

Abstract
Small conductance Ca2+-activated K+ channels (SK channels) have been reported in excitable cells, where they aid in integrating changes in intracellular Ca2+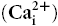 with membrane potential. We have recently reported the functional existence of SK2 channels in human and mouse cardiac myocytes. Moreover, we have found that the channel is predominantly expressed in atria compared to the ventricular myocytes. We hypothesize that knockout of SK2 channels may be sufficient to disrupt the intricate balance of the inward and outward currents during repolarization in atrial myocytes. We further predict that knockout of SK2 channels may predispose the atria to tachy-arrhythmias due to the fact that the late phase of the cardiac action potential is highly susceptible to aberrant excitation. We take advantage of a mouse model with genetic knockout of the SK2 channel gene. In vivo and in vitro electrophysiological studies were performed to probe the functional roles of SK2 channels in the heart. Whole-cell patch-clamp techniques show a significant prolongation of the action potential duration prominently in late cardiac repolarization in atrial myocytes from the heterozygous and homozygous null mutant animals. Morover, in vivo electrophysiological recordings show inducible atrial fibrillation in the null mutant mice but not wild-type animals. No ventricular arrhythmias are detected in the null mutant mice or wild-type animals. In summary, our data support the important functional roles of SK2 channels in cardiac repolarization in atrial myocytes. Genetic knockout of the SK2 channels results in the delay in cardiac repolarization and atrial arrhythmias.
with membrane potential. We have recently reported the functional existence of SK2 channels in human and mouse cardiac myocytes. Moreover, we have found that the channel is predominantly expressed in atria compared to the ventricular myocytes. We hypothesize that knockout of SK2 channels may be sufficient to disrupt the intricate balance of the inward and outward currents during repolarization in atrial myocytes. We further predict that knockout of SK2 channels may predispose the atria to tachy-arrhythmias due to the fact that the late phase of the cardiac action potential is highly susceptible to aberrant excitation. We take advantage of a mouse model with genetic knockout of the SK2 channel gene. In vivo and in vitro electrophysiological studies were performed to probe the functional roles of SK2 channels in the heart. Whole-cell patch-clamp techniques show a significant prolongation of the action potential duration prominently in late cardiac repolarization in atrial myocytes from the heterozygous and homozygous null mutant animals. Morover, in vivo electrophysiological recordings show inducible atrial fibrillation in the null mutant mice but not wild-type animals. No ventricular arrhythmias are detected in the null mutant mice or wild-type animals. In summary, our data support the important functional roles of SK2 channels in cardiac repolarization in atrial myocytes. Genetic knockout of the SK2 channels results in the delay in cardiac repolarization and atrial arrhythmias.
Atrial fibrillation (AF) is one of the most common arrhythmias seen clinically and is associated with a significant increase in morbidity and mortality (Chugh et al. 2001), yet, treatment strategies have so far proven largely inadequate. The mechanisms for AF are highly heterogeneous and are often related to underlying heart or pulmonary diseases. On the other hand, there is growing evidence to implicate cardiac ion channel mutations as possible causes of inherited AF (Brugada et al. 1997; Chen et al. 2003; Ellinor & Macrae, 2003; Hong et al. 2005). Specifically, mutations in several K+ channel genes, KCNQ1, KCNE2, KCNH2 and KCNA5, have been linked to human AF (Chen et al. 2003; Brugada et al. 2004; Yang et al. 2004; Hong et al. 2005; Olson et al. 2006).
We have recently reported the molecular identification and functional existence of Ca2+-activated K+ (KCa) channels in human and mouse cardiac myocytes (Xu et al. 2003; Tuteja et al. 2005; Lu et al. 2007). Indeed, KCa channels are present in most neurons and mediate the afterhyperpolarizations following AP. KCa channels can be divided into three main subfamilies (Kohler et al. 1996; Ishii et al. 1997; Vergara et al. 1998; Bond et al. 1999; Kaczorowski & Garcia, 1999; Bond et al. 2005). These include the large-conductance Ca2+- and voltage-activated K+ channels (BK), the intermediate-conductance KCa channels (IK), and the small-conductance KCa channels (SK), which are sensitive to apamin and scyllatoxin. Among the SK channels, they are encoded by at least three genes, KCNN1 (SK1), KCNN2 (SK2), and KCNN3 (SK3), with differential sensitivity toward apamin (Kohler et al. 1996; Stocker & Pedarzani, 2000).
Specifically, we have shown that the SK2 channel plays a crucial role in human and mouse cardiac repolarization, especially during the late phase of the cardiac action potential (AP) (Xu et al. 2003; Tuteja et al. 2005; Lu et al. 2007). Moreover, there is differential expression of SK2 channels with more abundant expression of SK2 channels in the atria compared to the ventricles (Xu et al. 2003; Tuteja et al. 2005; Lu et al. 2007). Taken together the findings that SK2 channels are predominantly expressed in atria and the roles of the channels in late repolarization, we hypothesize that knockout of SK2 channels may be sufficient to disrupt the intricate balance of the inward and outward currents during repolarization in atrial myocytes. We further predict that genetic knockout of SK2 channels may predispose the atria to tachy-arrhythmias due to the fact that the late phase of cardiac AP is highly susceptible to aberrant excitation, e.g. early afterdepolarization.
To directly test the hypothesis and to investigate the physiological roles of SK2 channels in intact animals, we take advantage of a mouse model with null mutation of SK2 channels (Bond et al. 2004). In addition, there are some questions which remain unsettled. In particular, our previous studies suggest that in contrast to the KCa channels described in neurons, which underlie the afterhyperpolarization, in the heart, the currents contribute mainly towards cardiac repolarization (Xu et al. 2003; Tuteja et al. 2005; Lu et al. 2007). We reason that careful examination of the SK2 null mutant mice will help to distinguish the roles of the SK2 channel during different phases of the cardiac AP. Our previous published study using this mouse model was focused mainly on the roles of the SK2 channel in atrioventricular nodes (Zhang et al. 2008). Here, we focused our study on the roles of SK2 channels in atrial myocytes using in vivo and in vitro electrophysiological studies. Specifically, the present study directly defines the functional roles of SK2 channels in whole animals using a genetically engineered mouse model, and provide the possible link between abnormalities in cardiac SK2 channels and cardiac arrhythmias.
Methods
All animal care and procedures were approved by the University of California, Davis Institutional Animal Care and Use Committee. Animal use was in accordance with National Institutes of Health and institutional guidelines.
SK2 null mutant mice (SK2+/Δ and SK2Δ/Δ mice)
SK2 null mutant mice were generated as previously described (Bond et al. 2004). A single loxP site was introduced into the 5′UTR 300 nucleotides 5′ of the initiator methionine and a cassette consisting of the neomycin-resistance gene flanked by loxP sites, and the coding sequence for enhanced green fluorescent protein (GFP) was inserted into an EcoRV site in intron 2, ∼2.3 kb from the end of exon 2. Recombination at the loxP sites yields an allele that is deleted for SK2 exons 1 and 2. The targeting construct was electroporated into embryonic stem cells (ES cells), and several properly recombined ES cell clones were injected into C57Bl/6 blastocysts. One chimera gave germline transmission of the targeted allele. Crosses between heterozygous-floxed SK2 and the Cre deleter mouse yielded offspring that are heterozygous for the SK2-deleted allele (+/Δ). All mice were housed and kept on a 12 h light/dark cycle with food and water available ad libitum. Mice were weaned at 3 weeks and genotyped. Mutant allele was confirmed using polymerase chain reaction of tail tissue as previously described (Bond et al. 2004). The SK2 null mutant mouse line was backcrossed more than seven generations onto the C57Bl/6J background. The number of surviving homozygous null mutant mice does not strictly obey the Mendelian ratio (5% instead of 25% for a heterozygous cross), which is likely to be due to partial embryonic lethality. There is additional attrition due to perinatal mortality in the newborn homozygous null mutant mice. In addition, the SK2 homozygous null mutant mice show neurological abnormalities with continuous trembling and some animals show circling behaviour. However, detailed neurological assessment was not performed in this study.
