

Abstract
Krabbe disease (KD) is an inherited neurological disorder caused by the deficiency of galactocerebrosidase activity resulting in accumulation of psychosine, which leads to energy depletion, loss of oligodendrocytes, induction of gliosis and inflammation by astrocytes in CNS. Here, for the first time, we report the regulation of “cellular energy switch”, AMP-activated protein kinase (AMPK), by psychosine in oligodendrocytes and astrocytes. Psychosine treatment significantly downregulated AMPK activity, resulting in increased biosynthesis of lipids including cholesterol and free fatty acid (FFA) in oligodendrocytes cell line (MO3.13) and primary astrocytes. Pharmacological activator of AMPK, 5-Aminoimidazole-4-carboxamide-1- β-4-ribofuranoside (AICAR) attenuated the psychosine-mediated down-regulation of AMPK and restored altered biosynthesis of lipids. AICAR treatment also downregulated psychosine induced expression of proinflammatory cytokines and iNOS in primary astrocytes. However, AICAR treatment had no effect on psychosine induced-ROS generation, arachidonic acid release and death of oligodendrocytes; suggesting the specific role of AMPK in regulation of psychosine mediated inflammatory response of astrocytes but not in cell death of oligodendrocytes. This study delineates an explicit role for AMPK in psychosine induced inflammation in astrocytes without directly affecting the cell death of oligodendrocytes. It also suggests that AMPK activating agents act as anti-inflammatory agents and can hold a therapeutic potential in KD/twitcher disease, particularly when used in combination with drugs, which protect oligodendrocyte cell loss, such as sPLA2 inhibitor (Giri et al. 2006a).
Keywords: Krabbe disease, psychosine, inflammation, AMPK, oligodendrocyte, astrocytes
Introduction
KD is an inherited demyelinating disease, caused by accumulation of a toxic lipid, psychosine, due to deficiency of a crucial enzyme, galactosylcerebrosidase (GalC) (Suzuki 1998). Accumulation of psychosine in oligodendrocytes, results in their death which leads to demyelination in KD. The same is also observed in Krabbe's animal model, the twitcher mice (D.A. Wenger 2001). Toxic levels of psychosine have been shown to induce several signaling responses in cell culture which mimic KD/twitcher disease condition in vivo, like oligodendrocyte cell death (Taniike et al. 1999; Jatana et al. 2002; Haq et al. 2003), induction of pro-inflammatory cytokines (TNFα and IL-6) and iNOS (Giri et al. 2002), activation of PLA2 activity and generation of LPC (Giri et al. 2006a), downregulation of PI3K-Akt survival pathway (Zaka et al. 2005), expression of prostaglandin D synthase (Mohri et al. 2006a; Mohri et al. 2006b), formation of multinuclear globoid like cells in U937 monocytic cells (Kanazawa et al. 2000) and NK cells (Maghazachi et al. 2004). Apoptotic cells and expression of apoptotic-related molecule such as TNFα and its receptor (TNFR1) have been observed in twitcher mice brains and in brains of KD patients, underlining the hypothesis that induction of inflammation during progressive accumulation of psychosine is one of the main biochemical and pathogenetic mechanism of cell death in the Krabbe brain (LeVine and Brown 1997; Taniike et al. 1999; Jatana et al. 2002).
AMP-activated protein kinase (AMPK) is a heterotrimeric enzyme consisting of a catalytic subunit (α) and regulatory subunits (β and γ), and gets activated under stress conditions that deplete cellular ATP levels including glucose deprivation and hypoxia/ischemia (Hardie 2004; Carling 2005). Upon activation, it phosphorylates and inactivates a number of metabolic enzymes involved in ATP-consuming pathways like acetyl-CoA carboxylase (ACC) and HMG-CoA reductase and activates ATP-generating processes such as glucose uptake and fatty acid oxidation (Hardie 2004; Carling 2005). The role of AMPK has been documented in regulating various cellular physiological processes such as proliferation and apoptosis (Rattan et al. 2005; Motoshima et al. 2006), inflammation and immune response (Giri et al. 2004; Pilon et al. 2004; McCullough et al. 2005) and adipocyte differentiation (Habinowski and Witters 2001; Hwang et al. 2005; Giri et al. 2006b). Because of its involvement in diverse physiological processes, it has been implicated in various metabolic and inflammatory diseases such as diabetes, EAE and Alzheimer's disease (Tschape et al. 2002; Ayasolla et al. 2005; Nath et al. 2005; Long and Zierath 2006; Paintlia et al. 2006; Prasad et al. 2006).
Here, for the first time, we document the role of AMPK in psychosine mediated dysregulation of lipid homeostasis involved in cell death pathways in oligodendrocytes and inflammatory pathways in astrocytes with implication in KD/twitcher disease.
MATERIALS AND METHODS
Reagents
Dulbecco's modified Eagle's medium/4.5 glucose medium, fetal bovine serum and Hank's balanced salt solutions were obtained from Life Technologies (Grand Island, NY, USA). Antibodies against p-AMPKα (T172), AMPKα and p-ACC (S79) were purchased from Cell Signaling Technology (Beverly, MA, USA). ACC recombinant protein was purchased from Upstate Biotech (Billerica, MA). 6-carboxy 2', 7'-dichlorodihydrofluorescein diacetate (DCFDA) was from Molecular Probes (Eugene, OR). Antibodies against iNOS and β-actin were from Santa Cruz Biotechnology (Santa Cruz, CA, USA). 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) was purchased from Sigma and lactic dehydrogenase (LDH) kits was obtained from Roche (Nutley, NJ). Psychosine was from Matreya (Pleasant Gap, PA, USA). MO3.13 oligodendroglial cells were a kind gift from Dr. Catherine Waters (Division of Neuroscience, Biological Sciences, University of Manchester, UK).
Cell culture
Rat primary astrocytes and primary oligodendrocytes were prepared in our laboratory as described previously (Giri et al. 2004; Giri et al. 2006a). The cells were maintained in DMEM (4.5gm glucose/l) containing 10% FBS and 10μg/mL gentamicin. All cultured cells (astrocytes and oligodendrocytes) were maintained at 37°C in 5% CO2/95% air. At 80% confluency the cells were incubated with serum-free DMEM medium for 24 hr prior to incubation with various treatments. MO3.13 and oligodendroglial cells and B12 were maintained in DMEM (4.5gm glucose/l) containing 10% FBS and 10μg/mL gentamicin. The oligodendrocyte cell line (MO3.13) was maintained under serum-free conditions for 24 h before treatment and observed that serum-free conditions led to the expression of oligodendrocyte marker (GST-π) (Haq et al. 2003). Psychosine was initially dissolved in DMSO and then diluted in media at a final concentration of 0.1%.
AMPK activity
AMPKα activity was assayed in treated and untreated MO3.13 cells with various concentrations of psychosine in presence or absence of AICAR as described in figure legends using anti-AMPKα antibody for immunoprecipitation. For this, cells were lysed in lysis buffer (50 mM Tris-HCl, pH 7.4, containing 50 mM NaCl, 1 mM EDTA, 0.5 mM EGTA, 10% glycerol, and protease inhibitor mixture). Approximately, 200 μg of cell lysate was incubated with anti-AMPKα antibody for 2 hr, then 30 μl of protein A/G plus agarose was added and incubated for an additional 1 hr at 4°C. The immune complexes were washed twice in lysis buffer and twice in kinase buffer (62.5 mM HEPES, pH 7.0, 62.5 mM NaCl, 62.5 mM NaF, 6.25 mM sodium pyrophosphate, 1.25 mM EDTA, 1.25 mM EGTA, and 1 mM dithiothreitol) incubated at 30°C in 30 μl of kinase assay buffer containing 200 μM AMP, ATP mixture (200 μM ATP and 1.5 μCi of [γ32P]ATP), with ACC recombinant protein (Upstate Biotech, Millipore, MA) for 20 min. The reaction was terminated by adding sample buffer followed by boiling for 3 min. Samples were loaded on SDS-PAGE 4-20% Tris-glycine gel and transferred on nitrocellulose membrane followed by exposure on X-ray film (ECL Hyper film, Amersham LifeScience, Arlington Heights, IL).
