

Abstract
Toll-like receptors (TLRs) are a family of evolutionarily conserved molecules that directly detect pathogen invasion or tissue damage and initiate a biological response. TLRs can signal through two primary intracellular pathways and as such can induce either immuno-stimulatory or immuno-modulatory molecules. Both sides of this twin-edged sword are being examined for their therapeutic potential in combating neurological disease. The immuno-stimulatory properties of TLRs are being used to generate tumor-specific immune responses to CNS tumors while the immuno-modulatory properties are being used to suppress damaging inflammatory responses to stroke. Recently, a third component of TLR signaling has begun to emerge—that of direct neuroprotection. Hence, the TLRs offer novel targets for the treatment of neurological disease.
Preface
In the decade since the initial characterization of the mammalian Toll-like receptor (TLR) family, studies on these innate immune receptors have generated fascinating insights into their multiple and complex functions. Most studies have focused on the role of TLRs in pathogen detection, however there is an emergent role for these receptors in non-pathogen-associated disease. This review will examine TLRs as potential targets for such neurological disease states.
An Introduction to Toll-like receptors
The Toll-like receptors, so-called because of their homology to the Drosophila Toll receptor, were first characterized in mammals by their ability to recognize pathogen-associated molecules such as the bacterial cell wall components peptidoglycan (TLR2) and lipopolysaccharide (TLR4), as well as dsRNA (TLR3), ssRNA (TLR7), and nonmethylated Cytosine-Guanosine (CpG) DNA (TLR9). TLRs, upon activation by specific ligands, induce downstream signals that lead to cytokine and chemokine production which can initiate a localized inflammatory response and function as a first-line defense against pathogen invasion. TLRs are located on antigen presenting cells such as B cells, dendritic cells, monocytes/macrophage and microglia within the central nervous system. In addition, these receptors can be expressed by the endothelium and by cells within the brain parenchyma such as astrocytes, oligodendrocytes, and neurons.
Each TLR family member, with the exception of TLR3, signals through the MyD88 dependent pathway, initiated by the MyD88 adaptor protein. Recruitment of MyD88 to the activated receptor initiates formation of the IL-1 receptor associated kinase (IRAK) complex resulting in phosphorylation of IKKa/b, activation of the transcription factors NFkB, IRF1, and IRF7, and generation of the pro-inflammatory cytokines IL-6 and TNFα, among others. TLR3, on the other hand, signals through the MyD88 independent pathway, initiated by the TRIF adaptor molecule. Recruitment of TRIF to the receptor initiates phosphorylation of IKKe, which activates the transcription factors IRF3 and IRF7, and generates anti-viral molecules such as IFNβ. Of the TLRs, only TLR4 can utilize either of these pathways.
In addition to their role in pathogen detection and defense, TLRs act as sentinels of tissue damage and mediate inflammatory responses to aseptic tissue injury. Host-endogenous molecules associated with damaged cells and tissues can also activate TLRs. Heat shock proteins, components of the extracellular matrix, and fibrinogen have all been shown to activate TLR4, while host DNA and mRNA are endogenous ligands of TLR9 and TLR3, respectively. It is through this dual role that TLRs offer therapeutic promise as targets in neurological disease.
TLRs and immune activation
TLR agonists have long been utilized as vaccine adjuvants, which stimulate and enhance adaptive anti-pathogen immune responses. An emerging strategy for the treatment of carcinogenic tumors involves the stimulation of an adaptive anti-tumor immune response. In the brain, immunotherapy of this kind offers the potential for highly specific tumor targeting with protection of normal brain structures. Multiple TLR agonists have been investigated in this regard. The protective response is initiated either within the systemic compartment or within the CNS itself.
One particularly attractive candidate for cancer immunotherapy is imiquimod, a synthetic TLR7 agonist that has been given FDA approval as a topical treatment for HSV-2 lesions. Recently, topical application of imiquimod has been shown to decrease intracranial tumor burden in a mouse model of malignant melanoma[1] –an effect thought to be due to increased survival of peptide-pulsed dendritic cells and increased CD8+ priming. However, this success did not translate into increased survival time, as imiquimod treatment increased hemorrhaging and inflammation-induced mortality, which highlights the delicate balance between potentiating a central immune response and overwhelming this confined anatomical locale.