Analysis of cardiac function by echocardiography
In order to directly define the effects of the SK2 channel knockout on cardiac function in the whole animals, echocardiograms using M-mode and two-dimensional measurements to assess systolic function were performed as described previously (Xu et al. 2006). The measurements represented the average of six selected cardiac cycles from at least two separate scans performed in random-blind fashion with papillary muscles used as a point of reference for consistency in level of scan. End diastole was defined as the maximal left ventricular (LV) diastolic dimension and end systole was defined as the peak of posterior wall motion. Fractional shortening (FS), a surrogate of systolic function, was calculated from LV dimensions as follows:
where EDD and ESD are LV end diastolic and end systolic dimension, respectively.
Immunofluorescence confocal microscopy
Immunofluorescence labelling was performed as described previously (Xu et al. 2003; Lu et al. 2007). Cells were fixed by 4% paraformadehyde in phosphate-buffered saline (PBS) for 30 min at room temperature, washed with PBS for 5 min ×3, treated with 0.4% Triton X-100 in PBS for 15 min, then washed and treated with −20°C methanol for 10 min. Finally, cells were washed and treated with anti-SK2 antibody (Sigma-Aldrich, 1: 100 dilution) and anti-α-actinin2 antibody (Sigma-Aldrich, 1: 800 dilution).
Immunofluorescence-labelled samples were examined using a Pascal Zeiss confocal laser scanning microscopy. For double staining, secondary antibody conjugated FITC was excited at 488 nm with the Ar laser and detected with a 505–530 nm band pass filter while conjugated Texas Red and Rhodamine were excited at 543 nm with the HeNe1 laser and detected at a 560 nm long pass filter. To ascertain that there were no overlap between the detection of FITC and Texas Red or Rhodamine, singly labelled cells were imaged under identical conditions as those used for dual-labelled probes to confirm proper signal isolation of each channel. Control experiments performed by preincubation of the primary antibody with the respective antigenic peptide (1: 1) did not show positive staining under the same experimental conditions used. Identical settings were used for all the specimens.
Cardiac myocyte isolation
Single mouse atrial and ventricular myocytes were isolated from hetero- and homozygous null mutant and WT animals from the same littermates at 12–14 weeks of age (Sambrano et al. 2002; Xu et al. 2003). Due to the known electrophysiological heterogeneity in various regions of the heart, ventricular myocytes were isolated from left ventricular free wall (LVFW). Briefly, mice were injected with 0.1 ml heparin (1000 units ml−1) 10 min prior to heart excision, then anesthetized with pentobarbital i.p. (80 mg kg−1). Hearts were removed, placed into ice-cold Tyrode solution (mmol l−1: NaCl 140, KCl 5.4, MgCl2 1, Hepes (N-2-hydroxyethylpiperazine-N′-2-ethanesulphonic acid) 10 and glucose 10; pH 7.4 with NaOH), cannulated under a dissecting microscope and mounted on a Langendorff apparatus. Hearts were perfused with Tyrode solution gassed with 100% O2 at 37 °C. The perfusion pressure was monitored and the flow rate (∼2 ml min−1) was adjusted to maintain perfusion pressure at ∼80 mmHg. After 5 min, the solution was switched to 30 ml of Tyrode solution containing 13 mg collagenase (type 2, 322 units mg−1, Worthington) and 1 mg protease (type XIV, 4.5 units mg−1, Sigma-Aldrich). After 30–45 min of enzyme perfusion, hearts were removed from the perfusion apparatus. LVFW or atrial tissue were collected into high-K+ solution (mmol l−1) (potassium glutamate 120, KCl 20, MgCl2 1, EGTA 0.3, glucose 10 and Hepes 10, pH 7.4 with KOH), gently teased using pipettes for 3 min. Cells were allowed to rest for 2 h before use for electrophysiological recording. This isolation procedure yields 60–80% of Ca2+-tolerant atrial and ventricular myocytes with clear striation. Electrophysiological recordings were performed within 8 h after cell isolation.
Action potential (AP) recordings from SK2+/Δ, SK2Δ/Δ and WT animals
APs were recorded at room temperature using the perforated-patch technique (Ahmmed et al. 2000). All other experiments were performed using the conventional whole-cell patch-clamp technique (Hamill et al. 1981) at room temperature.
For AP recordings, the patch-pipettes were back-filled with amphotericin (200 μg ml−1). The pipette solution contained (mm): potassium glutamate 120, KCl 25, MgCl2 1, CaCl2 1, Hepes 10, pH 7.4 with KOH. The external solution contained: NaCl 138, KCl 4, MgCl2 1, CaCl2 2, NaH2PO4 0.33, glucose 10, Hepes 10, pH 7.4 (NaOH).
To directly assess for the increase propensity in the development of early afterdepolarization (EAD), APs were recorded using external solution as indicated above (Control solution) as well as Cs+ solution containing 3 mm of CsCl and 2 mm of KCl (NaCl 138, CsCl 3 KCl 2, MgCl2 1, CaCl2 2, NaH2PO4 0.33, glucose 10, Hepes 10, pH 7.4 (NaOH)) as described previously by Nuss et al. (1999). To quantify the occurrence of EAD, we analysed incidence of EAD as a percentage of cells and frequency of EAD (EADs per AP). Incidence of EAD quantifies the fraction of cells that exhibited EAD, regardless of how often EAD occurred in a particular cell; while frequency of EAD represents the total number of EADs that occur per AP once a steady-state condition is achieved during continuous stimulation. Specifically, a train of 20 APs were stimulated from rest at 1 Hz. The first five APs were used to establish a steady-state condition and these were not analysed. Myocytes in which no EAD had occurred during a train of APs were excluded, so that this analysis quantifies the frequency of EADs only in the subgroup of cells that exhibited EADs.
Ca2+-activated K+ current (IK,Ca) recordings
Whole-cell Ca2+-activated K+ current (IK,Ca) was recorded using conventional voltage-clamp protocol as previously described (Xu et al. 2003). The extracellular solution contained (in mm): N-methylglucamine (NMG) 140, KCl 4, MgCl2 1, glucose 5, and Hepes 10 (pH 7.4 using HCl). The internal solution consisted of (in mm): potassium gluconate 144, MgCl2 1.15, EGTA 5, Hepes 10, and CaCl2 yielding a free (unchelated) [Ca2+] of 500 nmol l−1 using Calcium Titration Software (Robertson & Potter, 1984) to calculate free [Ca2+], bound and dissociated. The pH was adjusted to 7.25 using KOH. All experiments were performed using 3 m KCl agar bridges. Cell capacitance was also calculated as the ratio of total charge (the integrated area under the current transient) to the magnitude of the pulse (20 mV). Currents were normalized to cell capacity to obtain the current density. The series resistance was compensated electronically. In all experiments, a series resistance compensation of ≥90% was obtained. Currents were recorded using Axopatch 200B amplifier (Axon Instruments, Union City, CA, USA), filtered at 10 kHz using a 4-pole Bessel filter and digitized at sampling frequency of 50 kHz. Data analysis was carried out using custom-written software and commercially available PC-based spreadsheet and graphics software (Origin v. 6.0, OriginLab Corp, Northampton, MA, USA).
