: 10.1136/gut.30.6.835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Reinecker HC, Steffen M, Witthoeft T, et al. Enhanced secretion of tumor necrosis factor-alpha, IL-6, and IL-1 beta by isolated lamina propria mononuclear cells from patients with ulcerative colitis and Crohn's disease. Clin Exp Immunol. 1993;94:174–81. doi: 10.1111/j.1365-2249.1993.tb05997.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Breese EJ, Michie CA, Nicholls SW, et al. Tumor necrosis factor alpha-producing cells in the intestinal mucosa of children with inflammatory bowel disease. Gastroenterology. 1994;106:1455–66. doi: 10.1016/0016-5085(94)90398-0. [DOI] [PubMed] [Google Scholar]
- 7.Schmid RM, Adler G. NF-κB/Rel/IκB: implications in gastrointestinal diseases. Gastroenterology. 2000;118:1208–28. doi: 10.1016/s0016-5085(00)70374-x. [DOI] [PubMed] [Google Scholar]
- 8.Karin M, Ben Neriah Y. Phospholylation meets ubiquitination: the control of NF-kappa B activity. Annu Rev Immunol. 2000;18:621–63. doi: 10.1146/annurev.immunol.18.1.621. [DOI] [PubMed] [Google Scholar]
- 9.Ghosh S, Baltimore D. Activation in vitro of NF-kappa B by phosphorylation of its inhibitor I kappa B. Nature. 1990;344:678–82. doi: 10.1038/344678a0. [DOI] [PubMed] [Google Scholar]
- 10.Henkel T, Machleidt T, Alkalay I, Kronke M, Ben-Neriah Y, Baeuerle PA. Rapid proteolysis of I kappa B-alpha is necessary for activation of transcription factor NF-kappa B. Nature. 1993;365:182–5. doi: 10.1038/365182a0. [DOI] [PubMed] [Google Scholar]
- 11.Ghosh S, Karin M. Missing pieces in the NF-kappa B puzzle. Cell. 2002;109(Suppl):S81–96. doi: 10.1016/s0092-8674(02)00703-1. [DOI] [PubMed] [Google Scholar]
- 12.Hayden MS, Ghosh S. Signaling to NF-kappa B. Genes Dev. 2004;18:2195–224. doi: 10.1101/gad.1228704. [DOI] [PubMed] [Google Scholar]
- 13.Krappmann D, Scheidereit C. A pervasive role of ubiquitin conjugation in activation and termination of Ikappa B kinase pathways. EMBO Rep. 2005;6:321–6. doi: 10.1038/sj.embor.7400380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schreiber S, Nikolaus S, Hampe J. Activation of nuclear factor κB in inflammatory bowel disease. Gut. 1998;42:477–84. doi: 10.1136/gut.42.4.477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Neurath MF, Pettersson S, Meyer zum Buschenfelde KH, Strober W. Local administration of antisense phosphorothioate oligonucleotides to the p65 subunit of NF-κB abrogates established experimental colitis in mice. Nat Med. 1996;2:998–1004. doi: 10.1038/nm0996-998. [DOI] [PubMed] [Google Scholar]
- 16.Ciechanover A. The ubiquitin–proteasome proteolytic pathway. Cell. 1994;79:13–21. doi: 10.1016/0092-8674(94)90396-4. [DOI] [PubMed] [Google Scholar]
- 17.Adams J. The proteasome: a suitable antineoplastic target. Nat Rev Cancer. 2004;4:349–60. doi: 10.1038/nrc1361. [DOI] [PubMed] [Google Scholar]
- 18.Palombella VJ, Rando OJ, Goldberg AL, Maniatis T. The ubiquitin–proteasome pathway is required for processing the NF-kappa B1 precursor protein and the activation of NF-kappa B. Cell. 1994;78:773–85. doi: 10.1016/s0092-8674(94)90482-0. [DOI] [PubMed] [Google Scholar]
- 19.Traenckner EB, Wilk S, Baeuerle PA. A proteasome inhibitor prevents activation of NF-kappa B and stablizes a newly phosphorylated form I kappa B-alpha that is still bound to NF-kappa B. EMBO J. 1994;13:5433–41. doi: 10.1002/j.1460-2075.1994.tb06878.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Alkalay I, Yaron A, Hatzubai A, Orian A, Ciechanover A, Ben-Neriah Y. Stimulation-dependent I kappa B alpha phosphorylation marks the NF-kappa B inhibitor for degradation via the ubiquitin–proteasome pathway. Proc Natl Acad Sci USA. 1995;92:10599–603. doi: 10.1073/pnas.92.23.10599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Elliott PJ, Zollner TM, Boehncke WH. Proteasome inhibition: a new anti-inflammatory strategy. J Mol Med. 2003;81:235–45. doi: 10.1007/s00109-003-0422-2. [DOI] [PubMed] [Google Scholar]
- 22.Visekruna A, Joeris T, Seidel D, et al. Proteasome-mediated degradation of IκBα and processing of p105 in Crohn disease and ulcerative colitis. J Clin Invest. 2006;116:3195–203. doi: 10.1172/JCI28804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lawrence T, Gilroy DW, Colville-Nash PR, Willoughby DA. Possible new role for NF-κB in the resolution of inflammation. Nat Med. 2001;7:1291–7. doi: 10.1038/nm1201-1291. [DOI] [PubMed] [Google Scholar]
