

Abstract
The uptake of lipoproteins by macrophages is a critical step in the development of atherosclerotic lesions. Cultured monocyte-derived macrophages take up large amounts of native LDL by receptor-independent fluid-phase pinocytosis, either constitutively or in response to specific activating stimuli, depending on the macrophage phenotype. We therefore sought to determine whether fluid-phase pinocytosis occurs in vivo in macrophages in atherosclerotic lesions. We demonstrated that fluorescent pegylated nanoparticles similar in size to LDL (specifically nontargeted Qtracker quantum dot and AngioSPARK nanoparticles) can serve as models of LDL uptake by fluid-phase pinocytosis in cultured human monocyte–derived macrophages and mouse bone marrow–derived macrophages. Using fluorescence microscopy, we showed that atherosclerosis-prone Apoe-knockout mice injected with these nanoparticles displayed massive accumulation of the nanoparticles within CD68+ macrophages, including lipid-containing foam cells, in atherosclerotic lesions in the aortic arch. Similar results were obtained when atherosclerotic mouse aortas were cultured with nanoparticles in vitro. These results show that macrophages within atherosclerotic lesions can take up LDL-sized nanoparticles by fluid-phase pinocytosis and indicate that fluid-phase pinocytosis of LDL is a mechanism for macrophage foam cell formation in vivo.
Introduction
Macrophage uptake of lipoproteins, especially LDL, is considered a critical process in the development of atherosclerosis, causing transformation of the macrophages into foam cells, thus promoting cholesterol accumulation and inflammation in the atherosclerotic plaque (1, 2). Heretofore, researchers believed that macrophages could internalize only modified lipoproteins, typically through a variety of scavenger receptors (3, 4). In recent studies, we have demonstrated an alternative mechanism for macrophage foam cell formation that does not depend on LDL modification or macrophage receptors (5–7). In this mechanism of foam cell formation, macrophages take up LDL through receptor-independent fluid-phase pinocytosis. In contrast to receptor-mediated uptake of lipoproteins, receptor-independent fluid-phase uptake of [125I]LDL does not become saturated at high [125I]LDL concentrations (5, 7). Pinocytosis of fluid and solutes such as LDL can occur either through uptake of fluid by micropinocytosis within small vesicles (micropinosomes) less than 0.1 μm in diameter or by macropinocytosis within large vacuoles (macropinosomes) usually greater than 1 μm (8, 9). Macropinosomes form from extended plasma membrane folds that fuse back with the plasma membrane, trapping extracellular fluid with the intracellular vacuoles that are formed.
Macrophage pinocytosis of LDL is either constitutive or activated through a protein kinase C–dependent signaling pathway, depending on the macrophage differentiation phenotype. Differentiation of human monocytes in the presence of FBS and M-CSF produces an elongated macrophage phenotype (CD68 and CD14 positive) in which large amounts of LDL are taken up and high levels of cholesterol accumulate via constitutive pinocytosis (7). Alternatively, human monocytes can be differentiated with human serum or FBS and GM-CSF, producing a rounded, “fried egg” macrophage phenotype (CD68 positive but CD14 negative) characterized by LDL uptake and cholesterol accumulation following stimulation of pinocytosis by protein kinase C activation (5, 6, 10, 11). The pinocytosis of fluid that occurs with both macrophage phenotypes is accompanied by vigorous macropinocytosis, suggesting that this mechanism of fluid uptake contributes to the total pinocytotic fluid uptake observed in these macrophages.
In order to show that fluid-phase pinocytosis of lipoproteins such as LDL can occur within atherosclerotic lesions in vivo, it is necessary to have a tracer that does not bind to cells and that ideally would be similar in size to LDL. Toward this end, we have made use of fluorescent nanoparticles that do not bind cells as a tracer for qualitative and quantitative analysis of fluid-phase pinocytosis. AngioSPARK nanoparticles are highly near-infrared fluorescent nanoparticles specifically designed for in vivo imaging. These nanoparticles are pegylated to minimize their interaction with cells, and when introduced into the vasculature, AngioSPARK nanoparticles remain localized therein for extended periods of time, showing a half-life of 14 hours in blood of mice (12). Qtracker nontargeted quantum dots (see Methods) are also stable, bright fluorophores that have high quantum yields and narrow fluorescence emission bands. Qtracker nontargeted quantum dots are also pegylated and exhibit a circulating half-life of 18.5 hours (13). The two types of nanoparticles are similar in size (20–50 nm for AngioSPARK and 28–29 nm for the quantum dots) to LDL (22 nm). We now show that these nanoparticles can be used to monitor fluid-phase pinocytosis in cultured cells and by cells in vivo. Like LDL, these fluorescent nanoparticles were taken up by cultured macrophages by fluid-phase pinocytosis. Using both types of fluorescent nanoparticles, we now demonstrate fluid-phase pinocytosis by macrophages in vivo in atherosclerotic lesions using the Apoe-knockout mouse model of atherosclerosis.
Results
First, we examined uptake of fluorescent DiI-acetylated LDL (DiI-AcLDL), DiI-LDL, quantum dots, and AngioSPARK nanoparticles by human monocyte–derived macrophages differentiated 1 week with M-CSF in vitro. Following differentiation, macrophages were incubated for 5 hours with increasing concentrations of DiI-AcLDL (0–160 nM), DiI-LDL (0–320 nM), quantum dots (0–160 nM), or AngioSPARK nanoparticles (0–160 nM) (Figure 1). DiI-AcLDL uptake was saturated at 80 nM (44 μg/ml), consistent with scavenger receptor–mediated uptake, as previously described for AcLDL (14). In contrast, DiI-LDL, quantum dot, and AngioSPARK uptake was linearly related to the concentration of these particles in the medium, showing no saturation and therefore not mediated by cell binding. Microscopy revealed that DiI-LDL, quantum dots, and AngioSPARK nanoparticles accumulated within the macrophages (Figure 2).
Figure 1. Macrophages take up quantum dot and AngioSPARK nanoparticles and DiI-LDL by fluid-phase pinocytosis.

M-CSF–differentiated human monocyte–derived macrophages were incubated with increasing concentrations of fluorescent lipoproteins or nanoparticles (x axis) for 5 hours, and then macrophage uptake of fluorescent nanoparticles (y axis) was determined as described in Methods. DiI-AcLDL uptake showed saturation at 80 nM (i.e., 44 μg/ml), consistent with receptor-mediated endocytosis, while quantum dot and AngioSPARK nanoparticle and DiI-LDL uptake showed no saturation, consistent with fluid-phase pinocytosis. DiI-AcLDL and quantum dot uptake were analyzed in the same experiment, but DiI-LDL and AngioSPARK uptake were determined in separate experiments using different macrophage cultures.