Lipid biosynthesis
The biosynthesis of non-polar (cholesterol, triglycerides and fatty acids) lipids was examined in MO3.13 and primary astrocytes, using [14C]-acetate. Briefly, MO3.13 or astrocytes treated with psychosine in presence or absence of AICAR and pulsed with 1μCi of [14C]-acetate for final 2h of incubation. After incubation, incorporation of labeled acetate in lipids was examined by extracting lipids using Folch method. Incorporation of labeled acetate into non-polar lipids was examined by running HPTLC plates followed by exposing them for three days on X-ray film at -70°C.
Arachidonic acid (AA) release assay
Primary oligodendrocyte or MO3.13 cells were pre-labeled with 0.5 μCi/ml [3H]-arachidonic acid in DMEM medium containing 10% FBS for 12 h at 37°C in a humidified incubator supplied with 95% air and 5% CO2. The labeled cells were then washed twice with serum-free media and treated with indicated treatments in serum-free DMEM medium at 37°C. At various time periods, radioactivity in the medium (100μl) was measured using a liquid scintillation counter as described before (Giri et al. 2006a).
Measurement of ROS
ROS were determined using the membrane permeable fluorescent dye 6-carboxy 2', 7'-dichlorodihydrofluorescein diacetate (DCFH2-DA) in serum-free medium as described before (Giri et al. 2006a). The cultured cells with or without treatment were incubated with 5μM DCF dye in PBS for 2 hr at 37°C. The change in fluorescence was determined at excitation 485 nm and emission 530 nm using a Soft Max Pro spectrofluorometer (Molecular Devices, Sunnyvale, CA).
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT), and lactic dehydrogenase (LDH) Assays for Cell Growth and Viability
The viability of cells was evaluated using MTT and LDH assays. MTT is based on the cleavage of tetrazolium salt MTT to a dark blue formazan product by mitochondrial dehydrogenase in viable cells. The absorbance of viable cells was measured with a test wavelength of 570 nm and a reference wavelength of 630 nm. LDH activity was determined in cell supernatant using LDH kit (Roche, Nutley, NJ) as per the manufacturer's instruction.
DNA Fragmentation
After treatment, cells were harvested and then lysed with lysis buffer containing 20 mM NaCl, 10 mM Tris-HCl (pH 8.0), 25 mM EDTA, 1% sodium dodecyl sulfate, and 1 mg/ml proteinase K for 2 hr in a 55°C water bath. Standard phenol/chloroform/isoamyl alcohol method (25:24:1) was used to remove protein and extract nucleic acid. RNA was digested with RNase A (100 μg/ml) for 2 hr at 37°C, and DNA concentrations were determined. DNA extracts were electrophoresed on a 2% agarose gel at 50 V for 45 min and visualized with ethidium bromide staining under UV illumination (Giri et al. 2006a).
Activation of Caspase 3
Caspase 3 activation was determined using the substrates [acetyl-Asp-Glu-Val-Asp (Ac-DEVD)-7-amino-4-methyl-coumarin (AMC) for caspase 3 procured from Biomol (Plymouth Meeting, PA)], which are cleaved into fluorescent reaction products by the respective caspase action (Giri et al. 2006a). For analysis of caspase 3 activity, cells were collected and lysed in 100 μL of lysis buffer containing 10 mm Tris-HCl, pH 7.5, 130 mm NaCl, 1% (w/v) Triton X-100, and 10 mm sodium pyrophosphate. The lysate was incubated with 20μm substrate in reaction buffer [20 mm HEPES/10% (v/v) glycerol/2 mm dithiothreitol] at 37°C for 15 min. The fluorescence intensity (expressed as arbitrary U/μg protein) of liberated AMC was measured using spectrofluorometry (excitation wavelength = 380 nm; emission wavelength = 460 nm).
Western Blot Analysis
Western blot analysis for phospho-ACC, phospho-AMPKα, iNOS, Cox-2, Fra-1 and β-actin was performed as described before (Giri et al. 2004; Rattan et al. 2005; Giri et al. 2006b). Briefly, treated oligodendrocytes or primary astrocytes were harvested and lysed with lysis buffer (50 mM Tris-HCl [pH 7.5], 250 mM NaCl, 5 mM EDTA, 50 mM NaF, and 0.5% Nonidet P-40) containing a protease inhibitor cocktail (Sigma). The lysate was clarified by centrifugation at 10,000g for 15 min at 4°C. Equal amounts of total protein (50μg) was subjected to 4-20% SDS-PAGE and electrophoretically transferred to a High-Bond nitrocellulose membrane (Amersham LifeScience, Arlington Heights, IL). After blocking with Tween 20-Tris-buffered saline (TTBS; 20 mM Tris-HCl [pH 7.6], 137 mM NaCl, and 0.05% Tween 20) containing 5% nonfat milk for 1 hr at room temperature, the membranes were incubated overnight at 4°C with the primary antibodies at 1:1,000 dilution in bovine serum albumin (BSA) buffer (TTBS with 3% BSA). The membranes were then washed three times for 10 min each in TTBS and incubated with an appropriately diluted horseradish peroxidase-labeled secondary antibody (1:5,000) in blotting buffer for 1 hr at room temperature. The membranes were washed three times, reacted with ECL reagent (Amersham Life Science), and subjected to autoradiography.
Enzyme assay for β-oxidation of palmitic acid
The enzyme activity of [1-14C] palmitic acid β-oxidation to acetate was measured in intact cells suspended in HBSS. Briefly, the reaction mixture in 0.25 ml of HBSS contained 50-60 μg of protein and 6 μM [1-14C] palmitic acid. Fatty acid was solubilized with α-cyclodextrin and β-oxidation of [1-14C] palmitic acid was carried out as described previously (Khan et al. 1998).
Statistical Analysis
Data are expressed as mean ± SD for n experiments. Statistical comparisons were made using Student Newman-Keuls test as required. A P value < 0.05 was considered statistically significant.
RESULTS
AMPK activity is downregulated by psychosine
To examine the effect of psychosine on AMPK, oligodendrocyte cell line, MO3.13 was treated with various doses of psychosine (5-20μM) for different time periods (0-8h). Psychosine was observed to down regulate the phosphorylation of ACC, a substrate of AMPK, in a time (Fig. 1A) and dose dependent manner (Fig. 1B), indicating decreased activity of AMPK. In corroboration, decreased enzymatic activity of AMPK by psychosine was also observed as assessed by kinase assay using recombinant ACC as a substrate, in a dose and time dependent manner (Fig. 1C). These observations were found to be consistent in primary oligodendrocyte and in rat oligodendrocyte cell line (B12) as psychosine treatment (10-20μM) inhibits basal phospho- levels of ACC and AMPKα in both cells (Fig. D & E) suggesting that downregulation of AMPK by psychosine is general phenomenon in oligodendrocytes. Psychosine (20μM) does not induce cell death at 8h of incubation (data not shown).
FIGURE 1. Psychosine induces downregulation of AMPK activity in oligodendrocyte cell line.

Oligodendrocyte cell line, MO3.13 was treated with psychosine (20γM) for indicated time periods (1-8h). Cells were lysed in lysis buffer as mentioned in Material and methods followed by immunoblot analysis for p-ACC and β-actin for equal loading of protein. Numeric values are shown as a ratio of densitometry values of p-ACC/β actin (A). MO3.13 cells were treated with indicated concentration of psychosine (5-20γM) for 8h and processed for p-ACC and β-actin as described above (B). Blots are representative of two independent experiments. For AMPK kinase assay, MO3.13 cells were treated for indicated time periods (Ci) and concentrations of psychosine (Cii) as described above. AMPKα was immunoprecipitated with anti-AMPKα antibody followed by kinase assay as described in Material and Methods. Densitometry analysis was performed and represented as a bar graph, which are representative of three independent experiments. Data are mean × SD of three values. *** P< 0.001, ** P< 0.01, and * P< 0.05, compared to untreated cells. Primary oligodendrocyte and rat oligodendrocyte cell line (B12) were treated with psychosine (10-20γM) for 8h followed by immunoblot analysis of pACC, pAMPKα and β-actin (D-E). Blots are representative of two independent experiments.