Several TLR agonists have been explored as stand-alone cancer therapeutics, under the hypothesis that certain cytotoxic features of these molecules, which cause the release of tumor-specific antigens, may couple with their known adjuvant properties to activate antigen-presenting cells in the vicinity of the tumor (Figure1). Lipopolysaccharide (LPS), a potent agonist of TLR4, has been investigated as just such a multifunctional anti-tumor immune activator. In a mouse model of glioblastoma multiforme (GM), Won and colleagues [2] demonstrated that treatment of mice with the lipid A moiety of LPS decreases the mean tumor mass of primary subcutaneous GM tumors through a process that depends, in part, on lymphocytes. This initial response rendered mice partially resistant to secondary intracranial tumors, implicating an adaptive protective process. This group showed that direct intra-tumoral LPS administration increases survival of wild-type mice, but not TLR4 knockout mice, bearing intracranial GM tumors, suggesting that the cytotoxicity of LPS is not sufficient to clear tumors, but may indeed act as a tumor-specific immune adjuvant[3].
Figure 1.

TLR ligands stimulate specific anti-tumor immune responses.
Perhaps the most promising TLR ligands in the fight against cancers of the CNS are the TLR9 agonists, nonmethylated CpG oligodeoxynucleotides. Carpentier et al [4] demonstrated the chemotherapeutic use of intra-tumoral CpG administration in mice and rats with subcutaneous and intracranial neuroblastoma tumors by showing that intra-tumoral CpG administration increases survival time and protects against secondary tumor challenge. Macrophages and lymphocytes play a role in the tumor regression and long-term protection seen in these studies [5]. Direct CpG administration can induce apoptosis of intracranial glioblastoma cells [6] and increase the CD8+/CD4+ composition of local lymphocyte infiltrates while decreasing the CD4+/CD25+/Foxp3+ population, suggesting that CpG can alter the characteristics of glioma-infiltrating leukocytes [7]. Despite the apoptotic effects of direct CpG injection, the addition of radiotherapy to release tumor antigens enhances both tumor regression and survival [8]. These exciting results have generated the rationale for a Phase I clinical trial of CpG oligodeoxynucleotides for patients with recurrent glioblastomas [9]. Twenty-four patients suffering from their first, second, or third recurrence of glioblastoma were treated by direct infusion of CpG. Two of the 24 patients showed a minor therapeutic response, perhaps highlighting the differences in TLR9 distribution between humans and rodents. TLR9 is expressed by glial cells in mice, however it appears to be restricted largely to B cells and plasmacytoid dendritic cells in healthy humans subjects (reviewed in reference [10]. A Phase II clinical trial is now in progress and concurrent studies continue to explore the application of this TLR ligand in generating a specific and robust anti-tumor immune response in patients with brain tumors.
TLRs and immune suppression
While therapies that target CNS tumors take advantage of the immuno-stimulatory properties of TLR’s, therapies that target other neurological disorders take advantage of the immuno-modulatory properties of these receptors. Such immunosuppressive therapies are based on the phenomena of “tolerance”, in which pretreatment with a low dose of a noxious stimulus renders an animal tolerant to a subsequent, more intense exposure to either the same or different stimulus. For example, pretreatment with a low dose of LPS can protect an animal from the normally detrimental effects of a second, higher dose of LPS, termed “LPS-induced LPS tolerance”, or from the detrimental effects of tissue ischemia, termed “LPS-induced ischemic tolerance”.
LPS-induced tolerance to cerebral ischemia was first demonstrated by Tasaki et al [11], who used low dose systemic administration of LPS to render spontaneously hypertensive rats tolerant to ischemic brain damage induced by middle cerebral artery occlusion (MCAO). LPS induced tolerance to brain ischemia has since been demonstrated in a mouse model of stroke and in a porcine model of deep hypothermic circulatory arrest [12,13]. Tolerance induction has been shown to require a small inflammatory response to LPS, as it can be blocked by simultaneous administration of cyclohexamide, dexamethasone, and TNFα inhibitors [11,14,15], yet exposure to small doses of LPS does not cause overt brain injury.