Figure 2. Accumulation of quantum dot and AngioSPARK nanoparticles and DiI-LDL in cultured macrophages.

Phase and fluorescence merged photomicrographs of human macrophages cultured for 24 hours without fluorescent particles (A) or with DiI-LDL (80 nM) (B), quantum dot nanoparticles (80 nM) (C), and AngioSPARK nanoparticles (80 nM) (D) are shown. The inset in C shows a confocal image. Arrowheads indicate nuclei with accumulation of perinuclear nanoparticles. Scale bars: 50 μm (A–D), 10 μm (C, inset).
In another experiment, human macrophages were pretreated for 60 minutes without or with 2 μg/ml cytochalasin D (an inhibitor of actin microfilament function) and then incubated with AngioSPARK nanoparticles (160 nM) for 30 minutes without or with cytochalasin D, followed by rinsing of the cultures. Fluorescence microscopy showed that while macrophages accumulated fluorescent nanoparticles without cytochalasin D, macrophages did not accumulate nanoparticles in the presence of cytochalasin D (data not shown). In both cases, no nanoparticles were observed bound to the plasma membrane of the macrophages confirming that the nanoparticles did not bind cells.
We also assessed the possibility that the size of nanoparticles might change during incubation with macrophages. To test for this, we performed fast performance liquid chromatography of quantum dot nanoparticles (80 nM) incubated with human macrophages for 5 hours or immediately after the nanoparticles (as supplied by the manufacturer; see Methods) were diluted (Figure 3). Chromatograms showed that the quantum dot nanoparticles eluted in 2 identifiable peaks: a small fraction of nanoparticles eluting in fractions 25–29 and a large fraction of nanoparticles eluting in fractions 32–45. We calculated the percentage of total fluorescence for each peak and determined that fractions 25–29 represented 3% of the total fluorescence for both samples, and fractions 32–45 represented 97% of the total fluorescence for both samples. These data show that quantum dot nanoparticle size was not altered during incubation with cultured macrophages. The nanoparticles eluted slightly ahead of the elution position of LDL, indicating that the nanoparticles are slightly larger than 22 nm, the size of LDL. This finding is consistent with information provided by the manufacturer (see Methods) that the size of the quantum dot nanoparticles we used is 28–29 nm.
Figure 3. Incubation of quantum dot nanoparticles with macrophages does not alter nanoparticle size.

Quantum dot nanoparticles (80 nM) were immediately fractionated by fast performance liquid chromatography (A) or incubated for 5 hours with M-CSF–differentiated human monocyte–derived macrophages prior to fractionation by fast performance liquid chromatography (B). The peak LDL elution was observed in fraction 48 as indicated. Fluorescence (y axis) of fractions (x axis) was detected as described in Methods. Vo indicates the void volume.
Next, we assessed the uptake of nanoparticles by murine bone marrow–derived macrophages differentiated in vitro for 4 days with M-CSF. Because we sought to investigate fluid-phase pinocytosis of nanoparticles by macrophages in vivo, we tested the possibility that components within Apoe-knockout mouse plasma might interact with nanoparticles, potentially causing nanoparticle uptake by macrophages to occur in a receptor-dependent manner. To test for this, we incubated quantum dot nanoparticles for 24 hours in Apoe-knockout plasma and then incubated Apoe-knockout bone marrow–derived macrophages with 0–160 nM of plasma-treated nanoparticles (Figure 4). Uptake of nanoparticles by macrophages was linearly related to the concentration of nanoparticles in the medium, and was not saturated. Thus, similar to results obtained with human macrophages, uptake of plasma-treated nanoparticles by mouse macrophages was not due to macrophage binding of the nanoparticles.
Figure 4. Murine macrophages take up plasma-treated quantum dot nanoparticles by fluid-phase pinocytosis.

Quantum dots were incubated for 24 hours in Apoe-knockout mouse plasma. Subsequently, M-CSF–differentiated murine bone marrow–derived macrophages were incubated with increasing concentrations of plasma-treated fluorescent quantum dots (x axis) for 5 hours, and then macrophage uptake of fluorescent nanoparticles (y axis) was determined as described in Methods.
AngioSPARK nanoparticles (0.5 nmol/mouse) or normal saline as a control were injected into Apoe-knockout mice fed a high-cholesterol diet for 20 weeks and into C57BL/6 wild-type mice fed a normal chow diet. After 24 hours, aortas were harvested and imaged with a near-infrared fluorescence (NIRF) reflectance imaging system. Aortic arch atherosclerotic lesions were also examined by fluorescence microscopy. NIRF reflectance imaging of aortas showed more intense fluorescence signal in aortas from Apoe-knockout mice that were injected with AngioSPARK nanoparticles compared with control mice, and the most intense signal was mainly localized in aortic arch areas where atherosclerotic lesions are known to occur in Apoe-knockout mice (15) (Figure 5A). AngioSPARK-injected wild-type mice without atherosclerotic lesions showed much less aortic fluorescence compared with AngioSPARK-injected Apoe-knockout mice (Figure 5B). Uninjected Apoe-knockout and wild-type mice showed a small degree of aortic fluorescence due to intrinsic tissue autofluorescence (Figure 5, C and D). Quantitative analysis of infrared mean fluorescence intensity in the aortic arch regions captured by NIRF reflectance imaging showed a greater than 6-fold increase in fluorescent signal in AngioSPARK-injected Apoe-knockout mice compared with AngioSPARK-injected wild-type mice (Figure 5E). Fluorescence microscopic imaging of aortic arch sections revealed accumulation of AngioSPARK nanoparticles in cells of the atherosclerotic lesions (Figure 6).
Figure 5. Visualization and quantification of AngioSPARK nanoparticle fluorescence signal in atherosclerotic aortas.

Mouse aortas from Apoe-knockout (Apoe–/–) and wild-type mice injected 24 hours earlier with AngioSPARK nanoparticles or normal saline as control were imaged ex vivo by NIRF reflectance imaging, as described in Methods. NIRF images of aortas from Apoe-knockout mice with AngioSPARK injection (A) show substantially brighter NIRF signal than the aortas from wild-type mice (B). Aortas from Apoe-knockout (C) and wild-type mice (D) injected with normal saline as control show only a small fluorescent signal due to intrinsic tissue autofluorescence. The vessel wall NIRF signal is mainly localized in the aortic arch (aa) regions defined between the 2 black bars. h, heart; da, descending aorta. The color scale from black to white indicates NIRF arbitrary units from 70 to 300 and applies to all aorta images. (E) Quantitative analysis of infrared mean fluorescence intensity in the aortic arch regions from 5 AngioSPARK-injected mice minus the intrinsic mean fluorescence intensity of aortas from 2 uninjected control mice (mean fluorescence intensity on the y axis). Fluorescence of atherosclerotic aortic arches from injected Apoe-knockout mice was significantly greater (P = 0.04) than fluorescence of aortic arches with no lesions from wild-type mice. Data represent mean ± SEM of mean fluorescence intensity determinations for each group.