Lipid homeostasis is altered by psychosine in oligodendrocytes
Since psychosine inhibits AMPK activity in oligodendrocytes, biosynthesis of fatty acid and cholesterol synthesis are expected to be higher as ACC and HMG-CoA reductase will be in an increased activated status compared to untreated cells (basal level). To examine this, MO3.13 cells were treated with various concentrations of psychosine for various time points, followed by [14C]-acetate treatment. In accordance, psychosine treatment significantly induced the biosynthesis of cholesterol and FFA in a time and dose dependent manner (Fig. 2 A, B & C), reflecting the inhibition of AMPK activity.
FIGURE 2. Psychosine alters lipid homeostasis in MO3.13 cells.

MO3.13 cells were treated with psychosine for various time periods as indicated (A) and with different concentrations of psychosine (5-20γM) for 8h followed by [ 14C]-acetate pulse for 2 hr. Lipids were isolated and incorporation of labeled acetate in FFA and cholesterol was assayed by HP-TLC as described in Methods and Materials (B). Densitometry analysis of FFA and cholesterol is plotted as a graph bars. Data are mean × SD of four values. *** P< 0.001, ** P< 0.01, * P< 0.05, NS; not significant, compared to untreated cells.
Regulation of AMPand, lipid homeostasis by sphingolipids
Since psychosine inhibits AMPK activity and alters lipid homeostasis, we investigated the specificity of the observed phenomenon, whether this effect is due to psychosine only or by other sphingolipids also. For this, MO3.13 cells were treated with various sphingolipids like psychosine (galactopsychosine), glucopsychosine, ceramide and sphingosine (20μM) for 8h and then processed for detection of phosphorylation of ACC. We observed that glucopsychosine was equally potent in downregulating AMPK activity as psychosine, whereas, ceramide and sphingosine had no effect (Fig. 3A). To further corroborate this data, we assessed β-oxidation in MO3.13 cells treated with sphingolipids. As shown in figure 3B, psychosine and glucopsychosine both were potent in inhibiting the β oxidation in MO3.13 cells as expected while sphingosine showed no effect. However, ceramide significantly induced the β oxidation in MO3.13 cells (Fig. 3B). Consistent with decreased activities of AMPK and β oxidation, psychosine and glucopsychosine significantly induced biosynthesis of FFA and cholesterol in MO3.13 cells (Fig. 3Ci-iii). Ceramide treatment also induced the biosynthesis of FFA without modulation of cholesterol, however, sphingosine had no effect on any non-polar lipids compared to untreated cells.
FIGURE 3. Regulation of AMPK by various sphingolipids.
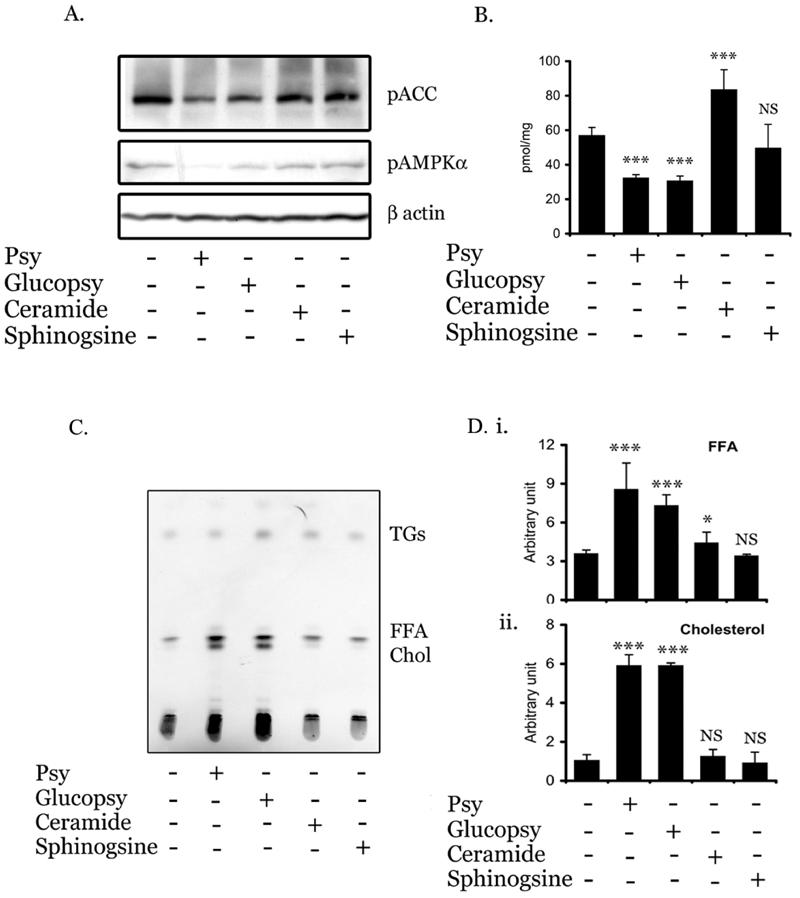
To examine the effect of various sphingolipids on AMPK in MO3.13 cells, MO3.13 cells were treated with 20γM of psychosine (galactopsychosine), glucospsychosine, ceramide and sphingosine for 8h followed by immunoblot analysis of pACC and β actin (A). MO3.13 cells were treated with various sphingolipids (20γM) for 8h and β oxidation of C16:0 was carried out as described in Methods and Materials (B). Data are expressed as mean ± SD of 6 different values. *** P<0.001, NS not significant as compared with untreated cells. To examine the effect of sphingolipids on lipid biosynthesis, MO3.13 cells were treated with various sphingolipids (20γM) for 6h followed by [ 14C]-acetate pulse for 2 hr. Lipids were isolated and incorporation of labeled acetate in FFA and cholesterol were assayed by HPTLC as described in Methods and Materials (C). Densitometry analysis of FFA and cholesterol is plotted as bar graphs (i, ii &iii). Data are means ± SD of three values. *** P< 0.001, ** P< 0.01, * P< 0.05, NS; not significant, compared to untreated cells.
AICAR restored psychosine mediated downregulation of AMPK activity and lipid alteration
It was of further interest to examine if forced activation of AMPK using its pharmacological activator, would over-ride the effects of psychosine on lipid metabolism. For this, we employed AICAR, an established AMPK activator in various cell types (Sullivan et al. 1994; Corton et al. 1995). Once taken up by the cell, AICAR is phosphorylated to AICA riboside monophosphate (ZMP), which functions as an AMP analog and activates AMPK (Corton et al. 1995). Treatment of MO3.13 cells with AICAR reversed the psychosine mediated downregulation of AMPK and ACC phosphorylation in a dose dependent manner (Fig. 4A&B). AICAR also restored the psychosine mediated inhibition of C16:0 β oxidation in MO3.13 cells (Fig. 4C). Treatment of AICAR also restored the altered lipid homeostasis induced by psychosine as observed by FFA and cholesterol levels in oligodendrocyte cell line (Fig. 4D&E).
FIGURE 4. Pharmacological activator of AMPK restores psychosine mediated downregulation of AMPK activity and lipid alteration.


MO3.13 cells were pretreated with AICAR (0.1-1.0mM) followed by incubation with psychosine (20γM). After 8h of incubation, cells were lysed in lysis buffer as mentioned in Material and methods followed by immunoblot analysis for p-ACC, p-AMPKα and β-actin for equal loading of protein (A). Densitometry values of ratio between p-AMPKα/β actin and p-ACC/β actin are plotted as bar graphs (B). Data are mean ± SD of three values. ** P< 0.01 as compared to untreated cells; *** P< 0.001, ### P< 0.001, as compared to untreated cells. MO3.13 cells were treated with psychosine (20γM) in the presence or absence of AICAR (1mM) for 8h and β oxidation of C16:0 was carried out as described in Methods and Materials (C). Data are expressed as mean ± SD of 6 different values. *** P<0.001, ## p<0.01, NS not significant as compared with untreated cells; ## P<0.01 as compared with psychosine treated cells. MO3.13 cells were pretreated with AICAR (0.1-1.0mM) and psychosine (20γM) for 6h followed by [14C]-acetate pulse for additional 2 hr. Lipids were isolated and incorporation of labeled acetate in cholesterol, fatty acids and triglycerides was assayed by HP-TLC as described in Methods and Materials (D). Densitometry values of psychosine treated samples in presence or absence of AICAR (1mM) FFA and cholesterol are plotted as bar graph (E). Data are means ± SD of four values. *** P< 0.001 compared to untreated cells; ### P< 0.001 as compared to psychosine treated cells.