In vivo electrophysiological studies in mice
In vivo electrophysiological studies were performed as previously described (Berul et al. 1996; Zhang et al. 2005). Standard pacing protocols were used to determine the electrophysiological parameters, including sinus node recovery time (SNRT), atrial, AV nodal, and ventricular effective refractory periods (AERP, AVNERP and VERP, respectively) and AV nodal conduction properties. Each animal underwent an identical pacing and programmed stimulation protocol.
To induce atrial and ventricular tachycardia and fibrillation, programmed extrastimulation techniques and burst pacing were utilized. Programmed right atrial and right ventricular double and triple extrastimulation techniques were performed at 100 ms drive cycle length, down to a minimum coupling interval of 10 ms. Right atrial and right ventricular burst pacing were performed as eight 50 ms and four 30 ms cycle length train episodes repeated several times, up to a maximum 1 min time limit of total stimulation. For comparison of the inducibility in each mouse, programmed extrastimulation techniques and stimulation duration of atrial and ventricular burst pacing were the same in all mice. Reproducibility was defined as greater than one episode of induced atrial or ventricular tachycardia.
Adenoviral construction
The full-length PCR amplified fragment of mouse cardiac SK2 channel was subcloned into TA vector as previously described (Invitrogen, Carlsbad, CA, USA). The dominant-negative (DN) construct of the mouse cardiac SK2 channel (SK2-DN) was generated using QuickChange™ Site-Directed Mutagenesis (Stratagene, La Jolla, CA, USA) to mutate the signature sequence (GYG) in the pore of the SK channel into alanine (GYG → AAA). All the clones were sequence verified.
A recombinant adenovirus containing SK2-DN channel (Ad-SK2-DN) was generated using the shuttle vector (pAdTrack–cytomegalovirus (CMV)) and pAdEasy-1 system (Stratagene). The plasmid also contains a reporter gene, GFP under the control of a separate CMV promoter. The titres of the viruses were assessed using plaque assay. A recombinant adenovirus containing GFP only was used as a control for our experiments.
Culture of adult mouse cardiac myocytes and adenoviral infection
Freshly isolated cardiac myocytes were allowed to sediment by gravity for 8–10 min and resuspended in modified Tyrode solution containing (mmol l−1): NaCl 113, KCl 4.7, KH2PO4 0.6, Na2HPO4, 0.6, MgSO4 1.2, NaHCO3 12, KHCO3 10, Hepes 10, taurine 30 and adjusted to a total volume of 10 ml containing 5% serum with the final Ca2+ concentration of 12.5 μm. The Ca2+ concentration was slowly raised using CaCl2 to a final concentration of 1 mm. All steps were conducted at room temperature.
The entire culture procedure was performed in a class II flow hood. Culture dishes were precoated for 1 h with 10 μg ml−1 mouse laminin (Invitrogen) in phosphate-buffered saline (PBS) with 1% penicillin–streptomycin (PS; Invitrogen) at room temperature. Cardiac myocytes were suspended in minimal essential medium (MEM; Sigma M1018) containing 1.2 mm Ca2+, 2.5% preselected fetal bovine serum (FBS; Invitrogen), and 1% PS (pH 7.35–7.45). After the myocytes were pelleted by gravity for ∼10 min, the supernatant was aspirated and the myocytes were washed two more times using the same protocol. The myocytes were then plated at 0.5–1 × 104 cells cm−2 in MEM containing 2.5% FBS and 1% PS. After 1 h in culture (5% CO2 incubator at 37°C), the medium was changed to FBS-free MEM.
Adenovirus-mediated gene transfer was implemented after 1 h of culture to achieve myocyte attachment. The culture medium was aspirated along with unattached myocytes, and a half-volume (e.g. 1 ml for a 35 mm petri dish) of the FBS-free MEM containing an appropriate titre of gene-carrying adenovirus was added to the dish. Another half-volume of the FBS-free MEM was added following an additional 1–2 h of culture.
Statistical analysis
IK,Ca density obtained from atrial myocytes among different groups of animals was compared by normalizing the currents with cell capacitance. Where appropriate, pooled data are presented as means ±s.e.m. Multiple comparisons were performed using one-way analysis of variance combined with Dunnett's test (SigmaStat, Systat Software Inc., San Jose, CA, USA). The null hypothesis was rejected when a two-tailed P value was < 0.05. Atrial arrhythmia inducibility was assessed using Fisher's exact test.
Results
SK2 null mutant mice
Figure 1A compared H&E histologic sections of WT, SK2+/Δ, and SK2Δ/Δ hearts. There was no evidence of cardiac abnormality in the heterozygous or homozygous null mutant animals. Fig. 1B shows examples of the M-mode echocardiogram obtained from the knockout animals compared to a WT littermate. Knockout of SK2 channel did not significantly change left ventricular systolic function. Summary data for the fractional shortening, end-systolic and end-diastolic dimensions (ESDs and EDDs) are shown in Table 1.
Figure 1. SK2+/Δ and SK2Δ/Δ mice.

A, histological sections (H&E-staining) of hearts obtained from WT, SK2+/Δ and SK2Δ/Δ mice. All histological sections are presented with the atria on top and the right ventricle to the left. Scale bar, 100 μm. B, examples of M-mode echocardiography in WT, SK2+/Δ and SK2Δ/Δ mice. SK2+/Δ and SK2Δ/Δ mice show no evidence of cardiac abnormalities with normal chamber size and cardiac function. Fractional shortening (FS) is summarized in Table 1. C, confocal photomicrographs of atrial myocytes isolated from WT, SK2+/Δ and SK2Δ/Δ mice. Cells were treated with anti-SK2 and anti-α-actinin2 antibodies. Immunofluorescence labelling was performed by treatment with secondary antibodies (fluorescein isothiocyanate-conjugated and Texas Red conjugated). The specificity of labelling was confirmed by elimination of immunoreactivity after preincubation of the primary antibody with the respective antigenic peptide (1: 1) labelled as control. The scale bar is 20 μm.
Table 1.
Echocardiographic studies in SK2+/Δ and SK2Δ/Δ mice compared to wild-type (WT) littermates
| WT (n= 12) | SK2+/Δ (n= 15) | SK2Δ/Δ (n= 10) | |
|---|---|---|---|
| LVEDD (cm) | 0.25 ± 0.02 | 0.29 ± 0.04 | 0.27 ± 0.02 |
| LVESD (cm) | 0.09 ± 0.01 | 0.12 ± 0.03 | 0.11 ± 0.01 |
| LVPWD (cm) | 0.08 ± 0.01 | 0.09 ± 0.02 | 0.05 ± 0.01 |
| LVPWS (cm) | 0.13 ± 0.01 | 0.13 ± 0.01 | 0.11 ± 0.01 |
| FS (%) | 65 ± 3 | 63 ± 4 | 62 ± 4 |
Data shown represent mean ±s.e.m.; LVEDD and LVESD, left-ventricular end-diastolic and end-systolic dimension, respectively; LVPWD and LVPWS, left-ventricular posterior wall thickness in end diastole and end systole, respectively; FS, fractional shortening; n refers to the number of the animals in the studies.
