

Summary
Gradual changes in steady-state levels of beta amyloid peptides (Aβ) in brain are considered an initial step in the amyloid cascade hypothesis of Alzheimer's disease. Aβ is a product of the secretase cleavage of amyloid precursor protein (APP). There is evidence that the membrane lipid environment may modulate secretase activity and alters its function. Cleavage of APP strongly depends on membrane properties. Since Aβ perturbs cell membrane fluidity, the cell membrane may be the location where the neurotoxic cascade of Aβ is initiated. Therefore, we tested effects of oligomeric Aβ on membrane fluidity of whole living cells, the impact of exogenous and cellular Aβ on the processing of APP and the role of GM-1 ganglioside. We present evidence that oligoAβ(1-40) stimulates the amyloidogenic processing of APP by reducing membrane fluidity and complexing with GM-1 ganglioside. This dynamic action of Aβ may start a vicious circle, where endogenous Aβ stimulates its own production. Based on our novel findings, we propose that oligoAβ(1-40) accelerates the proteolytic cleavage of APP by decreasing membrane fluidity.
Keywords: Beta Amyloid, membrane fluidity, GM-1 ganglioside, APP, Alzheimer
Introduction
Alzheimer's disease (AD) is the most common neurodegenerative disorder. A neuropathological hallmark of AD is amyloid plaques [1], which contain beta-amyloid peptides (Aβ). Amphipathic Aβ tends towards self-aggregation and accumulation, which initiates a cascade that triggers complex pathological reactions eventually leading to neuronal dysfunction and cell death [2-4]. Aβ aggregation is a concentration-dependent phenomenon, which is initiated via a seeded polymerization reaction [5]. Monomeric Aβ initially forms poorly characterized nuclei that assemble into larger aggregates. The nucleation of Aβ is followed by oligomer and protofibril formation, which ultimately leads to insoluble amyloid fibril assembly [3]. Current research suggests that oligomeric forms of Aβ may play a major role in AD pathophysiology [3, 4]. Soluble amyloid oligomers (oligoAβ) bind specifically to neurons, disrupt dendritic spines [6] and inhibit hippocampal long-term potentiation in vivo [7].
Most Aβ is composed of 38-43 amino acid residues, all deriving from the transmembrane amyloid precursor protein (APP) after the sequential proteolytic cleavage by different secretases. There are two concurrent cleavage pathways. In the non-amyloidogenic pathway, APP is first cleaved at the N-terminus by α-secretase within the Aβ sequence and precludes the formation of Aβ. α-Secretase is a member of the ADAM (a disintegrin and metalloprotease) family [8]. Via their metalloprotease domain, ADAMs are often implicated in ectodomain shedding, either to release e.g. growth factors or to initiate further intracellular signaling via regulated intramembrane proteolysis. ADAMs 9, 10 and 17, have been proposed to act as α-secretases for APP [9]. Proteolytic cleavage of APP by ADAM-enzymes produces a 105-125 kDa soluble N-terminal APP fragment (sAPPα) and an 83 residue membrane-associated C-terminal fragment (C83). The secreted sAPPα exert anti-apoptotic and neuroprotective effects [10]. Alternatively, Aβ is produced primarily in the trans-Golgi network and recycling compartments [12-14]. This amyloidogenic pathway involves first APP cleavage at the N-terminus by β-secretase to produce sAPPβ and a 99-residue membrane associated C-terminal fragment C99. β-Secretase is a membrane-anchored aspartyl protease with its active site in its ectodo-main and it was shown to be a member of the memapsin family. Subsequent cleavage of C99 by γ-secretase produces AICD along with varying lengths of extracellular Aβ, the most common being Aβ(1-40) and Aβ(1-42). This model of Aβ formation is now widely accepted. However, the presence and exact contribution of both intracellular and extra-cellular Aβ is still an issue of great controversy and interest in the field [15].
APP processing by α-, β-, and γ-secretase, which are strictly associated with cellular membranes, strongly depends on membrane fluidity. Modulation of membrane fluidity for example affects the accessibility of secretases acting on APP [16]. Kojro et al. showed that α-secretase activity increased with enhanced membrane fluidity [17]. Decreased membrane fluidity augments γ-secretase activity [18]. Moreover, processing of APP by β-secretase also might be explained by alterations in cell membrane fluidity [19].