Figure 6. Accumulation of AngioSPARK nanoparticles in mouse aortic arch atherosclerotic lesions.

Aortic arch sections from Apoe-knockout mice injected with AngioSPARK nanoparticles (A–C) or normal saline (D) are shown. Accumulation of AngioSPARK nanoparticles (red) within cells of atherosclerotic lesions is shown at higher magnification in C. Sections in A and B were not stained with DAPI, but nuclei of cells appear as small, nonfluorescent oval regions surrounded by red AngioSPARK nanoparticles. Cell nuclei stained with DAPI in C and D are shown in blue. Tissue autofluorescence is shown in green. Scale bars: 10 μm (A); 5 μm (B); 2 μm (C and D). i, intima; m, media. Horizontal bars on the left in C and D indicate intimal-medial borders.
Quantum dots (2 nmol/mouse) or normal saline as controls were also injected into Apoe-knockout mice. The concentration of injected quantum dot nanoparticles was such as to achieve an initial concentration of plasma nanoparticles equivalent to the concentration of LDL in humans (i.e., about 2 μM LDL). After 24 hours, aortas were harvested, and aortic arch atherosclerotic lesions were examined by fluorescence microscopy. Alternatively, aortic arches from uninjected mice were removed and incubated 24 hours with 0.8 μM quantum dots. In both cases, fluorescence imaging of aortic arch sections revealed accumulation of quantum dot nanoparticles in atherosclerotic lesions, mostly within cells that were identified with nuclear staining (Figure 7, A and B). Aortic arches incubated without quantum dots, used as controls, showed no fluorescent nanoparticles, as expected (Figure 7C). Quantum dots were localized within atherosclerotic lesion macrophages identified with anti-CD68 antibody (Figure 8, A and B) and foam cells identified with oil red O lipid staining (Figure 8, C and D). There were few or no quantum dots in the vascular media (data not shown).
Figure 7. Accumulation of quantum dots in mouse aorta atherosclerotic lesions occurs in vivo and in vitro.
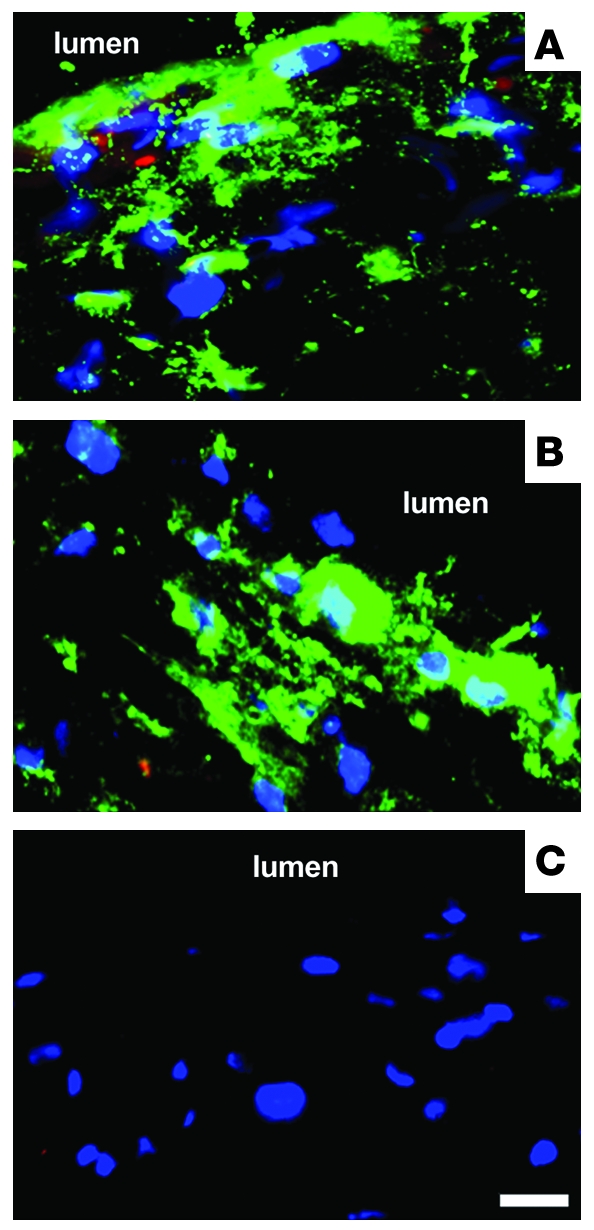
(A–C) Merged images of quantum dot (green) and nucleus (blue) fluorescence in the intima. Some red autofluorescence is present in the images in A and B. (A) Quantum dots (2 nmol) were injected into an Apoe-knockout mouse. After 24 hours, the aorta was harvested and processed for fluorescence microscopy. An aortic arch tissue section is shown, revealing accumulation of quantum dots in atherosclerotic lesions, mostly within cells. (B and C) Aortic arches from Apoe-knockout mice were cultured 24 hours with (B) or without (control) (C) 0.8 μM quantum dots. The accumulation of quantum dots within cells of atherosclerotic lesions was similar to that observed in vivo. The lumen region is indicated. Scale bar: 2 μm.
Figure 8. Accumulation of quantum dots in macrophages and foam cells of atherosclerotic lesions in vivo.

Quantum dots (2 nmol) were injected into an Apoe-knockout mouse. After 24 hours, the aorta was harvested, and the aortic arch was processed for fluorescence microscopy. Nuclei (blue) and quantum dots (green) in the intima were first imaged (A and C). Then the same respective tissue sections were either immunolabeled with anti-CD68 to detect macrophages (red) (B) or stained with oil red O to demonstrate lipid in foam cells (red) (D). Quantum dots are only visualized in A and C because they are lost from the tissue sections during subsequent oil red O lipid staining and macrophage immunostaining. These images show localization of quantum dots within macrophages and foam cells of atherosclerotic lesions. Scale bar: 2 μm.