We have previously published a detail study of the localization of the SK2 channel in atrial and ventricular myocytes (Lu et al. 2007). However, to directly document the lack of SK2 channels expression in the homozygous null mutant model, we performed immunofluorescence confocal microscopy as presented in Fig. 1C. Confocal scanning immunofluorescence microscopy was performed using anti-SK2 and anti-α-actinin2 antibodies in single atrial myocytes isolated from SK2Δ/Δ compared to WT illustrating lack of immunoreactivity to SK2 antibodies in atrial myocytes isolated from SK2Δ/Δ mice. Control experiments are shown using antibodies pretreated with antigenic peptides. Same settings were used for all experiments.
Action potentials recorded from atrial myocytes are prolonged in SK2+/Δ and SK2Δ/Δ mutant mice compared to WT littermates
We have previously documented the expression of SK2 channels predominantly in human and mouse atrial myocytes (Xu et al. 2003). To further test the functional roles of SK2 channels in atrial myocytes, we recorded APs from single atrial myocytes isolated from SK2+/Δ and SK2Δ/Δ mice and compared to their littermate controls (Fig. 2A and B). Figure 2A shows AP prolongation in SK2Δ/Δ mice compared to WT littermates. Summary data are shown in Fig. 2B. AP durations at 50 and 90% repolarization (APD50 and APD90) were significantly prolonged in atrial myocytes isolated from SK2+/Δ mice compared to their wild-type littermates. Moreover, APD were further prolonged in homozygous mutant mice compared to heterozygous animals consistent with a significant role of SK2 channel in the repolarization of the atrial myocytes. The degree of APD prolongation was more pronounced during late cardiac repolarization. In contrast, no significant changes were detected in the APD50 or APD90 for ventricular myocytes from heterozygous or homozygous mutant mice compared to the WT littermates (Fig. 2C and D). In addition, our previous data have shown that apamin significantly prolonged APD in atrial myocytes from WT mice (Xu et al. 2003). Therefore, we tested the effect of apamin (500 pm) on the APD in atrial myocytes isolated from SK2Δ/Δ animals. In contrast to the atrial myocytes from WT animals, there were no significant effects of apamin on APD (Fig. 2E). APD50 and APD90 in atrial myocytes isolated from SK2Δ/Δ mice are 24.7 ± 2.1 and 235 ± 19 ms at baseline and 23.8 ± 1.8 and 232 ± 20 after application of apamin, respectively (apamin 500 pm, NS, n= 9).
Figure 2. Action potentials recorded from atrial myocytes are prolonged in SK2+/Δ and SK2Δ/Δ mice.

A, examples of APs recorded using perforated patch-clamp techniques from atrial myocyte isolated from WT (continuous line), SK2+/Δ (dashes line) and SK2Δ/Δ mice (dotted line). B, summary data showing significant prolongation of APD50 and APD90 in SK2+/Δ mice compared to WT and SK2Δ/Δ compared to SK2+/Δ or WT (*P < 0.05). C, examples of APs recorded using perforated patch-clamp techniques from ventricular myocyte isolated from WT (continuous line), SK2+/Δ (dashes line) and SK2Δ/Δ mice (dotted line). D, summary data showing no significant differences in APD50 and APD90 in SK2+/Δ mice compared to WT and SK2Δ/Δ compared to SK2+/Δ or WT. E, example of action potentials recorded from atrial myocytes isolated from SK2Δ/Δ animals before and after application of apamin (500 pm).
Finally, there were no significant changes in the resting membrane potentials in the atrial or ventricular myocytes from heterozygous or homozygous mutant animals compared to WT littermates. Resting membrane potentials (RMPs) for atrial myocytes from WT, SK2+/Δ and SK2Δ/Δ mice were −71 ± 1, −72 ± 2 and −72 ± 1 mV, respectively (NS, n= 10) while RMPs for ventricular myocytes from WT, SK2+/Δ and SK2Δ/Δ mice were −82 ± 4, −79 ± 4 and −83 ± 4 mV, respectively (NS, n= 9–13).
Occurrences of early afterdepolarization (EAD)
In order to test whether the documented prolongation in APs in SK2 null mutant mice will result in an increase in the occurrences of early afterdepolarization and possibly contribute to cardiac arrhythmias, we recorded APs from atrial myocytes isolated from WT and SK2Δ/Δ animals using control solution as well as Cs+ solution (containing 3 mm CsCl and 2 mm KCl) (Fig. 3). Using control solution, EADs were observed in 14% of the atrial myocytes isolated from SK2Δ/Δ mice (n= 5 of 36 cells) but in none in the WT cells studied (n= 18, P < 0.01, Fisher's exact test). Exposure to Cs+ solution further provoked EADs resulting in an increase in the occurrences of EAD in both groups; however, EAD incidence remained significantly higher in SK2Δ/Δ myocytes (84%, n= 16 of 19 cells) compared with normal myocytes (32%, n= 8 of 25 cells, P < 0.01, Fisher's exact test). We further quantified the frequency of EAD (EADs per AP) in the subgroup of myocytes which exhibited EADs (Fig. 3F). Atrial myocytes isolated from SK2Δ/Δ mice showed an increase in the frequency of EADs in both control (0.34 ± 0.04, n= 5) and Cs+ solution (0.66 ± 0.03, n= 16) compared to WT myocytes (0 and 0.25 ± 0.07, n= 8, respectively, P < 0.01, t-test).
Figure 3. Representative recordings of steady-state action potentials from atrial myocytes isolated from WT and SK2Δ/Δ mice and quantification of occurrences of EADs.

A–D, steady-state action potentials (AP) recorded using stimulation frequency of 1 Hz in atrial myocytes isolated from WT (A and C) and SK2Δ/Δ mice (B and D) during perfusion with Control solution (A and B) and Cs+ solution (C and D). Arrows show occurrences of EADs. E, incidence of EADs (% of cells) in Control (left panel) compared to Cs+ solution (right panel). F, frequency of EADs (EADs per AP) in Control (left panel) compared to Cs+ solution (right panel).
IK,Ca is decreased in atrial myocytes isolated from SK2+/Δ and absent in SK2Δ/Δ mice
In order to further document the basis for the alteration in cardiac AP observed in the mutant animals, we examined whole-cell IK,Ca from single atrial myocytes from heterozygous and homozygous mutant mice and compared to WT controls. Figure 4A shows examples of current traces recorded from atrial myocytes isolated from WT, SK2+/Δ, and SK2Δ/Δ, respectively. Apamin-sensitive current (IK,Ca) was obtained from subtraction of total current recorded in control and after application of a low concentration of apamin (200 pmol l−1). Atrial myocytes isolated from SK2+/Δ showed a significant decrease in the IK,Ca density compared to the WT (Fig. 4A). Moreover, apamin-sensitive current was absent in atrial myocytes isolated from SK2Δ/Δ homozygous mutant animals. Summary data of the current density–voltage relations from the three different groups are shown in Fig. 4B (*P < 0.05, n= 10–12 cells for each group).
Figure 4. Whole-cell IK,Ca recordings.

A, examples of whole-cell IK,Ca recorded from atrial myocytes using voltage steps from a holding potential of −55 mV from WT, SK2+/Δ and SK2Δ/Δ mice. The current was recorded in control and after apamin (50 pmol l−1) and [Ca2+]i was 500 nmol l−1. The voltage protocol used is shown above the current traces. The apamin-sensitive current traces were obtained using digital subtraction and are shown in the right panel (note the change in the scale bars). B, summary of the current density–voltage relation of the apamin-sensitive current (*P < 0.05).