- 24.Begue B, Wajant H, Bambou JC, et al. Implication of TNF-related apoptosis-inducing ligand in inflammatory intestinal epithelial lesions. Gastroenterology. 2006;130:1962–74. doi: 10.1053/j.gastro.2006.03.022. [DOI] [PubMed] [Google Scholar]
- 25.Francis NM, Jin W, Wahl SM. Aberrant Toll receptor expression and endotoxin hypersensitivity in mice lacking a functional TGF-β1 signaling pathway. J Immunol. 2004;172:3814–21. doi: 10.4049/jimmunol.172.6.3814. [DOI] [PubMed] [Google Scholar]
- 26.Okayasu I, Hatakeyama S, Yamada M, Ohkusa T, Inagaki Y, Nakaya R. A novel method in the induction of reliable experimental acute and chronic ulcerative colitis in mice. Gastroenterology. 1990;98:694–702. doi: 10.1016/0016-5085(90)90290-h. [DOI] [PubMed] [Google Scholar]
- 27.Williams KL, Fuller CR, Dieleman LA, et al. Enhanced survival and mucosal repair after dextran sodium sulfate-induced colitis in transgenic mice that overexpress growth hormone. Gastroenterology. 2001;120:925–37. doi: 10.1053/gast.2001.22470. [DOI] [PubMed] [Google Scholar]
- 28.Silva HJ, Gatter KC, Millard PR, Kettlewell M, Mortensen NJ, Jewell DP. Crypt cell proliferation and HLA-DR expression in pelvic ileal pouches. J Clin Pathol. 1990;43:824–8. doi: 10.1136/jcp.43.10.824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gratzner HG. Monoclonal antibody to 5-bromo- and 5-iododeoxyuridine: a new reagent for detection of DNA replication. Science. 1982;218:474–5. doi: 10.1126/science.7123245. [DOI] [PubMed] [Google Scholar]
- 30.Lanier TL, Berger EK, Eacho PI. Comparison of 5-bromo-2-deoxyuridine and [3H]-thymidine for studies of hepatocellular proliferation in rodents. Carcinogenesis. 1989;10:1341–3. doi: 10.1093/carcin/10.7.1341. [DOI] [PubMed] [Google Scholar]
- 31.Mccormack SA, Viar MJ, Johnson LR. Migration of IEC-6 cells: a model for mucosal healing. Am J Physiol. 1992;263:G426, 435. doi: 10.1152/ajpgi.1992.263.3.G426. [DOI] [PubMed] [Google Scholar]
- 32.Ciacci C, Lind SE, Podolsky DK. Transforming growth factor beta regulation of migration in wounded rat intestinal epithelial monolayers. Gastroenterology. 1993;105:93–101. doi: 10.1016/0016-5085(93)90014-4. [DOI] [PubMed] [Google Scholar]
- 33.Targan SR, Hanauer SB, van Deventer SJ, et al. A short-term study of chimeric monoclonal antibody cA2 to tumor necrosis factor α for Crohn's disease. N Engl J Med. 1997;337:1029–35. doi: 10.1056/NEJM199710093371502. [DOI] [PubMed] [Google Scholar]
- 34.Ghosh S, Goldin E, Gordon FH, et al. Natalizumab for active Crohn's disease. N Engl J Med. 2003;348:24–32. doi: 10.1056/NEJMoa020732. [DOI] [PubMed] [Google Scholar]
- 35.Rogler G, Brand K, Vogl D, et al. Nuclear factor κB is activated in macrophages and epithelial cells of inflamed intestinal mucosa. Gastroenterology. 1998;115:357–69. doi: 10.1016/s0016-5085(98)70202-1. [DOI] [PubMed] [Google Scholar]
- 36.Ruiz PA, Shkoda A, Kim SC, Sartor RB, Haller D. IL-10 gene-deficient mice lack TGF-β/Smad signaling and fail to inhibit proinflammatory gene expression in intestinal epithelial cells after the colonization with colitogenic Enterococcus faecalis. Gastroenterology. 2005;174:2990–9. doi: 10.4049/jimmunol.174.5.2990. [DOI] [PubMed] [Google Scholar]
- 37.Cooper HS, Murthy SNS, Shah RS, Sedergran DJ. Clinicopathologic study of dextran sulfate sodium experimental murine colitis. Lab Invest. 1993;69:238–49. [PubMed] [Google Scholar]
- 38.Dieleman LA, Ridman BU, Tennyson GS, Beagley KW, Bucy RP, Elson CO. Dextran sulfate sodium-induced colitis occurs in severe combined immunodeficient mice. Gastroenterology. 1994;107:1643–52. doi: 10.1016/0016-5085(94)90803-6. [DOI] [PubMed] [Google Scholar]
- 39.Borst P, Elferink RO. Mammalian ABC transporters in health and disease. Annu Rev Biochem. 2002;71:537–92. doi: 10.1146/annurev.biochem.71.102301.093055. [DOI] [PubMed] [Google Scholar]
- 40.Ho GT, Moodie FM, Satsangi J. Multidrug resistance 1 gene (P-glycoprotein 170): an important determinant in gastrointestinal disease? Gut. 2003;52:759–66. doi: 10.1136/gut.52.5.759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Panwala CM, Jones JC, Viney JL. A novel model of inflammatory bowel disease: mice deficient for the multiple drug resistance gene, mdr1a, spontaneously develop colitis. J Immunol. 1998;161:5733–44. [PubMed] [Google Scholar]
- 42.Kuo MT, Liu Z, Wei Y, et al. Induction of human MDR1 gene expression by 2-acetylaminofluorene is mediated by effectors of the phosphoinositide 3-kinase pathway that activate NF-kappaB signaling. Oncogene. 2002;21:1945–54. doi: 10.1038/sj.onc.1205117. [DOI] [PubMed] [Google Scholar]