Discussion
We report the application of fluorescent pegylated nanoparticles for the visualization of fluid-phase pinocytosis in vivo. Previously, no suitable method existed for in vivo detection of this important endocytic process that, as we have shown here, functions in atherosclerotic lesion macrophages. The previous use of fluorescently labeled dextran to monitor fluid-phase pinocytosis has been found to be unsuitable because this probe can be internalized by receptor-mediated uptake in cells such as macrophages expressing the mannose receptor (16, 17). As discussed below, fluid-phase pinocytosis can mediate macrophage cholesterol accumulation, an important process in the development of atherosclerotic lesions. Furthermore, fluid-phase pinocytosis has been shown to function in vitro in dendritic cell and macrophage uptake of soluble antigens that are then processed for antigen presentation to T cells (9, 17). The techniques we report here make it possible now to study fluid-phase pinocytosis in vivo as it occurs in atherosclerosis and during the process of antigen presentation. Also, fluid-phase pinocytosis by other cell types in vivo can now be discovered and investigated.
Learning how macrophages accumulate cholesterol and transform into foam cells within atherosclerotic plaques has been a major goal of research directed at understanding the pathogenesis of atherosclerosis. The widely accepted hypothesis for foam cell formation in atherosclerotic lesions involves receptor-mediated endocytosis of blood-derived LDL that has entered the vessel wall and become modified presumably by oxidation or aggregation (3, 4). We have recently shown that modification of LDL is not necessary for its uptake and degradation by macrophages. Human monocyte–derived macrophages take up large amounts of native LDL by receptor-independent fluid-phase pinocytosis, constitutively or after activation with PMA, depending on the macrophage phenotype (5–7). We believe pinocytosis of fluid-phase rather than bound LDL to be a novel mechanism of lipoprotein-induced macrophage cholesterol accumulation. The current study has shown that this fluid phase–mediated mechanism of macrophage lipoprotein uptake could also occur in vivo in atherosclerotic plaques.
We encountered an unexpected technical problem with the use of pegylated nanoparticles to probe fluid pinocytosis in vivo. Because the nanoparticles do not bind to cell contents or matrix and cannot be fixed in place (by design, the nanoparticles lack reactive functional groups), the particles are not well retained in tissue sections that are rinsed. To overcome this problem, it was optimal to image the fluorescent nanoparticles in DAPI-stained frozen sections without rinsing, fixation, immunostaining, or mounting medium. Then further staining could be carried out, imaged, and superimposed on the nanoparticle image.
The culture model used for the research reported here is characterized by human monocyte–derived macrophages, differentiated in the presence of M-CSF, that constitutively take up large amounts of native LDL by fluid-phase pinocytosis (7). We showed that these macrophages can also take up by fluid-phase pinocytosis fluorescent nanoparticles similar in size to native LDL. DiI-LDL particles are LDL labeled with the fluorescent lipophilic dye DiI, which diffuses into the hydrophobic portion of LDL without affecting lipoprotein receptor binding (18–20). Thus, as expected from our previous studies with native LDL (5, 7), DiI-LDL, like the pegylated nanoparticles, showed nonsaturable fluid-phase uptake by macrophages. In contrast, DiI-AcLDL showed saturable receptor-mediated uptake by macrophages, consistent with its known interaction with macrophage scavenger receptors (21).
After injection of AngioSPARK and quantum dots into Apoe-knockout mice, both types of fluorescent nanoparticles could be localized within aortic arch atherosclerotic lesions that develop in these hypercholesterolemic mice. Macrophages within the atherosclerotic lesions accumulated the nanoparticles. We hypothesized that the mechanism underlying their uptake was fluid-phase pinocytosis, since this process was shown to occur in vitro with nanoparticles treated with plasma from Apoe-knockout mice that were then incubated with bone marrow–derived macrophages from Apoe-knockout mice. Two findings made it unlikely that the fluorescent nanoparticles were taken up by monocytes in the circulation and carried into the vessel wall. Rare CD68-labeled monocytes that were attached to the luminal surface of the vessel wall did not show accumulation of quantum dots. Moreover, when we exposed atherosclerotic aortas removed from Apoe-knockout mice to quantum dots in vitro rather than by injecting the quantum dots into mice, quantum dots also accumulated within atherosclerotic lesion macrophages. This shows that the quantum dots could enter the vessel wall and be taken up by macrophages already present within atherosclerotic lesions.
Our results showed that pinocytosis persists even in lipid-filled macrophage foam cells, because macrophages that showed extensive lipid deposits also accumulated substantial amounts of quantum dots (Figure 7). Thus, macrophage lipid accumulation does not downregulate fluid-phase pinocytosis, consistent with our in vitro findings that LDL-induced macrophage cholesterol accumulation does not downregulate macrophage pinocytosis (7, 22). Unregulated uptake of LDL could eventually lead to toxic levels of cholesterol accumulation within macrophages, contributing to macrophage death, since excess unesterified cholesterol accumulation is one factor shown to trigger macrophage apoptosis (23).
Previously, it was shown that a minimum of one-third of LDL catabolism in animals and humans occurs via a receptor-independent route, most prominently in the spleen, liver, small intestine, and kidney (24). These same tissues show the highest rates of fluid-phase pinocytosis (25), consistent with the suggestion that receptor-independent catabolism of plasma LDL is due to fluid-phase pinocytosis (26, 27). Also, consistent with the hypothesis that receptor-independent catabolism of plasma LDL is due to fluid-phase pinocytosis is the finding that receptor-independent catabolism of plasma LDL is a linear function of the plasma LDL concentration (24). Suppression of macrophage function results in decreased receptor-independent catabolism of LDL and raised LDL levels, suggesting that receptor-independent uptake of LDL is mediated in part by macrophages (26, 28). Furthermore, a recent study (27) has shown enhanced fluid-phase (i.e., bulk-phase) uptake of LDL cholesterol in those tissues infiltrated by macrophages in a mouse model of Niemann-Pick type C disease, a cholesterol storage disorder caused by a defect in cellular trafficking of cholesterol.
Our findings here show substantial fluid-phase pinocytosis by macrophages in atherosclerotic plaques. Thus, not only is macrophage fluid-phase pinocytosis of LDL a mechanism for foam cell formation that we have shown occurs with cultured macrophages (5–7), but it is also a plausible mechanism to explain foam cell formation of atherosclerotic plaque macrophages. Monocyte differentiation into plaque macrophages is well established. Also, M-CSF is present in plaques (29, 30), and we have shown that this factor differentiates monocytes into a macrophage phenotype showing constitutive pinocytosis (7, 11). LDL is present within atherosclerotic plaques at levels that are about twice the blood concentrations of LDL (31–33). These levels are greater than 1 mg/ml (i.e., ≅2 μM) LDL, and we showed these LDL levels can produce substantial macrophage cholesterol accumulation in human monocyte–derived macrophages (5, 7). Fluid-phase pinocytosis of LDL produces macrophage LDL uptake that occurs in a linear relationship with LDL concentration. Thus, fluid-phase pinocytosis is an uptake mechanism that can explain why plasma cholesterol concentration is an indicator of risk for the development of coronary artery disease (34, 35). Also, fluid-phase pinocytosis of LDL can explain why macrophage foam cell formation occurs even in the absence of the scavenger receptors that mediate uptake of oxidized LDL (36, 37). These findings indicate that future studies aimed at limiting macrophage cholesterol accumulation in atherosclerotic plaques should consider fluid-phase pinocytosis of LDL as a potentially important target pathway.