LPS-induced ischemic protection requires an inflammatory response prior to the ischemic event, yet paradoxically it protects, in part, by suppressing the inflammatory response following ischemia. For example, pretreatment enhances blood perfusion of the ischemic area at the time of ischemia and afterwards [16,17], thereby preventing the impairment of endothelial and smooth muscle relaxation [18] that is induced by ischemia-reperfusion injury. It has been postulated that preservation of microvascular function may be due to suppressed lymphocyte adhesion to activated endothelium, either by prevention of cellular inflammatory responses to ischemia [12] or by suppression of endothelial activation and adhesion molecules[19].
Suppression of the normal inflammatory responses to ischemia is a hallmark of the LPS-preconditioned brain. Rosenzweig and colleagues have shown that LPS preconditioning protects the brain against the cytotoxic effects of TNFα after cerebral ischemia. Mice that have been preconditioned with LPS prior to ischemia show a pronounced suppression of the TNFα pathway following stroke with reduced TNFα in the serum, decreased levels of cellular TNFR1 and enhanced levels of neutralizing soluble-TNFR1. Collectively, this leads to a muted TNFα response to ischemic injury and increased cell survival.
Just as LPS-pretreatment can prevent TNFα-induced exacerbation of ischemic damage, it may also prevent TLR-induced exacerbation of ischemic damage. Several recent studies implicate TLR activation as an endogenous and detrimental response to cerebral ischemia. Cerebral ischemia causes a marked upregulation of mRNA for TLRs 2, 4 and 9 [20]. Importantly, mice lacking either TLR2 or TLR4 suffer from significantly smaller infarcts than wild-type mice[20,21]. Host endogenous TLR ligands such as Hsp70 are upregulated in the brain following ischemia [22], suggesting that TLRs may initiate damaging inflammatory responses to tissue injured by ischemic insult. Hence, as with LPS-induced tolerance to subsequent LPS exposure, LPS-induced tolerance to subsequent ischemia may, in part, be a tolerance to the inflammatory effects that occur downstream of TLR ligation.
Cells that are tolerant to LPS are defined by their inability to generate TNFα in response to TLR4 activation. Upon TLR4 ligation, LPS tolerant cells, unlike naïve cells, do not recruit MyD88 to TLR4, and fail to activate IRAK-1and NFkB thereby leading to a block in TNFα expression [23]. Additionally, LPS tolerant cells upregulate inhibitors of MyD88-dependent signaling, such as Ship-1 and IRAK-M [24,25]. Recent evidence suggests that this signaling suppression is restricted to the MyD88-dependent pathway because upon restimulation with LPS, murine macrophage cell lines do not increase production of TNFα—a product of the MyD88 dependent pathway, yet show enhanced production of IFNβ which is a product of the MyD88 independent pathway [26] (Figure 2).