Psychosine mediated cell death was not rescued by AMPK activation
Oligodendrocyte cell death induced by progressive accumulation of psychosine, in vivo and in vitro, is a hallmark of demyelinating KD. Because AICAR restored psychosine mediated downregulation of AMPK and altered lipid homeostasis in oligodendrocytes, we next examined whether activation of AMPK will attenuate psychosine-induced oligodendrocyte cell death. For this, MO3.13 cells were pre-treated with different concentrations of AICAR (0.1-2mM) for 2h, followed by psychosine (20μM) treatment. After 24 h, MTT and LDH assays were performed to assess cell survival. Consistent with our previous observations, psychosine at a concentration of 20 μM, induced cell death (Haq et al. 2003; Giri et al. 2006a), but, treatment with AICAR did not rescue the oligodendrocytes from psychosine-mediated cell death (Fig. 5A & B).
FIGURE 5. AMPK activator does not restore/attenuate psychosine mediated activation of cell death in MO3.13.

To examine cell viability, MO3.13 cells were plated and treated with AICAR (0.1-2.0mM) followed by psychosine treatment (20 μM) for 24 h. Viable cells were determined by MTT assay (A) and cell supernatant was processed for LDH assay (B) as described in Material and Methods. Data are the mean ± SD of six values. *** P<0.001 as compared with untreated cells; and NS, not significant as compared with psychosine-treated cells. Cells were treated as shown in figure and lysates were prepared and subjected to caspase 3 activity assay determined by measuring the fluorescence intensity (expressed as arbitrary U/mg protein) of liberated amino-4-methyl-coumarin using spectrofluorometry as described as in Materials and Methods (C). Data are mean ± SD of six values. *** P<0.001 compared with untreated cells; and NS, not significant as compared with psychosine-treated cells. MO3.13 cells were treated as in Materials and Methods and at 24 h were lysed in DNA lysis buffer; the extracted DNA obtained was electrophoresed on a 2% agarose gel. H2O2 (200μM) was used to induce DNA fragmentation as a control (D). Gel is representative of two individual experiments.
To further substantiate the data, effect of AICAR on psychosine induced caspase 3 activity and DNA fragmentation was examined. Cells were treated with psychosine in the presence or absence of different concentrations of AICAR. After 24h incubation, cells were lysed and assayed for caspase 3 activity. As depicted in figure 5C, AICAR treatment did not attenuate psychosine-induced caspase 3 activity in the oligodendrocyte cell line. For DNA fragmentation, cytoplasmic DNA isolated from cells treated in a similar manner was subjected to agarose gel electrophoresis. A DNA ladder was observed in cells treated with psychosine but not in control cells, and this ladder was increased further by the treatment with AICAR at 24 h of treatment (Fig. 5D). Therefore, these studies document that AICAR did not protect from psychosine-induced oligodendrocyte apoptotic cell death, even though effectively correcting psychosine induced lipids alteration.
AICAR had no effect on psychosine mediated activation of AA release and reactive oxygen species (ROS) generation
Since, we have recently documented that psychosine induces apoptosis in primary oligodendrocytes and the MO3.13 cell line via induction of sPLA2 activity and ROS generation (Giri et al. 2006a), therefore, we further examined the effect of AMPK activation on psychosine induced AA release and ROS generation in primary oligodendrocytes and MO3.13 cells. For this, primary oligodendrocytes or MO3.13 cells were labeled with radioactive [3H]-arachidonic acid (AA). After 12 h of incubation, cells were washed followed by treatment with psychosine (20 μM) in the presence or absence of different concentrations of AICAR (0.1-1mM). As shown in figure 6A, primary oligodendrocytes treated with psychosine exhibited a significant release of labeled AA. A similar trend was observed when the oligodendrocyte cell line MO3.13 was used (Fig. 6B). Whereas, treatment with AICAR did not attenuate psychosine induced AA release at concentrations tested in primary oligodendrocytes and MO3.13 cells (Fig. 6A & B).
FIGURE 6. AMPK activator has no effect on psychosine mediated activation of AA release and ROS generation in oligodendrocytes.

Primary oligodendrocytes (A) or MO3.13 cells (B) were labeled with [3H]AA as described. Cells were treated with different concentrations of AICAR (0.1-1.0mM) for 2hrs, followed by psychosine (20μM) treatment. After 8 h, radioactivity released in the medium (100 μl) was measured. Data are the mean ± SD of six values. *** P< 0.001 as compared with untreated cells; NS, not significant as compared with psychosine-treated cells. Primary oligodendrocyes (C) or MO3.13 cells (D) were treated for 2hrs with different concentrations of AICAR (0.1-1.0mM) and psychosine (20μM) in the presence of 6-carboxy29, 79-dichlorodihydrofluoresceindiacetate (DCFDA) dye, and fluorescence was recorded as described in Material and Methods. Data are mean ± SD of six values. *** P<0.001 compared with untreated cells; and NS, not significant as compared with psychosine-treated cells.
To investigate whether AICAR affects ROS generation, primary oligodendrocyte and MO3.13 cells were treated with psychosine (20μM) along with or without AICAR (0.1-1.0mM). ROS generation was measured using DCFDA dye followed by fluorescence reading. As shown in figure 6 (C & D), AICAR did not inhibit the psychosine-induced ROS generation in oligodendrocytes. These observations indicate that psychosine induced generation of ROS and activation of PLA2 are either upstream of AMPK or a parallel signaling pathway.
AMPK activation inhibited LPS/IFNγ/psychosine mediated induction of NO/iNOS/Cox-2 and proinflammatory cytokines in primary astrocytes
Previously, we have demonstrated higher expression of iNOS in activated astrocytes in KD disease brain (Giri et al. 2002). Psychosine treatment has also been reported to induce LPS/IFNγ-mediated proinflammatory cytokines and NO production in C6 glial and primary astrocytes (Giri et al. 2002). Since, AICAR has been reported to be anti-inflammatory molecule in astrocytes (Giri et al. 2004), we investigated the effect of AICAR in this disease process. Primary astrocytes were pre-treated with AICAR followed by LPS and psychosine. As documented before (Giri et al. 2002), psychosine treatment upregulated LPS mediated NO release, TNFα and IL-6 production, which was completely blocked by AICAR treatment (Fig. 7 Ai, ii & iii). AICAR treatment also down-regulated the expression of pro-inflammatory mediators including iNOS and Cox-2 protein expression, induced by LPS/IFNγ/psychosine (Fig. 7B). Psychosine further induced the expression of cytokine-mediated Fra-1 protein (a FOS family member and an AP-1 transcription factor component,) which is inhibited by AICAR treatment (Fig. 7B), suggesting Fra-1 might be one of the mechanisms of AICAR mediated downregulation of psychosine induced pro-inflammation in astrocytes. To evaluate the effect of AICAR on cytokines/psychosine induced downregulation of β oxidation, we examined the C16:0 oxidation in primary astrocytes. Primary astrocytes were treated with LPS/IFNγ and/or psychosine in the presence or absence of AICAR for 12h followed by estimation of β oxidation of C16:0. As shown in figure 7C, treatment with LPS/IFNγ significantly inhibited β oxidation which was further potentiated by the addition of psychosine. Psychosine alone was also able to downregulate the β oxidation in primary astrocytes. However, treatment with AICAR significantly reversed the inhibition of β oxidation induced by cytokines, alone or in combination with psychosine (Fig. 7C).
FIGURE 7. AMPK activator (AICAR) inhibits LPS/psychosine mediated induction of NO and proinflammatory cytokines production in primary astrocytes.