Methods
Culture of human monocyte–derived macrophages.
Human monocytes were purified with counterflow centrifugal elutriation of mononuclear cells obtained by monocytopheresis of normal human donors. Monocytopheresis was carried out by the Department of Transfusion Medicine, Clinical Center, NIH, and the human research protocol was approved by the NIH Institutional Review Board. Written informed consent was received from donors. M-CSF monocyte-derived macrophage cultures were initiated with the elutriated human monocytes suspended in RPMI 1640 medium (MediaTech) with 10% FBS (GIBCO, Invitrogen) seeded in 6-well plates (Corning CellBIND) at a density of 2 × 105 monocytes/cm2. After 2 hours incubation in a cell culture incubator with 5% CO2/95% air at 37°C, the attached monocytes were rinsed 3 times with RPMI 1640 medium. Then the monocytes were differentiated into macrophages by culturing with RPMI 1640 medium containing 10% FBS, 50 ng/ml M-CSF (PeproTech), and 25 ng/ml IL-10 (PeproTech) (38). The medium was exchanged with fresh medium after 6 days of culture, and monocyte-derived macrophages were used for experiments the next day.
For experiments, monocyte-derived macrophage cultures (about 0.2 mg protein/well) were first rinsed 3 times with serum-free RPMI 1640 medium. Cultures were incubated in RPMI 1640 medium with the indicated concentration of fluorescent nanoparticles and lipoproteins specified in each experiment. Experimental incubations with these monocyte-derived macrophages were carried out in the presence of 10% FBS and the same concentration of cytokines used during differentiation of monocytes into macrophages.
Culture of murine bone marrow–derived macrophages.
Bone marrow from 8-month-old female Apoe-knockout mice (C57BL/6 strain) fed a chow diet was obtained by flushing femurs and humeri with Dulbecco’s PBS (DPBS; without Ca2+, Mg2+) containing 1 mM EDTA. Cells were centrifuged for 5 minutes at 2,000 g and resuspended in RPMI 1640 medium containing 100 U/ml penicillin, 100 μg/ml streptomycin (Sigma-Aldrich), 10% FBS, and 50 ng/ml M-CSF. Then cells were seeded at 2 × 105 marrow cells/cm2 in 6-well culture plates (Corning CellBIND). After 24 hours, adherent cells were washed 3 times, and fresh medium was added to cultures.
After 4 days of additional culture, murine bone marrow–derived macrophages were rinsed 3 times with RPMI 1640 medium containing 100 U/ml penicillin and 100 μg/ml streptomycin and then incubated in this medium with added M-CSF (50 ng/ml), 10% mouse plasma, and the indicated concentration of nanoparticles.
Preparation of nanoparticles and lipoproteins for use in experiments.
Before use with culture, Qtracker 565 nontargeted quantum dots provided in borate buffer (catalog Q21031MP), human DiI-AcLDL, and human DiI-LDL (all from Invitrogen) were dialyzed 24 hours against RPMI 1640 medium (2 changes, 1 liter/each change). When used for injection into mice, quantum dots were dialyzed against normal saline. Dialysis was carried out with Pierce Slide-A-Lyzer cassettes (10,000-MW cut-off). After dialysis, the nanoparticles and lipoproteins were sterilized by passage through a 0.45-μm (pore size) low-protein binding filter (Gelman Sciences). AngioSPARK 680-IVM nanoparticles (VisEn Medical) were provided in PBS and were not dialyzed prior to use.
Determination of fluorescent nanoparticle uptake by macrophages.
Human macrophages were incubated in 1 ml culture medium (as described above) with increasing concentrations of AngioSPARK nanoparticles, quantum dots, DiI-LDL, or DiI-AcLDL as indicated for 5 hours. Then macrophages were rinsed 3 times with serum-free RPMI 1640 medium, harvested by scraping into 1 ml distilled H2O at 4°C, and sonicated. A sonicated sample of 800 μl for each condition was then analyzed with a spectrofluorometer. DiI-lipoprotein fluorescence was determined using a 520-nm excitation wavelength and a 575-nm emission wavelength. Quantum dot fluorescence was determined using a 480-nm excitation wavelength and a 565-nm emission wavelength. AngioSPARK fluorescence was determined using a 665-nm excitation wavelength and a 680-nm emission wavelength. Macrophage uptake of lipoproteins and fluorescent nanoparticles was calculated from a standard curve relating particle fluorescence to particle concentration and was expressed as pmol/mg cell protein. Protein content of macrophages was determined by the Lowry method (39).
For nanoparticle uptake by murine bone marrow–derived macrophages, quantum dots were dialyzed against RPMI 1640 containing 100 U/ml penicillin and 100 μg/ml streptomycin (2 changes, 1 liter/each change) and then sterile-filtered (as described above). Then quantum dots were incubated for 24 hours in a final concentration of 10% Apoe-knockout mouse plasma that was sterile-filtered through a 0.45-μm low-protein binding filter. Macrophages were incubated with increasing concentrations of quantum dots in 1 ml of culture medium containing antibiotics, M-CSF, and 10% mouse plasma composed of 1% Apoe-knockout plasma and 9% wild-type plasma. After 5 hours, macrophages were washed and harvested, and fluorescence and protein levels were determined as described for human macrophages.
Fast performance liquid chromatography of quantum dot nanoparticles.
Human macrophages were incubated with 1 ml of culture medium (as described above) containing 80 nM quantum dot nanoparticles for 5 hours. After incubation, culture medium was harvested and then fractionated by fast performance liquid chromatography. As a control, quantum dots, as supplied by the manufacturer, were diluted to 80 nM in ISOTON II Diluent (Beckman Coulter) and immediately analyzed by fast performance liquid chromatography. Two hundred and seventy-five microliters of each sample was fractionated using a series of two Superose 6 10/30 columns (GE Healthcare), equilibrated with buffer (150 mM NaCl, 20 mM Tris-HCl, 0.5 mM EDTA, and 0.1% NaN3), and eluted at a flow rate of 0.5 ml/min. Fractions were collected in a 96-well plate (0.5 ml/fraction) and stored at 4°C for 24 hours before fluorescence analysis. Fluorescence was measured for each fraction with a spectrofluorometer using a 480-nm excitation wavelength and a 565-nm emission wavelength. The void volume was determined to be 11 ml (i.e., fraction 22) using Blue Dextran 2000 (GE Healthcare) as a standard.