Figure 2.

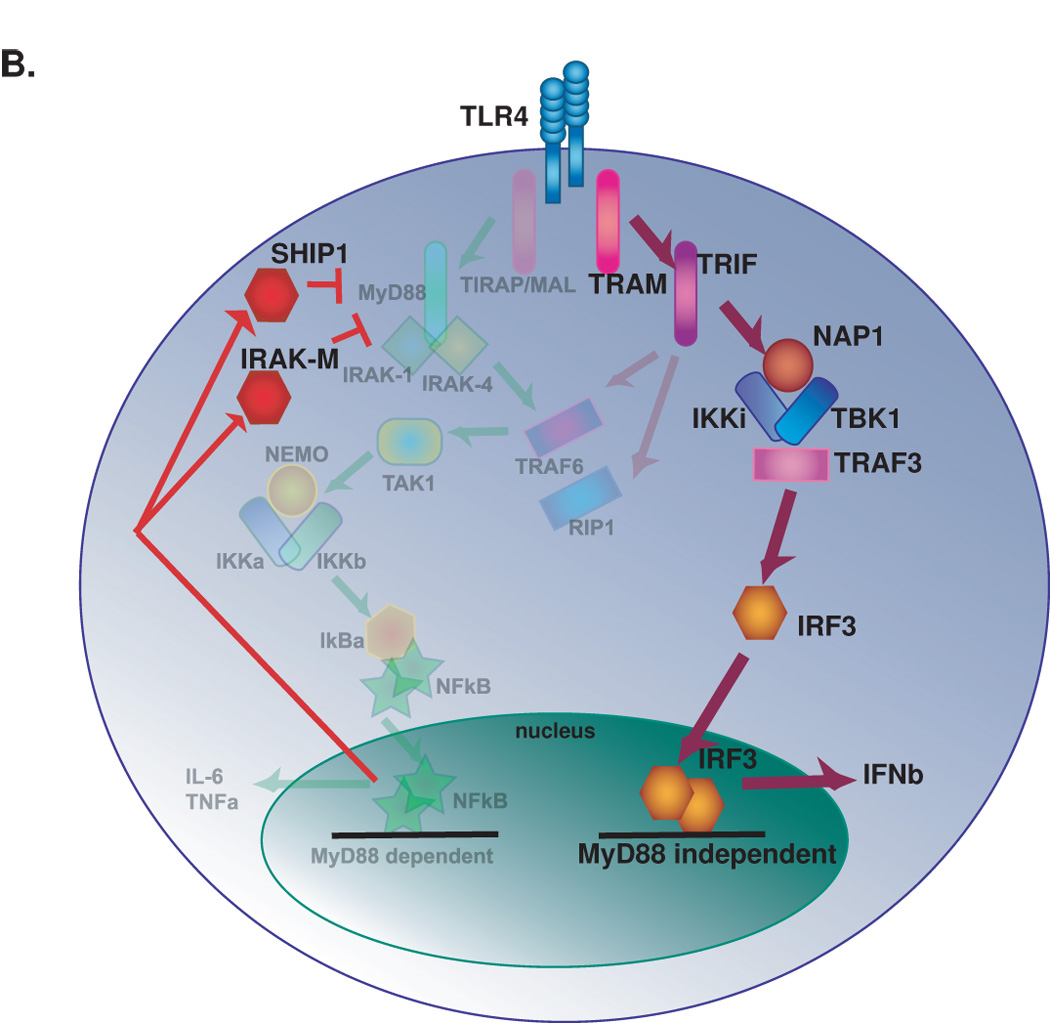
Tolerance to the downstream inflammatory effects of TLR4 activation. A. Intracellular signaling events downstream of initial TLR4 activation. Both the MyD88 dependent and MyD88 independent pathways are active. B. Signaling events upon subsequent TLR4 activation. Signaling through the MyD88 dependent pathway is inhibited and signaling through the MyD88 independent pathway is enhanced.
A similar scenario exists in LPS-induced neuroprotection against stroke injury. Our lab has shown that, following ischemia, LPS-pretreated mice display a suppressed TNFα response, and enhanced IFNβ production (Stevens, Stenzel-Poore, unpublished data). This suggests that LPS pretreatment may alter endogenous stroke-induced TLR4 signaling, much like it alters LPS-induced TLR4 signaling, by enhancing signaling down the MyD88-independent pathway. Evidence is emerging that the products of this pathway may be potent neuroprotectants.
TLRs and endogenous neuroprotection
Unique among the TLRs, TLR3 signals exclusively through the MyD88-independent pathway [27]. TLR3 detects viral dsRNA and can be activated by endogenous RNA released from necrotic cells [28]. TLR3 is expressed throughout the CNS on cerebral endothelium, microglia, oligodendrocytes, neurons, and most prominently on astrocytes [29–31].
Microglia, neurons and astrocytes all produce IFNβ upon stimulation with the synthetic TLR3 agonist polyinosinic-polycytidylic acid (poly(I:C)) [30,32,33]. IFNb is well known for its multiple antiviral functions yet, more recently, it has been shown to prevent cytokine-induced neutrophil infiltration into the brain and to attenuate blood-brain barrier disruption[34]; these latter effects could serve neuroprotective functions. Indeed, IFNβ administered subcutaneously after stroke has been shown to reduce infarct volume in rats and rabbits [35,36], and Phase I clinical testing is underway to determine whether IFNβ can be used as an acute stroke therapeutic.
IFNβ is not the only neuroprotective molecule generated from the MyD88-independent pathway. Astrocytes isolated from post mortem human adult subjects and stimulated with poly(I:C) induce the expression of multiple neuroprotective molecules (BDNF, neurotrophin 4, pleiotrophin, ephrin type B receptor1, TGFb2) and anti-inflammatory cytokines (IL-9, IL-10, IL-11) (Figure 3). Functionally, poly(I:C)-conditioned media from these cultures protects neuronal survival and suppresses astrocyte growth in organotypic human brain slice cultures [37]. There is evidence to suggest, however, that astrocytes activated by TLR3 suppress glutamate transport with potentially pathophysiological consequences [38].