Functional roles of SK2 channel in intact heart assessed using SK2 null mutant mice
Our previous data on the regional localization of SK2 channel transcript using in situ hybridization and functional studies using patch-clamp recordings are consistent with expression of SK2 channel mainly in atria (Xu et al. 2003; Tuteja et al. 2005). However, the functional roles for the differential expression of the SK2 channel in intact animals are completely unknown. We reason that SK2 null mutant mice will provide an ideal model to study the functional roles of the SK2 channel in atria in intact animals. We undertook in vivo electrophysiological studies comparing heterozygous, homozygous and WT animals. Homozygous null mutant mice show evidence of sinoatrial (SA) and atrioventricular (AV) node dysfunction as assessed by a significant prolongation of the sinus cycle length (SCL), corrected SNRT, Wenckebach cycle length (WCL) and AVNERP (see Table 2). Indeed, we have recently documented abnormalities in the AV node in this mutant model (Zhang et al. 2008).
Table 2.
In vivo electrophysiological studies in SK2+/Δ and SK2Δ/Δ mice compared to wild-type (WT) littermates
| WT (n= 10) | SK2+/Δ (n= 11) | SK2Δ/Δ (n= 8) | |
|---|---|---|---|
| SCL (ms) | 135 ± 8 | 155 ± 6 | 181 ± 9* |
| PR interval (ms) | 35 ± 2 | 40 ± 2 | 42 ± 3 |
| CSNRT (ms) | 50 ± 7 | 53 ± 8 | 75 ± 10* |
| WCL (ms) | 90 ± 1 | 102 ± 1* | 119 ± 2* |
| AVNERP (ms) | 65 ± 2 | 72 ± 3 | 80 ± 3* |
| AERP (ms) | 53 ± 2 | 56 ± 4 | 60 ± 6 |
| VERP (ms) | 37 ± 3 | 38 ± 2 | 41 ± 4 |
| Atrial arrhythmias | 0/10 | 5/11* | 7/8* |
| Ventricular arrhythmias | 0/10 | 0/11 | 0/8 |
Data shown represent mean ±s.e.m.; SCL, sinus cycle length, CSNRT, corrected sinus node recovery time (SNRT-SCL); WCL, Wenckebach cycle length; AVNERP, AERP and VERP refer to the effective refractory period for the atrioventricular node, atria and ventricles, respectively; AVNERP, AERP and VERP were performed using basic cycle-length of 120 ms; n refers to the number of the animals in the studies;
P < 0.05 comparing to WT.
Furthermore, atrial arrhythmias, mainly atrial fibrillation were induced in a significant number of heterozygous and homozygous animals. In contrast, atrial arrhythmias were induced in none of the wild-type littermates (P < 0.01 comparing wild-type and mutant animals using Fisher's exact test). Indeed, previous studies using the same background mouse model have shown that WT mice were not inducible for atrial arrhythmias in the absence of carbachol (Kovoor et al. 2001). Figure 5 shows an example of atrial fibrillation induced in SK2Δ/Δ mice (Fig. 5A) as well as evidence of type I second degree AV block in SK2Δ/Δ mice during sinus rhythm with typical group beating (Fig. 5B). In contrast, ventricular arrhythmias were not induced in either the homozygous or heterozygous null mutant animals, consistent with the notion that the channel is mainly expressed in atrial tissues. The in vivo electrophysiological parameters are summarized in Table 2.
Figure 5. Functional roles of SK2 channel in intact heart assessed using SK2 null mutant mice.

A, in vivo electrophysiological studies in SK2Δ/Δ mice showing evidence of inducible atrial fibrillation using atrial extrastimuli. Upper tracings are surface ECG (Lead I, aVF and III). Lower tracings are intracardiac electrograms showing atrial and ventricular electrograms with induced sustained rapid atrial fibrillation with relatively slow ventricular response. B, evidence of type I second degree AV block in SK2Δ/Δ mice during sinus rhythm.
AP shortening has previously been shown to predispose to AF in a large number of models (Nattel et al. 2007). On the other hand, the SK2 channel null mutant mouse model shows an increase in APD in atrial myocytes which was associated with an increase in AF inducibility. To further understand the cellular mechanisms contributing to the increase in propensity to AF, we documented a significant increase in the occurrences of early afterdepolarization (EAD) in atrial myocytes isolated from the homozygous null mutant mice compared to the WT animals (Fig. 3).
Recombinant adenovirus containing SK2-DN channel
As an additional test for the functional roles of SK2 channel in mouse atrial myocytes, we tested the effects of adenoviral mediated expression of SK2-DN channel in mouse atrial myocytes. K+ channels consist of four pore-forming subunits (α subunits) that co-assemble to form a functional channel together with smaller β subunits (Nerbonne & Kass, 2005). Previous studies have identified numerous K+ channel mutations that are capable of crippling channel activities in a dominant-negative manner when normal and defective subunits co-assemble to form multimeric complexes, most notably, mutations in KvLQT1 and HERG K+ channel causing congenital long QT syndrome (Keating & Sanguinetti, 2001). This four-subunit design for K+ channels can be utilized to generate a functional knockout of various K+ channels. Subunits can be generated that contain mutations in their pore forming region that render them incompetent to conduct current. The resulting subunit (DN subunit), when co-assembled with wild-type (WT) subunits, results in non-functional channels. Thus, over-expression of DN subunits results in a functional knockout of K+ current. Mutation of three amino acids (GYG → AAA) which are absolutely conserved in the pore-forming region of all known K+ channels has been successfully used to create a DN subunit (Tinker et al. 1996). The same signature sequence (GYG) is also conserved in all SK channels. Here, we utilized the same triple mutation GYG → AAA for functional knock-out strategy.
First, we directly document that the SK2-DN construct can indeed lead to a DN functional knockout of the WT current. CHO cells were transduced with Ad-SK2 or Ad-SK2-DN or in combination. GFP-positive cells were studied after 36–48 h of infection. Figure 6A shows an example of IK,Ca recorded in CHO cells transduced with adenovirus containing mouse cardiac SK2 channel. Recording conditions were the same as described in Fig. 4 with [Ca2+]i of 500 nm. The mutant channel (SK2-DN) was non-conductive (Fig. 6B). Furthermore, co-expression of the WT and mutant subunits resulted in DN suppression of the WT current (Fig. 6C). Summary data are shown in Fig. 6D.
Figure 6. Transduction of mouse atrial myocytes using recombinant adenovirus containing SK2-DN channel (Ad-SK-DN).

IK,Ca expressed in CHO cells. Current traces in CHO cells expressing WT SK2 channel (A) compared to SK2-DN channel (B). C, co-expression of the WT and mutant subunits resulted in the DN suppression of the WT current. D, summary data for the current density-voltage relations showing the DN effects of the WT current by SK2-DN construct. E, confocal photomicrographs showing expression of the green fluorescent protein (GFP) in cardiac myocyte culture 2 days after adenovirus infection. F, control experiments are shown (non-infected cells). The corresponding differential interference contrast (DIC) image is shown in the right panel. G, representative examples of AP recordings from mouse atrial myocytes transduced by Ad-SK2-DN compared to control cells (Ad-GFP). H, summary data for APD50 and APD90 comparing the two groups of cells, *P < 0.05. I, summary data for the resting membrane potential in the two groups of cells, P = NS.