Mouse aorta culture.
Apoe-knockout mice bred on a C57BL/6 background (The Jackson Laboratory) were euthanized by exsanguination at age 36–40 weeks. The arterial tree was perfused with DPBS plus Ca2+, Mg2+, and the perfused aorta was dissected from the heart to the iliac bifurcation. Then the aortic arch was separated from the remaining aorta and was placed into a 22-mm diameter culture dish and incubated with RPMI 1640 medium containing 10% autologous mouse serum without or with 0.8 μM quantum dots, in a total volume of 500 μl. After 24-hour incubation in a cell culture incubator with 5% CO2/95% air at 37°C, the aortic arch was rinsed 3 times in DPBS with Ca2+ and Mg2+ and then frozen in OCT medium for cryostat sectioning.
Intravenous injection of AngioSPARK and quantum dot nanoparticles into mice.
AngioSPARK nanoparticles (0.5 nmol/mouse) or an equivalent volume of normal saline solution (0.1 ml volume) were injected intravenously via tail vein into Apoe-knockout male mice fed a high-cholesterol diet for 20 weeks starting at 8 weeks of age (5 mice) and into C57BL/6 wild-type mice of the same age and sex fed a chow diet (2 mice) (carried out at VisEn Medical). For quantum dot injection, 40-week-old Apoe-knockout male mice fed only a chow diet were anesthetized using 2.5% Avertin (0.015 ml/g body weight, i.p.) before a saphenous vein cut-down procedure was performed. Then quantum dots (2 nmol/mouse) in normal saline solution (1 ml volume) were infused via saphenous vein (carried out at the NIH). Normal saline solution without nanoparticles was injected into control mice. All animal procedures at the NIH were performed according to a research protocol approved by the Animal Care and Use Committee of the NHLBI, and all animal procedures at VisEn Medical were performed according to a research protocol approved by the Animal Care and Use Committee of VisEn Medical in accordance with the Guide for the care and use of laboratory animals (NIH publication no. 85-23. Revised 1985). Twenty-four hours after injection, mice were euthanized by exsanguination, arterial trees were perfused, and aortic arches were prepared for cryosectioning as described above.
Ex vivo NIRF reflectance imaging.
Mouse aortas from Apoe-knockout and wild-type mice injected with AngioSPARK nanoparticles or normal saline as a control were imaged ex vivo by NIRF reflectance imaging at VisEn Medical. NIRF images with a 2-minute acquisition were captured with a 12-bit monochrome CCD camera equipped with an emission long-pass filter at a 700-nm wavelength (Kodak 2000MM reflectance imaging system). In order to quantify the infrared signal, regions of interest (ROIs) were drawn around aortic arch regions (defined from the aortic root to the origin of third artery branch (i.e., left subclavian artery) using Kodak Molecular Imaging Software version 4.0.1. Mean fluorescence was determined for each ROI as a sum of the signal intensities divided by the area of the selected region. Mean fluorescence of aortic arch regions from uninjected Apoe-knockout and wild-type mice due to intrinsic tissue autofluorescence was subtracted from the mean fluorescence of aortic regions from Apoe-knockout and wild-type AngioSPARK-injected mice, respectively.
Immunostaining and fluorescence microscopy.
Longitudinal aortic arch 10-μm frozen sections, which included greater and lesser aortic arch curvatures extending from the aortic root to beyond the origin of the left subclavian artery, were applied to Superfrost Plus slides (Daigger). Sections from Apoe-knockout mice injected with quantum dots or normal saline as control were stained, without fixation, with 1 μg/ml DAPI acetate (D-9564; Sigma-Aldrich) in DPBS with Ca2+ and Mg2+ to fluorescently label nuclei. Then sections were imaged with a digital camera attached to an Olympus IX81 fluorescence microscope at the NIH. Quantum dot fluorescence was imaged using Qdot565 Chroma Filter Set 32005 (exciter E460SPUVv2; emitter D565/20m) and pseudocolored green; DAPI staining was imaged using DAPI Chroma Filter Set U-N31000 (exciter D360/40; emitter D460/50) and pseudocolored blue; and tissue autofluorescence was imaged using the Texas Red Chroma Filter Set 41004 (exciter HQ560/55; emitter HQ645/75) and pseudocolored red. Merged color images of quantum dot fluorescence, cell nuclei DAPI fluorescence staining, and tissue autofluorescence were used to visualize localization of quantum dot nanoparticles in atherosclerotic lesion cells, as shown in Figure 6.
For some sections, after imaging of quantum dots and nuclei, macrophage immunostaining was performed by fixing tissue sections in ice-chilled acetone, rehydrating sections in DPBS, and sequential blocking with 10% normal goat serum in DPBS followed by treatment with avidin and biotin blocking solutions (Vector Laboratories). Then sections were immunostained using 10 μg/ml rat monoclonal anti-mouse CD68 antibody (AbD Serotec), followed by incubation with 20 μg/ml secondary biotinylated anti–rat IgG antibody (Vector Laboratories). Negative control staining was performed using rat IgG negative control (AbD Serotec) as primary antibody at the same concentration as the rat anti-CD68 antibody. Sections were then incubated with 5 μg/ml Alexa Fluor 594–conjugated streptavidin to image the macrophage immunolabeling using the Texas Red Chroma Filter Set 41004 (exciter HQ560/55; emitter HQ645/75). This immunolabeling was pseudocolored red. Following immunolabeling, sections were restained with DAPI as described above to label cell nuclei. This was necessary because acetone removed the initial DAPI staining. Merged color images of CD68-labeled macrophage fluorescence staining and restained cell nuclei DAPI fluorescence staining were obtained and compared with the initial merged color image of quantum dots and nuclei (Figure 8, A and B). Nuclear staining in both sets of merged images allowed for alignment of the sets of merged images.
In other sections, after imaging of quantum dots and nuclei, lipid staining was carried out with oil red O (40). Following oil red O staining, sections were restained with DAPI as described above to image cell nuclei. Oil red O lipid staining fluorescence was imaged using the Texas Red Chroma Filter Set 41004 (exciter HQ560/55; emitter HQ645/75) and was pseudocolored red. Merged color images of quantum dot fluorescence and cell nuclei DAPI staining (Figure 8C) were compared with merged color images of oil red O and cell nuclei DAPI staining (Figure 8D) showing the same area.