Figure 3.

Neuroprotective TLR signaling events downstream of TLR3 and TLR4 activation.
TLR3, with its unique property of signaling in a MyD88-independent manner, may represent an endogenous neuroprotective response to CNS damage. Necrotic cell products, such as mRNA, activate TLR3 on cells in the CNS, which leads to the production of neuroprotective cytokines (e.g. IFNb). Astrocytes are known to upregulate multiple neuroprotective cytokines in response to the TLR3 agonist, poly(I:C). Thus, TLR3 offers substantial promise as a novel therapeutic target in neuroprotection against CNS-related injury.
Conclusion and Prospective
The above studies demonstrate an important role for TLRs in the generation of protective immune responses to cancers, suppression of the damaging inflammatory responses to ischemia, and the endogenous protection of brain tissue from injury. While the use of TLR ligands to generate endogenous anti-tumor immunity is a well-practiced vaccination strategy, the use of these molecules in ischemic therapy provides a powerful new paradigm for stroke therapeutics. There is considerable potential for prophylactic ischemic treatment as ~30% of first-time stroke survivors will suffer from another stroke within their lifetime, almost 50% of patients undergoing coronary artery bypass grafting suffer neurological effects from perioperative emboli [39], and up to 1% of all surgical patients will suffer from perioperative strokes (reviewed in reference [40]). Systemic pretreatment of these patients, be it by repeated administration in anticipation of an impending stroke or by a single acute treatment in preparation for surgery, has the potential to improve the quality of life of thousands of high-risk patients each year. By setting the stage for improved ischemic outcome, should an ischemic event occur, TLR pre-stimulation offers a low-risk, high-benefit opportunity to combat cerebral disease and promises to generate new views about these ancient receptors and their role in health and disease.
Acknowledgements
The authors wish to acknowledge support from the National Institute of Neurological Disease and Stroke (NINDS), 2P01 NS35965 (MSP), 5R01 NS050567 (MSP), and support from the OHSU Foundation Tarter Trust Fellowship (BJM).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Prins RM, Craft N, Bruhn KW, Khan-Farooqi H, Koya RC, Stripecke R, Miller JF, Liau LM. The TLR-7 agonist, imiquimod, enhances dendritic cell survival and promotes tumor antigen-specific T cell priming: relation to central nervous system antitumor immunity. J Immunol. 2006;176:157–164. doi: 10.4049/jimmunol.176.1.157. [DOI] [PubMed] [Google Scholar]
- 2.Won EK, Zahner MC, Grant EA, Gore P, Chicoine MR. Analysis of the antitumoral mechanisms of lipopolysaccharide against glioblastoma multiforme. Anticancer Drugs. 2003;14:457–466. doi: 10.1097/00001813-200307000-00012. [DOI] [PubMed] [Google Scholar]
- 3.Chicoine MR, Zahner M, Won EK, Kalra RR, Kitamura T, Perry A, Higashikubo R. The in vivo antitumoral effects of lipopolysaccharide against glioblastoma multiforme are mediated in part by Toll-like receptor 4. Neurosurgery. 2007;60:372–380. doi: 10.1227/01.NEU.0000249280.61761.2E. discussion 381. [DOI] [PubMed] [Google Scholar]
- 4.Carpentier AF, Chen L, Maltonti F, Delattre JY. Oligodeoxynucleotides containing CpG motifs can induce rejection of a neuroblastoma in mice. Cancer Res. 1999;59:5429–5432. [PubMed] [Google Scholar]
- 5.Auf G, Carpentier AF, Chen L, Le Clanche C, Delattre JY. Implication of macrophages in tumor rejection induced by CpG-oligodeoxynucleotides without antigen. Clin Cancer Res. 2001;7:3540–3543. [PubMed] [Google Scholar]
- 6.El Andaloussi A, Sonabend AM, Han Y, Lesniak MS. Stimulation of TLR9 with CpG ODN enhances apoptosis of glioma and prolongs the survival of mice with experimental brain tumors. Glia. 2006;54:526–535. doi: 10.1002/glia.20401. [DOI] [PubMed] [Google Scholar]
- 7.Hussain SF, Yang D, Suki D, Aldape K, Grimm E, Heimberger AB. The role of human glioma-infiltrating microglia/macrophages in mediating antitumor immune responses. Neuro Oncol. 2006;8:261–279. doi: 10.1215/15228517-2006-008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Meng Y, Carpentier AF, Chen L, Boisserie G, Simon JM, Mazeron JJ, Delattre JY. Successful combination of local CpG-ODN and radiotherapy in malignant glioma. Int J Cancer. 2005;116:992–997. doi: 10.1002/ijc.21131. [DOI] [PubMed] [Google Scholar]
- *9. Carpentier A, Laigle-Donadey F, Zohar S, Capelle L, Behin A, Tibi A, Martin-Duverneuil N, Sanson M, Lacomblez L, Taillibert S, et al. Phase 1 trial of a CpG oligodeoxynucleotide for patients with recurrent glioblastoma. Neuro Oncol. 2006;8:60–66. doi: 10.1215/S1522851705000475. A Phase 1 clinical trial to define the safety profile of CpG-28, administered intratumorally to patients with recurrent glioblastoma.