Next, freshly isolated adult mouse cardiomyocytes from wild-type C57Bl/6J mice were transduced with Ad-GFP or Ad-SK2-DN. The transduction efficiency was determined using GFP as the reporter gene. As shown in Fig. 6E, more than 90% of cardiomyocytes can be transduced at 48 h. To directly examine the functional role of IK,Ca in cardiac myocytes, perforated patch-clamp techniques were used to record APs at 48 h after viral infection. Figure 6G shows examples of APs recorded from cells infected with Ad-SK2-DN compared to Ad-GFP. A significant prolongation of the terminal portion of the AP was observed in the cells infected with Ad-SK2-DN compared to Ad-GFP alone. Summary data (Fig. 6H) for action potential duration at 50% and 90% repolarization (APD50 and APD90) show significant differences in the AP duration in the cells infected by Ad-SK2-DN compared to Ad-GFP. No significant differences in the resting membrane potential (RMP) were noted in the two groups of myocytes.
Discussion
In this study, we directly tested the role of the SK2 channel in atrial myocytes as well as in the intact animals using a SK2 null mutant mouse model. The SK2+/Δ and SK2Δ/Δ mice showed normal cardiac chamber size and function as assessed by echocardiography with no evidence of structural cardiac abnormalities. On the other hand, APD was significantly prolonged especially during late repolarization in atrial myocytes isolated from SK2+/Δ and SK2Δ/Δ mice. In contrast, there were no significant differences in the APD in ventricular myocytes in the mutant mice compared to WT littermates. The findings are consistent with our previous published data that SK2 channels are highly expressed in atrial myocytes. On the other hand, our previous data did document the expression of SK2 channels in mouse ventricular myocytes, albeit at a much lower level than atrial myocytes (Xu et al. 2003; Lu et al. 2007). In the present study, APD from ventricular myocytes isolated from homozygous null mutant mice showed a small trend towards an increase over the WT animals; however, the differences were not statistically significant. The reasons for such discrepancies are likely to be secondary to the difficulty in establishing small differences when there are normal variations in APD observed in isolated cells.
To further document the differential expression level of the SK2 channel among the different groups of mice, we directly recorded IK,Ca using whole-cell patch-clamp techniques. Examination of Fig. 4 shows a significant difference in the apamin-sensitive current in the WT animals only at potentials more positive than 40 mV and less than −80 mV. The current was present at potentials positive to −80 mV; however, because of the cell to cell variation, there was no statistical significance when one compared to those recorded from the null mutant mice. Indeed, APD recorded from atrial myocytes in Fig. 2A shows significant increase in APD both at 50 and 90% repolarization in heterozygous and homozygous null mutant mice.
Finally, using in vivo electrophysiological recordings to examine the roles of the SK2 channel in intact animals, we demonstrated that knockout of the SK2 channel resulted in the occurrence of atrial arrhythmias associated with prolongation of the AVN conduction. Our in vivo electrophysiological data as well as patch-clamp recordings are consistent with the notion that SK2 channels are expressed in and contribute functionally and importantly to atrial myocytes. Indeed, this mutant mouse model represents one of the few genetic models of atrial fibrillation. Additionally, we tested the roles of the SK2 channel in isolated atrial myocytes using the SK2-DN construct. Transduction of the SK2-DN construct in mouse atrial myocytes resulted in the prolongation of the cardiac AP. In contrast to the Ca2+-activated K+ channels (KCa) described in neurons, which underlie the afterhyperpolarization, in the heart the currents contribute markedly towards the late phase of the cardiac repolarization. No significant changes in the resting membrane potentials were observed in the heterozygous or homozygous null mutant model or atrial myocytes transduced with the SK2-DN construct. Even though SK2 current represents only a small component of the total repolarization currents, the importance of the currents is underpinned by the fact that the late phase of the cardiac AP is susceptible to aberrant excitation, e.g. early after depolarization and arrhythmias. Our data showing the development of AF in the SK2 null mutant mice suggest a possible link between SK2 channel and atria arrhythmias. Indeed, recent study by Ozgen et al. have provided new exciting evidence that burst pacing may induce SK2 channel trafficking to the membrane leading to atrial electrical remodelling and provide a possible basis for an arrhythmogenic substrate (Ozgen et al. 2007). Nonetheless, the relevance of the SK2 channel in human AF remains only speculative at this time. Future studies are required to further assess this important question.
Atrial fibrillation (AF)
AF represents one of the most common atrial arrhythmias, and is associated with a significant risk of embolism and stroke. In addition, AF remains one of the most challenging arrhythmias to treat since treatment strategies have proven largely inadequate. The underlying mechanisms for AF are highly heterogeneous and are often related to underlying heart or pulmonary diseases. However, there is growing evidence showing that genetic factors are important in the pathogenesis of AF. Moreover, recent studies have identified mutations in several ion channel genes as possible causes of inherited AF (Brugada et al. 1997; Chen et al. 2003; Ellinor & Macrae, 2003; Ellinor et al. 2003; Hong et al. 2005). Specifically, in 1997, Brugada et al. identified the first locus for familial atrial fibrillation on chromosome 10q22–24. Since then, four relevant genes have been identified that encode K+ channel subunits (Lai et al. 2003; Brugada, 2005; Wiesfeld et al. 2005; Roberts, 2006). Indeed, exciting new evidence is accumulating to support the notion that AF can be a result of an ion channelopathy. Mutations in the K+ channel genes have been shown to result in a gain-of-function. The gain-of-function mutations are consistent with the decrease in APD and atrial refractory period which are thought to be the mechanisms for AF.
On the other hand, recent studies have demonstrated that at least two loss-of-function mutations in K+ channels are associated with clinical AF. A nonsense mutation in the KCNA5 gene that encodes the pore forming subunit, Kv1.5, a voltage-gated K+ channel expressed in human atria (Olson et al. 2006), has been identified which has been linked to human AF. The mutation results in a premature stop codon and a truncated Kv1.5 channel protein. It was demonstrated in the study that the loss-of-function mutation translated into AP prolongation and early after-depolarization (EAD) in human atrial myocytes. An additional study by Ehrlich et al. (2005) examined the electrophysiological effects of a single nucleotide polymorphism (SNP, A/G) at position 112 in the KCNE1 gene (encoding minK protein), resulting in a glycine/serine amino acid substitution at position 38 of the minK peptide (Ehrlich et al. 2005). The minK38G isoform was previously shown to be associated with AF occurrence. The minK38G isoform was found to be associated with a reduced slowly activating delayed rectifier K+ current (IKs). Moreover, it was suggested that the loss-of-function mutation may result in a prolongation of cardiac AP and occurrences of EAD under conditions of reduced repolarization reserve. Alternatively, it was suggested that the induction of electrical alternans may occur in the presence of minK38G (Ehrlich et al. 2005).
From the discussion above, it appears that both gain-of-function and loss-of-function mutations in different K+ channel genes among different families can result in AF from multiple mechanisms. One of the well documented cellular mechanisms for AF is the shortening of atrial refractory period as described above. However, our present study suggests that prolongation of the atrial APDs may also contribute to atrial arrhythmias but via different mechanisms, namely EAD. One additional recent study linking atrial stretch and KCNQ1 mutation in a family with AF (Otway et al. 2007) added yet another layer of complexity to the pathogenesis of AF.