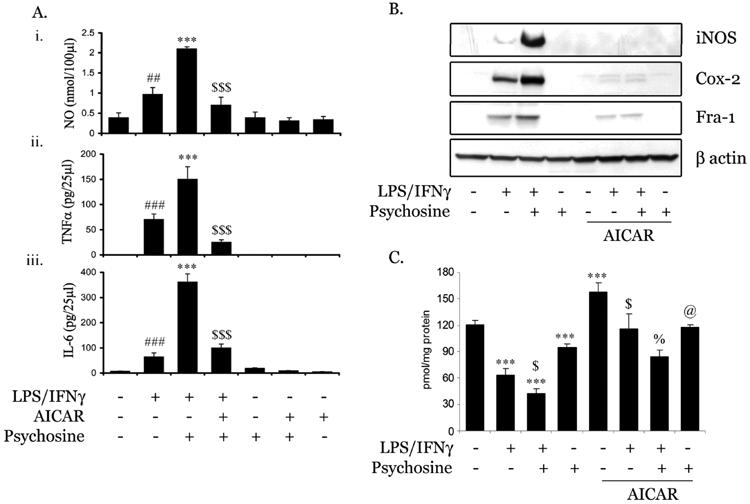
Under serum-free conditions, primary astrocytes were treated with AICAR (1mM) for 2h followed by psychosine (15μM) in the presence or absence of LPS/IFNγ (1μg/ml). After 24 h of incubation, media was removed and NO (i), TNFα (ii) and IL-6 (iii) levels estimated as described in Materials and Methods (A). Data are expressed as mean ± SD of 4 different values. ### P<0.001, ## P<0.01 as compared with untreated cells; ***P<0.001 as compared to LPS/IFNγ treated cells; $$$P<0.001 as compared to LPS/IFNγ/psychosine-treated cells. Cell lysate was used to detect levels of iNOS, Cox-2, Fra-1 and β-actin proteins expression using immunoblot analysis using their specific antibodies as described in Methods and Materials (B). Primary astrocytes were treated with AICAR and LPS/IFNγ as described before. After 12h incubation, β oxidation of C16:0 was carried out (C). Data are expressed as mean ± SD of 6 different values. *** P<0.001 as compared with untreated cells; $P<0.001 as compared to LPS/IFNγ treated cells; %P<0.01 as compared to LPS/IFNγ/psychosine-treated cells; @ P<0.05 as compared to psychosine-treated cells.
Lipid homeostasis in primary astrocytes is altered by psychosine
Further we examined the effect of psychosine on lipid biosynthesis in primary astrocytes. Primary astrocytes were treated with psychosine for various time points (2-8h) or various concentrations of psychosine and followed by [14C]-acetate pulse. In accordance, psychosine treatment induced the biosynthesis of cholesterol and FFA in primary astrocytes (Fig. 8 A-D) as in oligodendrocytes (Fig. 2).
FIGURE 8. Psychosine alters lipid homeostasis in primary astrocytes.

Primary astrocytes were treated with different concentration of psychosine (5-30μM) (A, C) and various time periods (psychosine, 20μM) as indicated and (B, D) for 8h followed by [14C]-acetate pulse for 2 hr. Lipids were isolated and incorporation of labeled acetate in cholesterol and FFA were assayed by HP-TLC as described in Methods and Materials. Densitometry analysis of cholesterol (A, B) and FFA (C, D) is plotted as bar graph. Data are mean ± SD of four values. NS; not significant, * P<0.05, ** P<0.01, ** P<0.001, as compared with untreated cells.
Psychosine also downregulates AMPK activity in primary astrocytes
Since psychosine inhibits lipid biosynthesis in primary astrocytes, we examined the effect of psychosine on AMPK activity in primary astrocytes. As shown in figure 9A, psychosine treatment at a concentration of 15μM partially inhibits the phosphorylation of ACC, while at 20μM concentration; it completely inhibits the phosphorylation of ACC (Fig. 9B). In contrast to other sphingolipids (ceramide and sphingosine), but similar to as seen in oligodendrocytes, glucopsychosine was equally potent in inhibiting AMPK in primary astrocytes as psychosine.
FIGURE 9. Psychosine induces downregulation of AMPK activity in Primary astrocytes.

Primary astrocytes were treated with psychosine (15μM) for various time periods (2-8h). Cells were lysed in lysis buffer as mentioned in Material and methods followed by immunoblot analysis for p-ACC and β actin for equal loading of protein. Numeric values were shown as a ratio of densitometry values of p-ACC/β actin (A). Primary astrocytes were treated with various concentrations of psychosine (5-20μM), glucopsychosine (20μM), ceramide (20μM) and sphingosine (20μM) for 8h and processed for p-ACC and β actin as described above (B). Blots are representative of three individual experiments.
These observations document that psychosine down-regulates AMPK in oligodendrocytes and in astrocytes. Activation of AMPK by AICAR reverses the psychosine-induced inflammatory signaling pathways for induction of proinflammatory cytokines, iNOS and Cox-2 in astrocytes but has no effect in psychosine-induce ROS-sPLA2-caspase 3 activation and cell death signaling pathway in oligodendrocyte cell death.
DISCUSSION
Accumulation of psychosine, generation of proinflammatory cytokines, loss of oligodendrocytes and demyelination are a hallmark of KD, however, the mechanism of psychosine-induced inflammation and loss of oligodendrocytes resulting in demyelination is not well understood (Suzuki 1998; Wenger 2001). In the present study, we have shown that AMPK activity plays differential role in oligodendrocyte and astrocytes in psychosine mediated inflammation and toxicity.
Oxidative stress is one of the key components in psychosine mediated toxicity in oligodendrocytes. Inhibition of generation of psychosine mediated ROS by anti-oxidants such as NAC inhibited ROS generation, LPC generation, AA release, sPLA2 activity, caspase 3 activity and finally restored psychosine mediated oligodendrocytes cell death (Giri et al. 2006a) and provided a rational for potential of anti-oxidant(s) as a therapeutic drug/s for Krabbe/twitcher. On one hand, psychosine induces oxidative stress and apoptotic oligodendrocytes cell death by activating JNK-AP-1 pathway (Haq et al. 2003) and on other hand, it down regulates cell survival pathways including NF-κB (Haq et al. 2003) and PI-3K-Akt (Zaka et al. 2005). However the source of oxidative stress by psychosine treatment is still not known.
AMP-activated protein kinase (AMPK) is recently identified as a energy sensor in cells, which is tightly regulated by the cellular AMP:ATP ratio (Hardie 2004; Carling 2005). It has been shown to play a central role in the regulation of energy homeostasis and metabolic stress (Hardie 2004; Carling 2005). It has been suggested that the AMPK system is evolved to protect the cell against ATP depletion by inhibiting biosynthetic pathways and stimulating energy-generating pathways (Hardie et al. 1998). It plays a major role in the regulation of lipid metabolism and phosphorylates key regulatory enzymes, including ACC and HMG-CoA reductase (Munday et al. 1988; Clarke and Hardie 1990). AMPK, a heterotrimeric complex comprising a catalytic subunit (α1 or α2), a regulatory subunit (β1, β2, γ1, γ2, or γ3) (Hardie et al. 2003). AMPKα1 and α2 are expressed through out the brain. The basal expression of α2 is low in astrocytes, however, but is increased significantly in activated astrocytes (Turnley et al. 1999). The oligodendrocyte cell line (MO3.13) shows more prominent expression of α1 compared to α2 (data not shown) subunits. The differential distribution of α1 and α2 in cytosol and nucleus, respectively, plays an important role in its action. Although the role of nuclear localized AMPK α2 is not yet known but it can be speculated that it might regulate transcription, as it has been shown to phosphorylate/regulate various transcription factors such as carbohydrate responsive element-binding protein (ChREBP) (Kawaguchi et al. 2002), HNF4α (Leclerc et al. 2001), coactivator p300 (Yang et al. 2001) and AICAR responsive element binding protein (AREBP) (Inoue and Yamauchi 2006). Accordingly, downregulation of AMPK should lead to the alteration in lipid homeostasis and its biosynthesis. Our observation in psychosine treated oligodendrocytes is consistent with observed psychosine induced biosynthesis of cholesterol and FFA in astrocytes.