- **10. Iwasaki A, Medzhitov R. Toll-like receptor control of the adaptive immune responses. Nat Immunol. 2004;5:987–995. doi: 10.1038/ni1112. A review of TLR expression and function in cells of the innate and adaptive immune systems.
- 11.Tasaki K, Ruetzler CA, Ohtsuki T, Martin D, Nawashiro H, Hallenbeck JM. Lipopolysaccharide pre-treatment induces resistance against subsequent focal cerebral ischemic damage in spontaneously hypertensive rats. Brain Research. 1997;748:267–270. doi: 10.1016/s0006-8993(96)01383-2. [DOI] [PubMed] [Google Scholar]
- 12.Rosenzweig HL, Lessov NS, Henshall DC, Minami M, Simon RP, Stenzel-Poore MP. Endotoxin preconditioning prevents the cellular inflammatory response during ischemic neuroprotection in mice. Stroke. 2004;35:2576–2581. doi: 10.1161/01.STR.0000143450.04438.ae. [DOI] [PubMed] [Google Scholar]
- 13.Hickey EJ, You X, Kaimaktchiev V, Stenzel-Poore M, Ungerleider RM. Lipopolysaccharide preconditioning induces robust protection against brain injury resulting from deep hypothermic circulatory arrest. J Thorac Cardiovasc Surg. 2007;133:1588–1596. doi: 10.1016/j.jtcvs.2006.12.056. [DOI] [PubMed] [Google Scholar]
- 14.Bordet R, Deplanque D, Maboudou P, Puisieux F, Pu Q, Robin E, Martin A, Bastide M, Leys D, Lhermitte M, et al. Increase in endogenous brain superoxide dismutase as a potential mechanism of lipopolysaccharide-induced brain ischemic tolerance. J Cereb Blood Flow Metab. 2000;20:1190–1196. doi: 10.1097/00004647-200008000-00004. [DOI] [PubMed] [Google Scholar]
- *15. Rosenzweig HL, Minami M, Lessov NS, Coste SC, Stevens SL, Henshall DC, Meller R, Simon RP, Stenzel-Poore MP. Endotoxin preconditioning protects against the cytotoxic effects of TNFa after stroke: a novel role for TNFa in LPS-ischemic tolerance. J Cereb. Blood Flow & Metab. 2007 doi: 10.1038/sj.jcbfm.9600464. on line publication. The authors demonstrate that LPS pretreatment protects the brain from the injurious effects of ischemia-induced TNFα.