Taken together, our study supports the important functional roles of SK2 channel in cardiac atrial myocytes. Moreover, null mutation of the SK2 channel resulted in an increase in the atrial AP which was associated with an increased inducibility for atrial arrhythmias possibly via an increase in the occurrences of EAD. The findings may have important clinical implications since pharmacological inhibitors of K+ channels are typically used to treat arrhythmias and may potentially contribute to relevant pro-arrhythmia.
Finally, the significance of our findings may go beyond its novelty, because the KCa channel is differentially expressed in atria vs ventricles, and the use of specific ligands for SK channels may offer a unique therapeutic opportunity to directly modify the atrial cells without interfering with ventricular myocytes. This may have important therapeutic implications in treatment of patients with atrial arrhythmias, common problems encountered in our patient population.
Acknowledgments
This work was supported by NIH/HL75274, HL85844, HL85727 and the VA Merit Review Grant (NC) and NIH grants to JPA and MP. The authors are in debt to Dr. E. N. Yamoah for helpful suggestions and comments.
References
- Ahmmed GU, Dong PH, Song G, Ball NA, Xu Y, Walsh RA, Chiamvimonvat N. Changes in Ca2+ cycling proteins underlie cardiac action potential prolongation in a pressure-overloaded guinea pig model with cardiac hypertrophy and failure. Circ Res. 2000;86:558–570. doi: 10.1161/01.res.86.5.558. [DOI] [PubMed] [Google Scholar]
- Berul CI, Aronovitz MJ, Wang PJ, Mendelsohn ME. In vivo cardiac electrophysiology studies in the mouse. Circulation. 1996;94:2641–2648. doi: 10.1161/01.cir.94.10.2641. [DOI] [PubMed] [Google Scholar]
- Bond CT, Herson PS, Strassmaier T, Hammond R, Stackman R, Maylie J, Adelman JP. Small conductance Ca2+-activated K+ channel knock-out mice reveal the identity of calcium-dependent afterhyperpolarization currents. J Neurosci. 2004;24:5301–5306. doi: 10.1523/JNEUROSCI.0182-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bond CT, Maylie J, Adelman JP. Small-conductance calcium-activated potassium channels. Ann N Y Acad Sci. 1999;868:370–378. doi: 10.1111/j.1749-6632.1999.tb11298.x. [DOI] [PubMed] [Google Scholar]
- Bond CT, Maylie J, Adelman JP. SK channels in excitability, pacemaking and synaptic integration. Curr Opin Neurobiol. 2005;15:305–311. doi: 10.1016/j.conb.2005.05.001. [DOI] [PubMed] [Google Scholar]
- Brugada R. Is atrial fibrillation a genetic disease? J Cardiovasc Electrophysiol. 2005;16:553–556. doi: 10.1111/j.1540-8167.2005.40703.x. [DOI] [PubMed] [Google Scholar]
- Brugada R, Hong K, Dumaine R, Cordeiro J, Gaita F, Borggrefe M, Menendez TM, Brugada J, Pollevick GD, Wolpert C, Burashnikov E, Matsuo K, Wu YS, Guerchicoff A, Bianchi F, Giustetto C, Schimpf R, Brugada P, Antzelevitch C. Sudden death associated with short-QT syndrome linked to mutations in HERG. Circulation. 2004;109:30–35. doi: 10.1161/01.CIR.0000109482.92774.3A. [DOI] [PubMed] [Google Scholar]
- Brugada R, Tapscott T, Czernuszewicz GZ, Marian AJ, Iglesias A, Mont L, Brugada J, Girona J, Domingo A, Bachinski LL, Roberts R. Identification of a genetic locus for familial atrial fibrillation. N Engl J Med. 1997;336:905–911. doi: 10.1056/NEJM199703273361302. [DOI] [PubMed] [Google Scholar]
- Chen YH, Xu SJ, Bendahhou S, Wang XL, Wang Y, Xu WY, Jin HW, Sun H, Su XY, Zhuang QN, Yang YQ, Li YB, Liu Y, Xu HJ, Li XF, Ma N, Mou CP, Chen Z, Barhanin J, Huang W. KCNQ1 gain-of-function mutation in familial atrial fibrillation. Science. 2003;299:251–254. doi: 10.1126/science.1077771. [DOI] [PubMed] [Google Scholar]
- Chugh SS, Blackshear JL, Shen WK, Hammill SC, Gersh BJ. Epidemiology and natural history of atrial fibrillation: clinical implications. J Am Coll Cardiol. 2001;37:371–378. doi: 10.1016/s0735-1097(00)01107-4. [DOI] [PubMed] [Google Scholar]
- Ehrlich JR, Zicha S, Coutu P, Hebert TE, Nattel S. Atrial fibrillation-associated minK38G/S polymorphism modulates delayed rectifier current and membrane localization. Cardiovasc Res. 2005;67:520–528. doi: 10.1016/j.cardiores.2005.03.007. [DOI] [PubMed] [Google Scholar]
- Ellinor PT, Macrae CA. The genetics of atrial fibrillation. J Cardiovasc Electrophysiol. 2003;14:1007–1009. doi: 10.1046/j.1540-8167.2003.03307.x. [DOI] [PubMed] [Google Scholar]
- Ellinor PT, Shin JT, Moore RK, Yoerger DM, MacRae CA. Locus for atrial fibrillation maps to chromosome 6q14–16. Circulation. 2003;107:2880–2883. doi: 10.1161/01.CIR.0000077910.80718.49. [DOI] [PubMed] [Google Scholar]
- Hamill OP, Marty A, Neher E, Sakmann B, Sigworth FJ. Improved patch-clamp techniques for high-resolution current recording from cells and cell-free membrane patches. Pflugers Arch. 1981;391:85–100. doi: 10.1007/BF00656997. [DOI] [PubMed] [Google Scholar]
- Hong K, Bjerregaard P, Gussak I, Brugada R. Short QT syndrome and atrial fibrillation caused by mutation in KCNH2. J Cardiovasc Electrophysiol. 2005;16:394–396. doi: 10.1046/j.1540-8167.2005.40621.x. [DOI] [PubMed] [Google Scholar]
- Ishii TM, Silvia C, Hirschberg B, Bond CT, Adelman JP, Maylie J. A human intermediate conductance calcium-activated potassium channel. Proc Natl Acad Sci U S A. 1997;94:11651–11656. doi: 10.1073/pnas.94.21.11651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaczorowski GJ, Garcia ML. Pharmacology of voltage-gated and calcium-activated potassium channels. Curr Opin Chem Biol. 1999;3:448–458. doi: 10.1016/S1367-5931(99)80066-0. [DOI] [PubMed] [Google Scholar]
- Keating MT, Sanguinetti MC. Molecular and cellular mechanisms of cardiac arrhythmias. Cell. 2001;104:569–580. doi: 10.1016/s0092-8674(01)00243-4. [DOI] [PubMed] [Google Scholar]
- Kohler M, Hirschberg B, Bond CT, Kinzie JM, Marrion NV, Maylie J, Adelman JP. Small-conductance, calcium-activated potassium channels from mammalian brain. Science. 1996;273:1709–1714. doi: 10.1126/science.273.5282.1709. [DOI] [PubMed] [Google Scholar]