Membrane fluidity may contribute to the processing of APP and the cleavage product Aβ may in turn perturb the membrane environment. Studies have examined the effects of Aβ peptides on membrane fluidity in model membranes and biological membranes of mice, rats and humans (summarized in [20]). It is well recognized that changes in the physico-chemical state of the membrane can markedly alter activity of various membrane proteins, including α-, β-, and γ-secretase. The cell membrane represents the first site of interaction between Aβ and neurons, and may be the location where the neurotoxic cascade of Aβ is initiated. The mechanism by which Aβ affects membrane fluidity is not well-understood. Yanagiswa et al. discovered that Aβ binds to monosialoganglioside (GM-1 ganglioside) and induces the assembly of Aβ in the brain and in cultural cells [21]. GM-1 ganglioside reduces membrane fluidity of PC12 cells and alters the localization of receptors within the plasma membrane [43]. We examined the hypothesis that Aβ triggers the amyloidogenic processing of APP by decreasing membrane fluidity, which in turn stimulates its own production. Aβ perturbation of the membrane may involve complexing with GM-1 ganglioside.
Materials and Methods
Materials
Unless otherwise stated, all cell culture reagents were obtained from Gibco/Invitrogen. The inhibitors and substances applied were human Aβ(1-40) and rodent Aβ(1-42) (Bachem, Heidelberg, Germany), Pluronic F68 (Sigma-Aldrich, Taufkirchen, Germany, Taufkirchen, Germany), Benzyl alcohol (Fluka, Neu-Ulm, Germany, Neu-Ulm, Germany), lovastatin (MSD SHARP & DOME, Haar, Germany) and DAPT (Sigma-Aldrich, Taufkirchen, Germany). The Western blot reagents and Cholera Toxin Subunit B were obtained from Invitrogen (Karlsruhe, Germany); the PVDF membranes, ECL detection kit and hyper films were obtained from GE Healthcare (München, Germany). Trimethylamimmoniumdiphenylhexatrien (TMA-DPH), 1,1,1,3,3,3- and hexafluoro-2-propanol (HFIIP) were obtained from Sigma-Aldrich (Taufkirchen, Germany).
Antibodies
Full length APP and C-terminal fragments (CTF) were detected using mouse IgG C1/6.1 (kindly provided by Dr. Paul M. Mathews, Nathan Kline Institute, Orangeburg, NY, USA) which was raised against the conserved carboxyl-terminal 20 residues of APP (residues 676-695 of APP695)[22]. The secreted sAPPα was detected using monoclonal mouse IgG 6E10 (Signet Laboratories, Cat.9320-02), which recognizes the residues 1-17 of Aβ. Mouse anti-GAP-DH was obtained from Chemicon, Hofheim, Germany (Cat.MAB374). Anti-mouse HRP-conjugated secondary antibody was purchased from Calbiochem (Bad-Soden, Germany; (CAT.401253).
Cell culture
HEK293-cells and neuroblastoma SH-SY5Y-cells stable transfected with human APP695 were cultured in Dulbecco's modified Eagle's medium (DMEM) at 37° C and 5% CO2. The HEK-medium was supplemented with 10% FCS and penicillin/streptomycin. Geniticin (G418) was added at 3μg/ml as a selective antibiotic. For incubation with lovastatin, cells were grown in DMEM medium supplemented with 2% v/v Ultroser G instead of FCS. Ultroser G is free of cholesterol, requiring the cells to rely upon their own de-novo synthesis. Cells were treated for 24 h with 1, 2 or 4 μM) lovastatin. Untransfected HEK293-cells were cultured in the same medium without G418. Antibiotic treatments per se had no impact on the effects reported (data not shown). Medium for SH-SY5Y-APP695 cells was supplemented with 1% of glutamine, MEM-vitamins, Hygromycin B (3μg/ml), pyruvate and non-essential amino-acids plus 10% FCS. For lovastatin treatment, cells were maintained in serum-free OptiMEM medium supplemented with Hygromycin B. The cells were treated for 24 hr with lovastatin (1, 2, or 4 μM), Pluronic F68 (4.5 or 7.5 μM) and benzyl alcohol (5 or 10 mM). DAPT [1μM] was added to the culture medium for at least 10 passages.