- 16.Kunz A, Park L, Abe T, Gallo EF, Anrather J, Zhou P, Iadecola C. Neurovascular protection by ischemic tolerance: role of nitric oxide and reactive oxygen species. J Neurosci. 2007;27:7083–7093. doi: 10.1523/JNEUROSCI.1645-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dawson DA, Furuya K, Gotoh J, Nakao Y, Hallenbeck JM. Cerebrovascular hemodynamics and ischemic tolerance: lipopolysaccharide-induced resistance to focal cerebral ischemia is not due to changes in severity of the initial ischemic insult, but is associated with preservation of microvascular perfusion. Journal of Cerebral Blood Flow and Metabolism. 1999;19:616–623. doi: 10.1097/00004647-199906000-00004. [DOI] [PubMed] [Google Scholar]
- 18.Bastide M, Gele P, Petrault O, Pu Q, Caliz A, Robin E, Deplanque D, Duriez P, Bordet R. Delayed cerebrovascular protective effect of lipopolysaccharide in parallel to brain ischemic tolerance. J Cereb. Blood Flow & Metab. 2003;23:399–405. doi: 10.1097/01.WCB.0000050064.57184.F2. [DOI] [PubMed] [Google Scholar]
- 19.Ginis I, Schweizer U, Brenner M, Liu J, Azzam N, Spatz M, Hallenbeck J. TNF-alpha pretreatment prevents subsequent activation of cultured brain cells with TNF-alpha and hypoxia via ceramide. Am J Physiol. 1999;276:C1171. doi: 10.1152/ajpcell.1999.276.5.C1171. [DOI] [PubMed] [Google Scholar]
- 20.Ziegler G, Harhausen D, Schepers C, Hoffmann O, Rohr C, Prinz V, Konig J, Lehrach H, Nietfeld W, Trendelenburg G. TLR2 has a detrimental role in mouse transient focal cerebral ischemia. Biochem Biophys Res Commun. 2007;359:574–579. doi: 10.1016/j.bbrc.2007.05.157. [DOI] [PubMed] [Google Scholar]
- *21. Cao CX, Yang QW, Lv FL, Cui J, Fu HB, Wang JZ. Reduced cerebral ischemia-reperfusion injury in Toll-like receptor 4 deficient mice. Biochem Biophys Res Commun. 2007;353:509–514. doi: 10.1016/j.bbrc.2006.12.057. The authors implicate endogenous TLR4 signaling in the exacerbation of cerebral ischemic damage.
- 22.Kinouchi H, Sharp FR, Hill MP, Koistinaho J, Sagar SM, Chan PH. Induction of 70-kDa heat shock protein and hsp70 mRNA following transient focal cerebral ischemia in the rat. J Cereb Blood Flow Metab. 1993;13:105–115. doi: 10.1038/jcbfm.1993.13. [DOI] [PubMed] [Google Scholar]
- 23.Medvedev AE, Lentschat A, Wahl LM, Golenbock DT, Vogel SN. Dysregulation of LPS-induced Toll-like receptor 4-MyD88 complex formation and IL-1 receptor-associated kinase1 activation in endotoxin-tolerant cells. J Immunol. 2002;169:5209–5216. doi: 10.4049/jimmunol.169.9.5209. [DOI] [PubMed] [Google Scholar]
- 24.Sly LM, Rauh MJ, Kalesnikoff J, Song CH, Krystal G. LPS-induced upregualtion of SHIP is essential for endotoxin tolerance. Immunity. 2004;21:227–239. doi: 10.1016/j.immuni.2004.07.010. [DOI] [PubMed] [Google Scholar]
- 25.Kobayashi K, Hernandex L, Galan J, CA Janeway J, Medzhitov R, Flavell R. IRAK-M is a negative regulator of Toll-like receptor signaling. Cell. 2002;110:191–202. doi: 10.1016/s0092-8674(02)00827-9. [DOI] [PubMed] [Google Scholar]
- *26. Broad A, Kirby JA, Jones DE. Toll-like receptor interactions: tolerance of MyD88-dependent cytokines but enhancement of MyD88-independent interferon-beta production. Immunology. 2007;120:103–111. doi: 10.1111/j.1365-2567.2006.02485.x. The authors demonstrate a shift in TLR4 signaling, away from the MyD88 dependent pathway towards the MyD88 independent pathway, in endotoxin-tolerant cells.