The observed higher expression of AMPK in reactive astrocytes and identification of co-localization of AMPK isoforms in the nucleus indicate that AMPK may also have functions in addition to the regulation of energy metabolism (Salt et al. 1998; Turnley et al. 1999). These findings are consistent with the role of AMPK in the inflammatory process reported in this study. Recently, AMPK has been reported to have a protective function during glucose deprivation in neurons (Culmsee et al. 2001) and in APPL/APP processing (Tschape et al. 2002). All of these previous studies along with our study suggest a critical role for AMPK in the CNS that remains to be established.
Several studies including ours, have reported the expression of proinflammatory cytokines (IL-6, TNF α) and chemokines (MCP-1, IP-10, MIP-1α MIP-1β and RANTES) in cell culture and in twitcher mice brain (LeVine and Brown 1997; Wu et al. 2000; Wu et al. 2001; Giri et al. 2002; Haq et al. 2006). Also, expression of iNOS and GFAP in activated astrocytes in brains of KD patients, indicates the involvement of an inflammatory process in the pathogenesis of KD (Giri et al. 2002). Psychosine induced cytokine-mediated nuclear translocation of AP-1 and C/EBP by potentiating the expression of Fra-1 and C/EBP-δ proteins. This suggests that psychosine maintained or sustained the cytokine-primed expression of inflammatory mediator (iNOS) by further potentiating the nuclear translocation of AP-1 and C/EBP without modulating the cytokine-mediated transcription activity of NF-κB (Giri et al. 2002). Targeting the C/EBP-δ in psychosine mediated inflammation may be one of the important aspect as it is involved in the regulation of various genes including inflammatory genes (IL-6, IL-1β, TNFα, IL-8 and IL-12), and genes encoding proteins important for macrophagic or granulocytic functions such as iNOS, lysozyme, myeloperoxidase, and neutrophil elastase, the gene encoding the granulocyte colony stimulatory factor, and the macrophage, granulocyte, and granulocyte- macrophage receptor genes (Poli 1998). Since AMPK activator (AICAR) downregulates the expression of C/EBP-δ (Giri et al. 2004) and psychosine induced expression of iNOS and production of cytokines (Giri et al. 2002), therefore, AMPK activator may be a potential therapeutic anti-inflammatory agent for KD/twitcher. AICAR also inhibits inflammatory response by downregulating MAPKs activation (Giri et al. 2004) and PI3K-Akt pathway (Jhun et al. 2004), however, psychosine induces MAPKs activity (Giri et al. 2002; Haq et al. 2003; Giri et al. 2006a), may be one of the protective mechanism of AICAR.
These studies raise a hope that pharmacological drugs having anti-inflammatory effect and/or the drugs that protect oligodendrocyte from psychosine mediated death may have potential to be therapeutic drug for KD/twitcher whether it affect accumulated psychosine levels or not. At present, there is no treatment for KD except hematopoietic stem cell transplantation, which is not a cure but does slow down the course of the disease in humans as well as in twitcher mice (Eto et al. 2004; Yagi et al. 2004; Escolar et al. 2005; Kondo et al. 2005; Luzi et al. 2005). Our previous study using sPLA2 inhibitor (DEDA) provided protection to oligodendrocytes from psychosine mediated toxicity, therefore combination with AMPK activator with inhibitors of sPLA2 may be an attractive option for therapy. We have already documented AICAR as a therapeutic agent in cell culture and various inflammatory models (EAE, endotoximia, transplantation and diet induce obesity) (Giri et al. 2004; Lin et al. 2004; Ayasolla et al. 2005; Nath et al. 2005; Giri et al. 2006b; Paintlia et al. 2006; Prasad et al. 2006). Moreover, AICAR has been used previously as a drug for treating Lesch-Nyhan syndrome at a relatively high dose (100 mg/kg body weight) safely and with no side effects (Page et al. 1994). The safety, tolerance, and pharmacokinetics of intravenous doses of 10-100 mg/kg of AICAR in healthy men have been reported previously (Dixon et al. 1991). However, AICAR has a high clearance and poor bioavailability following oral administration.
In summary, present study documents that psychosine mediates inactivation of AMPK which results in lipid alteration both in astrocytes and oligodendrocytes. Although, AMPK activator did not rescue oligodendrocytes from psychosine mediated cell death, but it down regulated psychosine mediated production of NO, expression of proinflammatory cytokines and mediators (iNOS and Cox-2) in primary astrocytes suggests specific role of AMPK in oligodendrocytes and astrocytes. The observed loss of oligodendrocytes and induction of inflammatory disease in KD/twitcher brain suggest that drugs that attenuates loss of oligodendrocytes such as inhibitor of sPLA2 and inhibitor of inflammatory response such as activator of AMPK, in combination, may prove to be potential therapeutic for KD.
Acknowledgment
We would like to thank Dr. Catherine Waters (University of Manchester, UK) for the gift of MO 3.13 cells. We acknowledge the contribution of Drs. Je-Song Won and Jinsu Kim for providing primary astrocytes for this study. We thank Dr. Miguel Contreras for critical suggestions for this study and Joyce Bryan and Carrie Barnes for laboratory assistance. This work was supported by grants (NS-22576, NS-34741, NS 37766 and NS-40810) from the National Institutes of Health. This work was conducted in a facility constructed with support from the National Institutes of Health, Grant Number C06 RR018823 from the Extramural Research Facilities Program of the National Center for Research Resources.
REFERENCES
- Ayasolla KR, Giri S, Singh AK, Singh I. 5-aminoimidazole-4-carboxamide-1-beta-4-ribofuranoside (AICAR) attenuates the expression of LPS- and Abeta peptide-induced inflammatory mediators in astroglia. J Neuroinflammation. 2005;2:21. doi: 10.1186/1742-2094-2-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carling D. AMP-activated protein kinase: balancing the scales. Biochimie. 2005;87:87–91. doi: 10.1016/j.biochi.2004.10.017. [DOI] [PubMed] [Google Scholar]
- Clarke PR, Hardie DG. Regulation of HMG-CoA reductase: identification of the site phosphorylated by the AMP-activated protein kinase in vitro and in intact rat liver. Embo J. 1990;9:2439–2446. doi: 10.1002/j.1460-2075.1990.tb07420.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corton JM, Gillespie JG, Hawley SA, Hardie DG. 5-aminoimidazole-4-carboxamide ribonucleoside. A specific method for activating AMP-activated protein kinase in intact cells? Eur J Biochem. 1995;229:558–565. doi: 10.1111/j.1432-1033.1995.tb20498.x. [DOI] [PubMed] [Google Scholar]
- Culmsee C, Monnig J, Kemp BE, Mattson MP. AMP-activated protein kinase is highly expressed in neurons in the developing rat brain and promotes neuronal survival following glucose deprivation. J Mol Neurosci. 2001;17:45–58. doi: 10.1385/JMN:17:1:45. [DOI] [PubMed] [Google Scholar]