Sections from Apoe-knockout mice injected with AngioSPARK nanoparticles or normal saline as a control were stained with 1 μg/ml DAPI acetate (D-9564; Sigma-Aldrich) in DPBS to label cell nuclei. Then, sections were imaged, without fixation, with a digital camera attached to an AxioVision Zeiss fluorescence microscope at VisEn Medical. AngioSPARK fluorescence was imaged using a NIRF Filter Set and pseudocolored red; DAPI staining was imaged using a UV filter set and pseudocolored blue; and tissue autofluorescence was imaged using a FITC filter set and pseudocolored green. Merged color images of AngioSPARK fluorescence, cell nuclei DAPI fluorescence staining, and tissue autofluorescence were used to visualize localization of AngioSPARK nanoparticles in atherosclerotic lesion cells as shown in Figure 6.
Cultured macrophages incubated with AngioSPARK nanoparticles or quantum dots were imaged with the same microscope filters, as described above for imaging of these particles in tissue sections.
Statistics.
Results from the ex vivo NIRF reflectance imaging are expressed as mean ± SEM and were analyzed by the 2-tailed unpaired Student’s t test for statistical significance. A P value less than 0.05 was considered significant.
Acknowledgments
We thank the Department of Transfusion Medicine, Clinical Center, NIH, for providing elutriated monocytes; Rani Rao and Janet Chang of the Section of Experimental Atherosclerosis, NHLBI, NIH; and John Stonik of the Molecular Disease Section, NHLBI, NIH, for technical assistance. We also thank Sylvie Kossodo and Jeff Peterson for advice and support for the portion of the study carried out at VisEn Medical. This research was supported in part by the Intramural Research Program of the NHLBI, NIH.
Footnotes
Conflict of interest: Chiara Buono is an employee of VisEn Medical, the manufacturer of AngioSPARK nanoparticles used in this research.
Nonstandard abbreviations used: AcLDL, acetylated LDL; DPBS, Dulbecco’s PBS; NIRF, near-infrared fluorescence.
Citation for this article: J. Clin. Invest. 119:1373–1381 (2009). doi:10.1172/JCI35548
References
- 1.Bobryshev Y.V. Monocyte recruitment and foam cell formation in atherosclerosis. Micron. 2006;37:208–222. doi: 10.1016/j.micron.2005.10.007. [DOI] [PubMed] [Google Scholar]
- 2.Hansson G.K. Inflammation, atherosclerosis, and coronary artery disease. N. Engl. J. Med. 2005;352:1685–1695. doi: 10.1056/NEJMra043430. [DOI] [PubMed] [Google Scholar]
- 3.Kunjathoor V.V., et al. Scavenger receptors class A-I/II and CD36 are the principal receptors responsible for the uptake of modified low density lipoprotein leading to lipid loading in macrophages. J. Biol. Chem. 2002;277:49982–49988. doi: 10.1074/jbc.M209649200. [DOI] [PubMed] [Google Scholar]
- 4.Steinberg D. Lewis A. Conner Memorial Lecture. Oxidative modification of LDL and atherogenesis. Circulation. 1997;95:1062–1071. doi: 10.1161/01.cir.95.4.1062. [DOI] [PubMed] [Google Scholar]
- 5.Kruth H.S., Huang W., Ishii I., Zhang W.Y. Macrophage foam cell formation with native low density lipoprotein. J. Biol. Chem. 2002;277:34573–34580. doi: 10.1074/jbc.M205059200. [DOI] [PubMed] [Google Scholar]
- 6.Kruth H.S., et al. Macropinocytosis is the endocytic pathway that mediates macrophage foam cell formation with native low density lipoprotein. J. Biol. Chem. 2005;280:2352–2360. doi: 10.1074/jbc.M407167200. [DOI] [PubMed] [Google Scholar]
- 7.Zhao B., et al. Constitutive receptor-independent low density lipoprotein uptake and cholesterol accumulation by macrophages differentiated from human monocytes with macrophage-colony-stimulating factor (M-CSF). J. Biol. Chem. 2006;281:15757–15762. doi: 10.1074/jbc.M510714200. [DOI] [PubMed] [Google Scholar]
- 8.Amyere M., et al. Origin, originality, functions, subversions and molecular signalling of macropinocytosis. Int. J. Med. Microbiol. 2002;291:487–494. doi: 10.1078/1438-4221-00157. [DOI] [PubMed] [Google Scholar]
- 9.Swanson J.A., Watts C. Macropinocytosis. Trends Cell Biol. 1995;5:424–428. doi: 10.1016/S0962-8924(00)89101-1. [DOI] [PubMed] [Google Scholar]
- 10.Ma H.T., et al. Protein kinase C beta and delta isoenzymes mediate cholesterol accumulation in PMA-activated macrophages. Biochem. Biophys. Res. Commun. 2006;349:214–220. doi: 10.1016/j.bbrc.2006.08.018. [DOI] [PubMed] [Google Scholar]
- 11.Waldo S.W., et al. Heterogeneity of human macrophages in culture and in atherosclerotic plaques. Am. J. Pathol. 2008;172:1112–1126. doi: 10.2353/ajpath.2008.070513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Groves, K., Kossodo, S., Naranayan, N., Peterson, J., and Rajopadhye, M. 2006. A new platform of superbright fluorescent nanoparticles for in vivo imaging. Paper presented at the 5th Annual Meeting of the Society for Molecular Imaging. August 30–September 2. Waikoloa, Hawaii, USA. [Google Scholar]
- 13.Yang R.S., et al. Persistent tissue kinetics and redistribution of nanoparticles, quantum dot 705, in mice: ICP-MS quantitative assessment. Environ. Health Perspect. 2007;115:1339–1343. doi: 10.1289/ehp.10290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Goldstein J.L., Ho Y.K., Basu S.K., Brown M.S. Binding site on macrophages that mediates uptake and degradation of acetylated low density lipoprotein, producing massive cholesterol deposition. Proc. Natl. Acad. Sci. U. S. A. 1979;76:333–337. doi: 10.1073/pnas.76.1.333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Plump A.S., et al. Severe hypercholesterolemia and atherosclerosis in apolipoprotein E-deficient mice created by homologous recombination in ES cells. Cell. 1992;71:343–353. doi: 10.1016/0092-8674(92)90362-G. [DOI] [PubMed] [Google Scholar]