- 27.Yamamoto M, Sato S, Mori K, Hoshino K, Takeuchi O, Takeda K, Akira S. Cutting edge: a novel Toll/IL-1 receptor domain-containing adapter that preferentially activates the IFN-beta promoter in the Toll-like receptor signaling. J Immunol. 2002;169:6668–6672. doi: 10.4049/jimmunol.169.12.6668. [DOI] [PubMed] [Google Scholar]
- 28.Kariko K, Ni H, Capodici J, Lamphier M, Weissman D. mRNA is an endogenous ligand for Toll-like receptor 3. J Biol Chem. 2004;279:12542–12550. doi: 10.1074/jbc.M310175200. [DOI] [PubMed] [Google Scholar]
- 29.Bsibsi M, Ravid R, Gveric D, Noort JMv. Broad expression of Toll-like receptors in the human central nervous system. J Neuropathol Exp Neurol. 2002;61:1013–1021. doi: 10.1093/jnen/61.11.1013. [DOI] [PubMed] [Google Scholar]
- 30.Lafon M, Megret F, Lafage M, Prehaud C. The innate immune facet of brain: human neurons express TLR-3 and sense viral dsRNA. J Mol Neurosci. 2006;29:185–194. doi: 10.1385/JMN:29:3:185. [DOI] [PubMed] [Google Scholar]
- 31.Farina C, Krumbholz M, Giese T, Hartmann G, Aloisi F, Meinl E. Preferential expression and function of Toll-like receptor 3 in human astrocytes. J Neuroimmunol. 2005;159:12–19. doi: 10.1016/j.jneuroim.2004.09.009. [DOI] [PubMed] [Google Scholar]
- 32.Olson JK, Miller SD. Microglia initiate central nervous system innate and adaptive immune responses through multiple TLRs. J Immunol. 2004;173:3916–3924. doi: 10.4049/jimmunol.173.6.3916. [DOI] [PubMed] [Google Scholar]
- 33.Krasowska-Zoladek A, Banaszewska M, Kraszpulski M, Konat GW. Kinetics of inflammatory response of astrocytes induced by TLR 3 and TLR4 ligation. J Neurosci Res. 2007;85:205–212. doi: 10.1002/jnr.21088. [DOI] [PubMed] [Google Scholar]
- *34. Veldhuis WB, Floris S, van der Meide PH, Vos IM, de Vries HE, Dijkstra CD, Bar PR, Nicolay K. Interferon-beta prevents cytokine-induced neutrophil infiltration and attenuates blood-brain barrier disruption. J Cereb Blood Flow Metab. 2003;23:1060–1069. doi: 10.1097/01.WCB.0000080701.47016.24. The authors show that direct IFNβ administration maintains the integrity of the blood-brain barrier after ischemic insult and reduces inflammatory cell infiltration.
- 35.Veldhuis W, Derksen J, Floris S, van der Meide P, de Vries H, Schepers J, Vos I, Dijkstra C, Kappelle L, Nicolay K, et al. Interferon-beta blocks infiltration of inflammatory cells and reduces infarct volume after ischemic stroke in the rat. J Cereb. Blood Flow & Metab. 2003;23:1029–1039. doi: 10.1097/01.WCB.0000080703.47016.B6. [DOI] [PubMed] [Google Scholar]
- 36.Liu H, Xin L, Chan BPL, Teoh R, Tang BL, Tan YH. Interferon beta administration confers a beneficial outcome in a rabbit model of thromboembolic cerebral ischemia. Neurosci Lett. 2002;327:146–148. doi: 10.1016/s0304-3940(02)00371-3. [DOI] [PubMed] [Google Scholar]
- *37. Bsibsi M, Persoon-Deen C, Verwer RW, Meeuwsen S, Ravid R, Van Noort JM. Toll-like receptor 3 on adult human astrocytes triggers production of neuroprotective mediators. Glia. 2006;53:688–695. doi: 10.1002/glia.20328. The authors show that astrocytes produce neuroprotective molecules in response to Poly(I:C) administration which enhance neuronal survival in human brain slice cultures.
- 38.Scumpia PO, Kelly KM, Reeves WH, Stevens BR. Double-stranded RNA signals antiviral and inflammatory programs and dysfunctional glutamate transport in TLR3-expressing astrocytes. Glia. 2005;52:153–162. doi: 10.1002/glia.20234. [DOI] [PubMed] [Google Scholar]
- 39.Newman MF, Kirchner JL, Phillips-Bute B, Gaver V, Grocott H, Jones RH, Mark DB, Reves JG, Blumenthal JA. Longitudinal assessment of neurocognitive function after coronary-artery bypass surgery. N Engl J Med. 2001;344:395–402. doi: 10.1056/NEJM200102083440601. [DOI] [PubMed] [Google Scholar]
- **40. Selim M. Perioperative stroke. N Engl J Med. 2007;356:706–713. doi: 10.1056/NEJMra062668. A clinical review of the pathophysiology of perioperative stroke with emphasis on risk stratification, risk modification, and patient management.