- Wenger DA, S. K, Suzuki Y, Suzuki K. Galactosylceramide lipidosis: globoid cell leukodystrophy (Krabbe disease) McGraw-Hill; New York: 2001. pp. 3669–3694. [Google Scholar]
- Dixon R, Gourzis J, McDermott D, Fujitaki J, Dewland P, Gruber H. AICA-riboside: safety, tolerance, and pharmacokinetics of a novel adenosine-regulating agent. J Clin Pharmacol. 1991;31:342–347. doi: 10.1002/j.1552-4604.1991.tb03715.x. [DOI] [PubMed] [Google Scholar]
- Escolar ML, Poe MD, Provenzale JM, Richards KC, Allison J, Wood S, Wenger DA, Pietryga D, Wall D, Champagne M, Morse R, Krivit W, Kurtzberg J. Transplantation of umbilical-cord blood in babies with infantile Krabbe's disease. N Engl J Med. 2005;352:2069–2081. doi: 10.1056/NEJMoa042604. [DOI] [PubMed] [Google Scholar]
- Eto Y, Shen JS, Meng XL, Ohashi T. Treatment of lysosomal storage disorders: cell therapy and gene therapy. J Inherit Metab Dis. 2004;27:411–415. doi: 10.1023/B:BOLI.0000031170.69676.68. [DOI] [PubMed] [Google Scholar]
- Giri S, Khan M, Rattan R, Singh I, Singh AK. Krabbe disease: psychosine-mediated activation of phospholipase A2 in oligodendrocyte cell death. J Lipid Res. 2006a;47:1478–1492. doi: 10.1194/jlr.M600084-JLR200. [DOI] [PubMed] [Google Scholar]
- Giri S, Jatana M, Rattan R, Won JS, Singh I, Singh AK. Galactosylsphingosine (psychosine)-induced expression of cytokine-mediated inducible nitric oxide synthases via AP-1 and C/EBP: implications for Krabbe disease. Faseb J. 2002;16:661–672. doi: 10.1096/fj.01-0798com. [DOI] [PubMed] [Google Scholar]
- Giri S, Nath N, Smith B, Viollet B, Singh AK, Singh I. 5-aminoimidazole-4-carboxamide-1-beta-4-ribofuranoside inhibits proinflammatory response in glial cells: a possible role of AMP-activated protein kinase. J Neurosci. 2004;24:479–487. doi: 10.1523/JNEUROSCI.4288-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giri S, Rattan R, Haq E, Khan M, Yasmin R, Won JS, Key L, Singh AK, Singh I. AICAR inhibits adipocyte differentiation in 3T3L1 and restores metabolic alterations in diet-induced obesity mice model. Nutr Metab (Lond) 2006b;3:31. doi: 10.1186/1743-7075-3-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Habinowski SA, Witters LA. The effects of AICAR on adipocyte differentiation of 3T3-L1 cells. Biochem Biophys Res Commun. 2001;286:852–856. doi: 10.1006/bbrc.2001.5484. [DOI] [PubMed] [Google Scholar]
- Haq E, Giri S, Singh I, Singh AK. Molecular mechanism of psychosine-induced cell death in human oligodendrocyte cell line. J Neurochem. 2003;86:1428–1440. doi: 10.1046/j.1471-4159.2003.01941.x. [DOI] [PubMed] [Google Scholar]
- Haq E, Contreras MA, Giri S, Singh I, Singh AK. Dysfunction of peroxisomes in twitcher mice brain: a possible mechanism of psychosine-induced disease. Biochem Biophys Res Commun. 2006;343:229–238. doi: 10.1016/j.bbrc.2006.02.131. [DOI] [PubMed] [Google Scholar]
- Hardie DG. The AMP-activated protein kinase pathway--new players upstream and downstream. J Cell Sci. 2004;117:5479–5487. doi: 10.1242/jcs.01540. [DOI] [PubMed] [Google Scholar]
- Hardie DG, Carling D, Carlson M. The AMP-activated/SNF1 protein kinase subfamily: metabolic sensors of the eukaryotic cell? Annu Rev Biochem. 1998;67:821–855. doi: 10.1146/annurev.biochem.67.1.821. [DOI] [PubMed] [Google Scholar]
- Hardie DG, Scott JW, Pan DA, Hudson ER. Management of cellular energy by the AMP-activated protein kinase system. FEBS Lett. 2003;546:113–120. doi: 10.1016/s0014-5793(03)00560-x. [DOI] [PubMed] [Google Scholar]
- Hwang JT, Park IJ, Shin JI, Lee YK, Lee SK, Baik HW, Ha J, Park OJ. Genistein, EGCG, and capsaicin inhibit adipocyte differentiation process via activating AMP-activated protein kinase. Biochem Biophys Res Commun. 2005;338:694–699. doi: 10.1016/j.bbrc.2005.09.195. [DOI] [PubMed] [Google Scholar]
- Inoue E, Yamauchi J. AMP-activated protein kinase regulates PEPCK gene expression by direct phosphorylation of a novel zinc finger transcription factor. Biochem Biophys Res Commun. 2006;351:793–799. doi: 10.1016/j.bbrc.2006.10.124. [DOI] [PubMed] [Google Scholar]
- Jatana M, Giri S, Singh AK. Apoptotic positive cells in Krabbe brain and induction of apoptosis in rat C6 glial cells by psychosine. Neurosci Lett. 2002;330:183–187. doi: 10.1016/s0304-3940(02)00655-9. [DOI] [PubMed] [Google Scholar]
- Jhun BS, Jin Q, Oh YT, Kim SS, Kong Y, Cho YH, Ha J, Baik HH, Kang I. 5-Aminoimidazole-4-carboxamide riboside suppresses lipopolysaccharide-induced TNF-alpha production through inhibition of phosphatidylinositol 3-kinase/Akt activation in RAW 264.7 murine macrophages. Biochem Biophys Res Commun. 2004;318:372–380. doi: 10.1016/j.bbrc.2004.04.035. [DOI] [PubMed] [Google Scholar]
- Kanazawa T, Nakamura S, Momoi M, Yamaji T, Takematsu H, Yano H, Sabe H, Yamamoto A, Kawasaki T, Kozutsumi Y. Inhibition of cytokinesis by a lipid metabolite, psychosine. J Cell Biol. 2000;149:943–950. doi: 10.1083/jcb.149.4.943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawaguchi T, Osatomi K, Yamashita H, Kabashima T, Uyeda K. Mechanism for fatty acid “sparing” effect on glucose-induced transcription: regulation of carbohydrate-responsive element-binding protein by AMP-activated protein kinase. J Biol Chem. 2002;277:3829–3835. doi: 10.1074/jbc.M107895200. [DOI] [PubMed] [Google Scholar]
- Khan M, Pahan K, Singh AK, Singh I. Cytokine-induced accumulation of very long-chain fatty acids in rat C6 glial cells: implication for X-adrenoleukodystrophy. J Neurochem. 1998;71:78–87. doi: 10.1046/j.1471-4159.1998.71010078.x. [DOI] [PubMed] [Google Scholar]
- Kondo Y, Wenger DA, Gallo V, Duncan ID. Galactocerebrosidase-deficient oligodendrocytes maintain stable central myelin by exogenous replacement of the missing enzyme in mice. Proc Natl Acad Sci U S A. 2005;102:18670–18675. doi: 10.1073/pnas.0506473102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leclerc I, Lenzner C, Gourdon L, Vaulont S, Kahn A, Viollet B. Hepatocyte nuclear factor-4alpha involved in type 1 maturity-onset diabetes of the young is a novel target of AMP-activated protein kinase. Diabetes. 2001;50:1515–1521. doi: 10.2337/diabetes.50.7.1515. [DOI] [PubMed] [Google Scholar]
- LeVine SM, Brown DC. IL-6 and TNFalpha expression in brains of twitcher, quaking and normal mice. J Neuroimmunol. 1997;73:47–56. doi: 10.1016/s0165-5728(96)00166-x. [DOI] [PubMed] [Google Scholar]
- Lin A, Sekhon C, Sekhon B, Smith A, Chavin K, Orak J, Singh I, Singh A. Attenuation of ischemia-reperfusion injury in a canine model of autologous renal transplantation. Transplantation. 2004;78:654–659. doi: 10.1097/01.tp.0000131664.18670.17. [DOI] [PubMed] [Google Scholar]
- Long YC, Zierath JR. AMP-activated protein kinase signaling in metabolic regulation. J Clin Invest. 2006;116:1776–1783. doi: 10.1172/JCI29044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luzi P, Rafi MA, Zaka M, Rao HZ, Curtis M, Vanier MT, Wenger DA. Biochemical and pathological evaluation of long-lived mice with globoid cell leukodystrophy after bone marrow transplantation. Mol Genet Metab. 2005;86:150–159. doi: 10.1016/j.ymgme.2005.06.023. [DOI] [PubMed] [Google Scholar]