- 16.Stahl P., Gordon S. Expression of a mannosyl-fucosyl receptor for endocytosis on cultured primary macrophages and their hybrids. J. Cell Biol. 1982;93:49–56. doi: 10.1083/jcb.93.1.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Norbury C.C. Drinking a lot is good for dendritic cells. Immunology. 2006;117:443–451. doi: 10.1111/j.1365-2567.2006.02335.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pitas R.E., Innerarity T.L., Mahley R.W. Foam cells in explants of atherosclerotic rabbit aortas have receptors for beta-very low density lipoproteins and modified low density lipoproteins. Arteriosclerosis. 1983;3:2–12. doi: 10.1161/01.atv.3.1.2. [DOI] [PubMed] [Google Scholar]
- 19.Stephan Z.F., Yurachek E.C. Rapid fluorometric assay of LDL receptor activity by DiI-labeled LDL. J. Lipid Res. 1993;34:325–330. [PubMed] [Google Scholar]
- 20.Reynolds G.D., St Clair R.W. A comparative microscopic and biochemical study of the uptake of fluorescent and 125I-labeled lipoproteins by skin fibroblasts, smooth muscle cells, and peritoneal macrophages in culture. Am. J. Pathol. 1985;121:200–211. [PMC free article] [PubMed] [Google Scholar]
- 21.Brown M.S., Basu S.K., Falck J.R., Ho Y.K., Goldstein J.L. The scavenger cell pathway for lipoprotein degradation: specificity of the binding site that mediates the uptake of negatively-charged LDL by macrophages. J. Supramol. Struct. 1980;13:67–81. doi: 10.1002/jss.400130107. [DOI] [PubMed] [Google Scholar]
- 22.Buono C., Li Y., Waldo S.W., Kruth H.S. Liver X receptors inhibit human monocyte-derived macrophage foam cell formation by inhibiting fluid-phase pinocytosis of LDL. J. Lipid Res. 2007;48:2411–2418. doi: 10.1194/jlr.M700170-JLR200. [DOI] [PubMed] [Google Scholar]
- 23.Tabas I. Consequences of cellular cholesterol accumulation: basic concepts and physiological implications. J. Clin. Invest. 2002;110:905–911. doi: 10.1172/JCI16452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Spady D.K., Huettinger M., Bilheimer D.W., Dietschy J.M. Role of receptor-independent low density lipoprotein transport in the maintenance of tissue cholesterol balance in the normal and WHHL rabbit. J. Lipid Res. 1987;28:32–41. [PubMed] [Google Scholar]
- 25.Horbach G.J., van Leeuwen R.E., Yap S.H., van Bezooijen C.F. Changes in fluid-phase endocytosis in the rat with age and their relation to total albumin elimination. Mech. Ageing Dev. 1986;33:305–312. doi: 10.1016/0047-6374(86)90055-2. [DOI] [PubMed] [Google Scholar]
- 26.Slater H.R., Packard C.J., Shepherd J. Receptor-independent catabolism of low density lipoprotein. Involvement of the reticuloendothelial system. J. Biol. Chem. 1982;257:307–310. [PubMed] [Google Scholar]
- 27.Liu B., Xie C., Richardson J.A., Turley S.D., Dietschy J.M. Receptor-mediated and bulk-phase endocytosis cause macrophage and cholesterol accumulation in Niemann-Pick C disease. J. Lipid Res. 2007;48:1710–1723. doi: 10.1194/jlr.M700125-JLR200. [DOI] [PubMed] [Google Scholar]
- 28.Packard C.J., Slater H.R., Shepherd J. The reticuloendothelial system and low density lipoprotein metabolism in the rabbit. Biochim. Biophys. Acta. 1982;712:412–419. doi: 10.1016/0005-2760(82)90361-7. [DOI] [PubMed] [Google Scholar]
- 29.Clinton S.K., et al. Macrophage colony-stimulating factor gene expression in vascular cells and in experimental and human atherosclerosis. Am. J. Pathol. 1992;140:301–316. [PMC free article] [PubMed] [Google Scholar]
- 30.Rosenfeld M.E., et al. Macrophage colony-stimulating factor mRNA and protein in atherosclerotic lesions of rabbits and humans. Am. J. Pathol. 1992;140:291–300. [PMC free article] [PubMed] [Google Scholar]
- 31.Hoff H.F., Gaubatz J.W., Gotto A.M., Jr. Apo B concentration in the normal human aorta. Biochem. Biophys. Res. Commun. 1978;85:1424–1430. doi: 10.1016/0006-291X(78)91162-2. [DOI] [PubMed] [Google Scholar]
- 32.Smith E.B. Transport, interactions and retention of plasma proteins in the intima: the barrier function of the internal elastic lamina. Eur. Heart J. 1990;11(Suppl. E):72–81. doi: 10.1093/eurheartj/11.suppl_e.72. [DOI] [PubMed] [Google Scholar]
- 33.Smith E.B., Ashall C. Low-density lipoprotein concentration in interstitial fluid from human atherosclerotic lesions. Relation to theories of endothelial damage and lipoprotein binding. Biochim. Biophys. Acta. 1983;754:249–257. doi: 10.1016/0005-2760(83)90139-x. [DOI] [PubMed] [Google Scholar]
- 34.Kannel W.B., Castelli W.P., Gordon T. Cholesterol in the prediction of atherosclerotic disease. New perspectives based on the Framingham study. Ann. Intern. Med. 1979;90:85–91. doi: 10.7326/0003-4819-90-1-85. [DOI] [PubMed] [Google Scholar]
- 35.Lewington S., et al. Blood cholesterol and vascular mortality by age, sex, and blood pressure: a meta-analysis of individual data from 61 prospective studies with 55,000 vascular deaths. Lancet. 2007;370:1829–1839. doi: 10.1016/S0140-6736(07)61778-4. [DOI] [PubMed] [Google Scholar]
- 36.Kuchibhotla S., et al. Absence of CD36 protects against atherosclerosis in ApoE knock-out mice with no additional protection provided by absence of scavenger receptor A I/II. Cardiovasc. Res. 2008;78:185–196. doi: 10.1093/cvr/cvm093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Moore K.J., et al. Loss of receptor-mediated lipid uptake via scavenger receptor A or CD36 pathways does not ameliorate atherosclerosis in hyperlipidemic mice. J. Clin. Invest. 2005;115:2192–2201. doi: 10.1172/JCI24061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hashimoto S., Yamada M., Motoyoshi K., Akagawa K.S. Enhancement of macrophage colony-stimulating factor-induced growth and differentiation of human monocytes by interleukin-10. Blood. 1997;89:315–321. [PubMed] [Google Scholar]
- 39.Lowry O.H., Rosebrough N.J., Farr A.L. Protein measurement with the folin phenol reagent. J. Biol. Chem. 1951;193:265–275. [PubMed] [Google Scholar]
- 40.Kruth H.S. Histochemical detection of esterified cholesterol within human atherosclerotic lesions using the fluorescent probe filipin. Atherosclerosis. 1984;51:281–292. doi: 10.1016/0021-9150(84)90175-8. [DOI] [PubMed] [Google Scholar]