Preparation of Aβ(1-40) peptides
The preparation of monomeric, oligomeric and fibrilar Aβ(1-40) was performed as previously reported [23, 24]. Briefly, 1mg of Aβ(1-40) was dissolved in 200μl 1,1,1,3,3,3-hexafluoro-2-propanol and the peptide solution was evaporated using a speed vacuum for 45 minutes. Preparation of monomeric Aβ(1-40) peptides. The dried film was re-suspended in 2μl DMSO and diluted in 98μl DMEM medium to achieve a working solution of 100μM. The solution was vortexed for 30 sec. and immediately used for the experiments. Preparation of oligomeric Aβ(1-40) peptides. The dried film was re-suspended in 2μl DMSO and diluted in 98μl DMEM medium to achieve a working solution of 100μM. The solution was vortexed for 30 sec. and incubated at 4°C for 24 h. Preparation of fibrillar Aβ(1-40) peptides. The dried film was re-suspended in 2μl DMSO and diluted in 98μl HCl 0,01 N to achieve a working solution of 100μM. The solution was vortexed for 30 sec. and incubated at 37°C for 24 h. Aβ(1-40) peptides were characterized using native gel electrophoresis, followed by silver staining and electron microscopy as described below.
Characterization of Aβ(1-40) peptides
Blue native SDS-PAGE gel electrophoresis and silver staining was performed according procedures previously published [25]. Prepared Aβ(1-40) samples (see above) were mixed with 6× Laemmli buffer in a sample/buffer ratio of 5:1 (Boston bioproduct, Cat # ADP-111R) and placed in wells of 13 % polyacrylamide gel (0.75mm). The gel was run in a 1× trisglycerine-SDS buffer from Bio-Rad at a constant voltage (90mV for 10 min then at 180 mV for another 60 min). Finally, the gel was silver stained (Silver Stain Plus Kit from Bio-Rad (cat # 161-0449) according to the manufacture's instructions.
Transmission electron microscopy was used for visualization of Aβ peptides as previously published described [26]. Aliquots of Aβ(1-40) samples (10 μl) were pipetted on to the surface of coated copper microscope grids. The grids were air-dried and the samples were then stained with 1% uranyl acetate. Grids were examined in a Electron EM109 transmission microscope (Zeiss, Oberkochen, Germany).
Western blot analysis
Total protein levels were determined by the Lowry method. The samples were prepared by diluting 20μg protein with the reducing agent (10×) and loading buffer (4×). After denaturation for 10 minutes at 90°C, the samples were electrophoresed on a 4-12% NuPage Bis/Tris gel for 40 minutes at 200V and then transferred on a PVDF membrane for 90 minutes at 30V. For the detection of secreted sAPPα, the conditioned medium was collected and the values were normalized to the cell lysate protein concentration. The membranes were blocked overnight and incubated with the primary antibody for 1h. After washing, the blot was hybridized with an HRP-conjugated secondary antibody for 30 minutes. Visualization was done using an ECL detection kit from GE Healthcare. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) served as a loading control. Band analysis was performed using BioRad's Quantity One software.
ELISA detection of Aβ(1-42) and sAPPα
The Aβ(1-42) levels were determined using ELISA-kits from Biosource, Solingen, Germany (Cat.KHB3482 and KHB3544) according to the manufacturer's instructions. Briefly, the conditioned medium was collected and supplemented with PMSF. The cells were collected and washed twice with 1× PBS. After centrifugation (22,000 × rpm, 2h, 4°C), the cells were resuspended in guanidine hydrochloride (HCL, 5M) and incubated for 4h. The suspension was diluted and centrifuged at 16,000 × g for 20 minutes. The supernatant was used for the ELISA.
In those experiments where oligoAβ(1-40) was added to cells, cellular Aβ(1-40) levels could not be determined in parallel, since the antibody used in the ELISA-kit for Aβ(1-40) detects both, cellular endogenous and added exogenous Aβ(1-40) (data not shown).
For the quantitative analysis of sAPPα, wild type APP over-expressing HEK and control cells were plated at equal density in 24 well plates. Conditioned, serum-free media were collected after 24h and supernatants were centrifuged at 200 × g for 4 minutes to eliminate cell debris. Soluble sAPPα was quantified using the IBL human sAPPα colorimetric sandwich ELISA Kit (IBL Immuno-Biological Laboratories, Hamburg, Germany) according to the supplier's instructions.
Determination of membrane fluidity in living cells
The membrane fluidity of living cells was determined as earlier described [24]. The fluorescence polarization probe TMA-DPH incorporates very rapidly into plasma membranes of cells, and is specifically localized on the cell surface making its use particularly appropriate for determining plasma membrane fluidity in living cells [28]. Cells were washed twice with warm HBSS and supplemented with 2μM TMA-DPH. 700,000 cells per vial were incubated at 37° C for 20 minutes. The membrane fluorescence polarization was determined using a SLM Aminco Bowman Series 2 luminescence spectrometer. The cuvette temperature was 37°C. The steady-state fluorescence polarization (Ps) was expressed as the anisotropy (rs) of the probe, using the following equation: rS = 2PS/3 - PS. The rS is inversely correlated to the membrane fluidity, particularly to the acyl-chain flexibility of phospholipids.