- Maghazachi AA, Knudsen E, Jin Y, Jenstad M, Chaudhry FA. D-galactosyl-beta1-1'-sphingosine and D-glucosyl-beta1-1'-sphingosine induce human natural killer cell apoptosis. Biochem Biophys Res Commun. 2004;320:810–815. doi: 10.1016/j.bbrc.2004.06.027. [DOI] [PubMed] [Google Scholar]
- McCullough LD, Zeng Z, Li H, Landree LE, McFadden J, Ronnett GV. Pharmacological inhibition of AMP-activated protein kinase provides neuroprotection in stroke. J Biol Chem. 2005;280:20493–20502. doi: 10.1074/jbc.M409985200. [DOI] [PubMed] [Google Scholar]
- Mohri I, Taniike M, Okazaki I, Kagitani-Shimono K, Aritake K, Kanekiyo T, Yagi T, Takikita S, Kim HS, Urade Y, Suzuki K. Lipocalin-type prostaglandin D synthase is up-regulated in oligodendrocytes in lysosomal storage diseases and binds gangliosides. J Neurochem. 2006a;97:641–651. doi: 10.1111/j.1471-4159.2006.03753.x. [DOI] [PubMed] [Google Scholar]
- Mohri I, Taniike M, Taniguchi H, Kanekiyo T, Aritake K, Inui T, Fukumoto N, Eguchi N, Kushi A, Sasai H, Kanaoka Y, Ozono K, Narumiya S, Suzuki K, Urade Y. Prostaglandin D2-mediated microglia/astrocyte interaction enhances astrogliosis and demyelination in twitcher. J Neurosci. 2006b;26:4383–4393. doi: 10.1523/JNEUROSCI.4531-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motoshima H, Goldstein BJ, Igata M, Araki E. AMPK and cell proliferation--AMPK as a therapeutic target for atherosclerosis and cancer. J Physiol. 2006;574:63–71. doi: 10.1113/jphysiol.2006.108324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munday MR, Campbell DG, Carling D, Hardie DG. Identification by amino acid sequencing of three major regulatory phosphorylation sites on rat acetyl-CoA carboxylase. Eur J Biochem. 1988;175:331–338. doi: 10.1111/j.1432-1033.1988.tb14201.x. [DOI] [PubMed] [Google Scholar]
- Nath N, Giri S, Prasad R, Salem ML, Singh AK, Singh I. 5-aminoimidazole-4-carboxamide ribonucleoside: a novel immunomodulator with therapeutic efficacy in experimental autoimmune encephalomyelitis. J Immunol. 2005;175:566–574. doi: 10.4049/jimmunol.175.1.566. [DOI] [PubMed] [Google Scholar]
- Page T, Barshop B, Yu AL, Nyhan WL. Treatment of Lesch-Nyhan syndrome with AICAR. Adv Exp Med Biol. 1994;370:353–356. doi: 10.1007/978-1-4615-2584-4_76. [DOI] [PubMed] [Google Scholar]
- Paintlia AS, Paintlia MK, Singh I, Singh AK. Immunomodulatory effect of combination therapy with lovastatin and 5-aminoimidazole-4-carboxamide-1-beta-D-ribofuranoside alleviates neurodegeneration in experimental autoimmune encephalomyelitis. Am J Pathol. 2006;169:1012–1025. doi: 10.2353/ajpath.2006.051309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pilon G, Dallaire P, Marette A. Inhibition of inducible nitric-oxide synthase by activators of AMP-activated protein kinase: a new mechanism of action of insulin-sensitizing drugs. J Biol Chem. 2004;279:20767–20774. doi: 10.1074/jbc.M401390200. [DOI] [PubMed] [Google Scholar]
- Poli V. The role of C/EBP isoforms in the control of inflammatory and native immunity functions. J Biol Chem. 1998;273:29279–29282. doi: 10.1074/jbc.273.45.29279. [DOI] [PubMed] [Google Scholar]
- Prasad R, Giri S, Nath N, Singh I, Singh AK. 5-aminoimidazole-4-carboxamide-1-beta-4-ribofuranoside attenuates experimental autoimmune encephalomyelitis via modulation of endothelial-monocyte interaction. J Neurosci Res. 2006;84:614–625. doi: 10.1002/jnr.20953. [DOI] [PubMed] [Google Scholar]
- Rattan R, Giri S, Singh AK, Singh I. 5-Aminoimidazole-4-carboxamide-1-beta-D-ribofuranoside inhibits cancer cell proliferation in vitro and in vivo via AMP-activated protein kinase. J Biol Chem. 2005;280:39582–39593. doi: 10.1074/jbc.M507443200. [DOI] [PubMed] [Google Scholar]
- Salt I, Celler JW, Hawley SA, Prescott A, Woods A, Carling D, Hardie DG. AMP-activated protein kinase: greater AMP dependence, and preferential nuclear localization, of complexes containing the alpha2 isoform. Biochem J. 1998;334(Pt 1):177–187. doi: 10.1042/bj3340177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan JE, Brocklehurst KJ, Marley AE, Carey F, Carling D, Beri RK. Inhibition of lipolysis and lipogenesis in isolated rat adipocytes with AICAR, a cell-permeable activator of AMP-activated protein kinase. FEBS Lett. 1994;353:33–36. doi: 10.1016/0014-5793(94)01006-4. [DOI] [PubMed] [Google Scholar]
- Suzuki K. Twenty five years of the “psychosine hypothesis”: a personal perspective of its history and present status. Neurochem Res. 1998;23:251–259. doi: 10.1023/a:1022436928925. [DOI] [PubMed] [Google Scholar]
- Taniike M, Mohri I, Eguchi N, Irikura D, Urade Y, Okada S, Suzuki K. An apoptotic depletion of oligodendrocytes in the twitcher, a murine model of globoid cell leukodystrophy. J Neuropathol Exp Neurol. 1999;58:644–653. doi: 10.1097/00005072-199906000-00009. [DOI] [PubMed] [Google Scholar]
- Tschape JA, Hammerschmied C, Muhlig-Versen M, Athenstaedt K, Daum G, Kretzschmar D. The neurodegeneration mutant lochrig interferes with cholesterol homeostasis and Appl processing. Embo J. 2002;21:6367–6376. doi: 10.1093/emboj/cdf636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turnley AM, Stapleton D, Mann RJ, Witters LA, Kemp BE, Bartlett PF. Cellular distribution and developmental expression of AMP-activated protein kinase isoforms in mouse central nervous system. J Neurochem. 1999;72:1707–1716. doi: 10.1046/j.1471-4159.1999.721707.x. [DOI] [PubMed] [Google Scholar]
- Wenger DA, Suzuki K, Suzuki Y, Suzuki K. Galactosylceramide lipidosis: globoid cell leukodystrophy (Krabbe disease) McGraw-Hill; New York: 2001. pp. 3669–3694. [Google Scholar]
- Wu YP, Matsuda J, Kubota A, Suzuki K, Suzuki K. Infiltration of hematogenous lineage cells into the demyelinating central nervous system of twitcher mice. J Neuropathol Exp Neurol. 2000;59:628–639. doi: 10.1093/jnen/59.7.628. [DOI] [PubMed] [Google Scholar]
- Wu YP, McMahon EJ, Matsuda J, Suzuki K, Matsushima GK, Suzuki K. Expression of immune-related molecules is downregulated in twitcher mice following bone marrow transplantation. J Neuropathol Exp Neurol. 2001;60:1062–1074. doi: 10.1093/jnen/60.11.1062. [DOI] [PubMed] [Google Scholar]
- Yagi T, McMahon EJ, Takikita S, Mohri I, Matsushima GK, Suzuki K. Fate of donor hematopoietic cells in demyelinating mutant mouse, twitcher, following transplantation of GFP+ bone marrow cells. Neurobiol Dis. 2004;16:98–109. doi: 10.1016/j.nbd.2004.01.002. [DOI] [PubMed] [Google Scholar]
- Yang W, Hong YH, Shen XQ, Frankowski C, Camp HS, Leff T. Regulation of transcription by AMP-activated protein kinase: phosphorylation of p300 blocks its interaction with nuclear receptors. J Biol Chem. 2001;276:38341–38344. doi: 10.1074/jbc.C100316200. [DOI] [PubMed] [Google Scholar]
- Zaka M, Rafi MA, Rao HZ, Luzi P, Wenger DA. Insulin-like growth factor-1 provides protection against psychosine-induced apoptosis in cultured mouse oligodendrocyte progenitor cells using primarily the PI3K/Akt pathway. Mol Cell Neurosci. 2005;30:398–407. doi: 10.1016/j.mcn.2005.08.004. [DOI] [PubMed] [Google Scholar]