- Kovoor P, Wickman K, Maguire CT, Pu W, Gehrmann J, Berul CI, Clapham DE. Evaluation of the role of IKACh in atrial fibrillation using a mouse knockout model. J Am Coll Cardiol. 2001;37:2136–2143. doi: 10.1016/s0735-1097(01)01304-3. [DOI] [PubMed] [Google Scholar]
- Lai LP, Lin JL, Huang SK. Molecular genetic studies in atrial fibrillation. Cardiology. 2003;100:109–113. doi: 10.1159/000073910. [DOI] [PubMed] [Google Scholar]
- Lu L, Zhang Q, Timofeyev V, Zhang Z, Young JN, Shin HS, Knowlton AA, Chiamvimonvat N. Molecular coupling of a Ca2+-activated K+ channel to L-type Ca2+ channels via. α-actinin2. Circ Res. 2007;100:112–120. doi: 10.1161/01.RES.0000253095.44186.72. [DOI] [PubMed] [Google Scholar]
- Nattel S, Maguy A, Le Bouter S, Yeh YH. Arrhythmogenic ion-channel remodeling in the heart: heart failure, myocardial infarction, and atrial fibrillation. Physiol Rev. 2007;87:425–456. doi: 10.1152/physrev.00014.2006. [DOI] [PubMed] [Google Scholar]
- Nerbonne JM, Kass RS. Molecular physiology of cardiac repolarization. Physiol Rev. 2005;85:1205–1253. doi: 10.1152/physrev.00002.2005. [DOI] [PubMed] [Google Scholar]
- Nuss HB, Kaab S, Kass DA, Tomaselli GF, Marban E. Cellular basis of ventricular arrhythmias and abnormal automaticity in heart failure. Am J Physiol Heart Circ Physiol. 1999;277:H80–91. doi: 10.1152/ajpheart.1999.277.1.H80. [DOI] [PubMed] [Google Scholar]
- Olson TM, Alekseev AE, Liu XK, Park S, Zingman LV, Bienengraeber M, Sattiraju S, Ballew JD, Jahangir A, Terzic A. Kv1.5 channelopathy due to KCNA5 loss-of-function mutation causes human atrial fibrillation. Hum Mol Genet. 2006;15:2185–2191. doi: 10.1093/hmg/ddl143. [DOI] [PubMed] [Google Scholar]
- Otway R, Vandenberg JI, Guo G, Varghese A, Castro ML, Liu J, Zhao J, Bursill JA, Wyse KR, Crotty H, Baddeley O, Walker B, Kuchar D, Thorburn C, Fatkin D. Stretch-sensitive KCNQ1 mutation A link between genetic and environmental factors in the pathogenesis of atrial fibrillation? J Am Coll Cardiol. 2007;49:578–586. doi: 10.1016/j.jacc.2006.09.044. [DOI] [PubMed] [Google Scholar]
- Ozgen N, Dun W, Sosunov EA, Anyukhovsky EP, Hirose M, Duffy HS, Boyden PA, Rosen MR. Early electrical remodeling in rabbit pulmonary vein results from trafficking of intracellular SK2 channels to membrane sites. Cardiovasc Res. 2007;75:758–769. doi: 10.1016/j.cardiores.2007.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts R. Mechanisms of disease: Genetic mechanisms of atrial fibrillation. Nat Clin Pract Cardiovasc Med. 2006;3:276–282. doi: 10.1038/ncpcardio0509. [DOI] [PubMed] [Google Scholar]
- Robertson S, Potter JD. The regulation of free Ca2+ ion concentration by metal chelators. Methods Pharmacol. 1984;5:63–75. [Google Scholar]
- Sambrano GR, Fraser I, Han H, Ni Y, O'Connell T, Yan Z, Stull JT. Navigating the signalling network in mouse cardiac myocytes. Nature. 2002;420:712–714. doi: 10.1038/nature01306. [DOI] [PubMed] [Google Scholar]
- Stocker M, Pedarzani P. Differential distribution of three Ca2+-activated K+ channel subunits, SK1, SK2, and SK3, in the adult rat central nervous system. Mol Cell Neurosci. 2000;15:476–493. doi: 10.1006/mcne.2000.0842. [DOI] [PubMed] [Google Scholar]
- Tinker A, Jan YN, Jan LY. Regions responsible for the assembly of inwardly rectifying potassium channels. Cell. 1996;87:857–868. doi: 10.1016/s0092-8674(00)81993-5. [DOI] [PubMed] [Google Scholar]
- Tuteja D, Xu D, Timofeyev V, Lu L, Sharma D, Zhang Z, Xu Y, Nie L, Vazquez AE, Young JN, Glatter KA, Chiamvimonvat N. Differential isoform expression of small conductance Ca2+-activated K+ channels, SK1, SK2 and SK3 channels in mouse atrial and ventricular myocytes. Am J Physiol Heart Circ Physiol. 2005;289:H2714–2723. doi: 10.1152/ajpheart.00534.2005. [DOI] [PubMed] [Google Scholar]
- Vergara C, Latorre R, Marrion NV, Adelman JP. Calcium-activated potassium channels. Curr Opin Neurobiol. 1998;8:321–329. doi: 10.1016/s0959-4388(98)80056-1. [DOI] [PubMed] [Google Scholar]
- Wiesfeld AC, Hemels ME, Van Tintelen JP, Van den Berg MP, Van Veldhuisen DJ, Van Gelder IC. Genetic aspects of atrial fibrillation. Cardiovasc Res. 2005;67:414–418. doi: 10.1016/j.cardiores.2005.04.033. [DOI] [PubMed] [Google Scholar]
- Xu D, Li N, He Y, Timofeyev V, Lu L, Tsai HJ, Kim IH, Tuteja D, Mateo RK, Singapuri A, Davis BB, Low R, Hammock BD, Chiamvimonvat N. Prevention and reversal of cardiac hypertrophy by soluble epoxide hydrolase inhibitors. Proc Natl Acad Sci U S A. 2006;103:18733–18738. doi: 10.1073/pnas.0609158103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y, Tuteja D, Zhang Z, Xu D, Zhang Y, Rodriguez J, Nie L, Tuxson HR, Young JN, Glatter KA, Vazquez AE, Yamoah EN, Chiamvimonvat N. Molecular identification and functional roles of a Ca2+-activated K+ channel in human and mouse hearts. J Biol Chem. 2003;278:49085–49094. doi: 10.1074/jbc.M307508200. [DOI] [PubMed] [Google Scholar]
- Yang Y, Xia M, Jin Q, Bendahhou S, Shi J, Chen Y, Liang B, Lin J, Liu Y, Liu B, Zhou Q, Zhang D, Wang R, Ma N, Su X, Niu K, Pei Y, Xu W, Chen Z, Wan H, Cui J, Barhanin J, Chen Y. Identification of a KCNE2 gain-of-function mutation in patients with familial atrial fibrillation. Am J Hum Genet. 2004;75:899–905. doi: 10.1086/425342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q, Timofeyev V, Lu L, Li N, Singapuri A, Long MK, Bond CT, Adelman JP, Chiamvimonvat N. Functional roles of a Ca2+-activated K+ channel in atrioventricular nodes. Circ Res. 2008;102:465–471. doi: 10.1161/CIRCRESAHA.107.161778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, He Y, Tuteja D, Xu D, Timofeyev V, Zhang Q, Glatter KA, Xu Y, Shin HS, Low R, Chiamvimonvat N. Functional roles of Cav1.3(a1D) calcium channels in atria: insights gained from gene-targeted null mutant mice. Circulation. 2005;112:1936–1944. doi: 10.1161/CIRCULATIONAHA.105.540070. [DOI] [PubMed] [Google Scholar]