Fluorescence & laser scanning fluorescence microscopy
Aβ was stained using Fluoro-Aβ(1-40) conjugated with Fluorescein (Advanced Bioconcept, Cat. AB2012). GM-1 ganglioside was visualized using fluorescent labeled cholera toxin subunit B. Briefly, 100,000 cells were plated overnight on cover slips coated with polylysin. Next, the medium was supplemented with 2μM oligoAβ(1-40) containing 25 nM of Fluoro-Aβ(1-40) for 24h. After incubation, 2μl of cholera toxin subunit B conjugated with Alexa Fluor 555 were added to the culture medium, which was then incubated for 10 minutes. The cells were washed three times with PBS and incubated with 1.990 ml medium plus 10μl of the anti-cholera toxin antibody (Calbiochem, Darmstadt, Germany; Cat. 227040) for 15 minutes. Again, the cells were washed three times with PBS and fixed with 4% paraformaldehyde for 30 minutes at 4°C. Cholesterol was stained in fixed cells using filipin (50 μg/ml in PBS) (Sigma-Aldrich, Taufkirchen, Germany; Cat. F9765).
For fluorescence microscopy, Gel Mount was used to fix the object slide. The fluorescent images were acquired on a Nikon ECLIPSE E800 microscope equipped with a Nikon DXM1200C digital camera connected to a PC. The images were evaluated using the Nikon NIS-Elements Imaging software package.
For confocal laser-scanning fluorescence microscopy, the samples were embedded in Mowiol and analyzed using a confocal laser-scanning microscope (Leica TCS SP5, Wetzlar, Germany), equipped with a 63×1,4 oil-immersion objective. The images were evaluated using the Imaris Imaging and ImarisColocSoftware (Version 6.2.0).
Determination of unesterified cholesterol
Unesterfied cholesterol levels were determined using the CHOD-PAP method (Cholesteroloxidase-Peroxidase-Aminophenazon-Phenol) developed in our lab as reported earlier [29]. Cells were centrifuged and washed twice with 1× PBS. The pellets were re-suspended in PBS with a protease-inhibitor cocktail (Roche) and frozen at -80°C. Before use, the cells were homogenized using a Dounce homogenizer fitted with a Teflon pestle and a Brandson sonicator.
Cytotoxicity
The cytotoxicity of AB, benzyl alcohol, pluronic F68 and lovastatin used in the current study was determined using the MTT assay as previously reported [30]. No cytotoxicity was observed for the concentrations of inhibitors and substances used in the current study (data not shown).
Statistics
All experiments were done at least in three independent experiments and all assays were done in trriplicates. Statistical analyses were performed using one-way ANOVA followed by a Tukey comparison test. All data were expressed as the means ± SD. The correlations were calculated using the Pearson using GraphPad Prism 4.0 software package (San Diego, USA).
Results
Oligomeric Aβ(1-40) reduces membrane fluidity and enhances β-secretase cleavage of APP
Aβ interacts with neuronal membranes and decreases fluidity [20]. We tested if Aβ induced changes in membrane fluidity alters processing of APP in living cells. Cells stably transfected with human APP695 were incubated with oligomeric Aβ(1-40) (oligoAβ(1-40)) and fluorescence polarization of TMA-DPH, Aβ(1-42) levels, C-terminal fragments of APP (CTF) and sAPPα levels were determined. Transfecting the APP gene induces an overproduction of the protein, and APP overproduction increases Aβ peptide secretion [31]. In the following experiments non-neuronal human embryonic kidney cells stably transfected with human APP695 (HEK293-APP695) were used, which provides expression levels that are sufficient for adequate assessment of APP cleavage products, such as sAPPα (supplementary figure 3).
Treatment with oligoAβ(1-40) significantly decreased plasma membrane fluidity of living cells (Fig. 1A) and increased levels of secreted cellular Aβ(1-42) (Fig.1B). Western blot analysis revealed that the levels of C99, the β-secretase related C-terminal fragment (CTF), are significantly elevated by oligoAβ(1-40) (Fig.1C). α-Secretase cleaved CTF C83 and secreted levels of sAPPα (Fig.1C) remained unchanged.
Figure 1. Effects of oligoAβ(1-40) on membrane properties the and processing of APP.
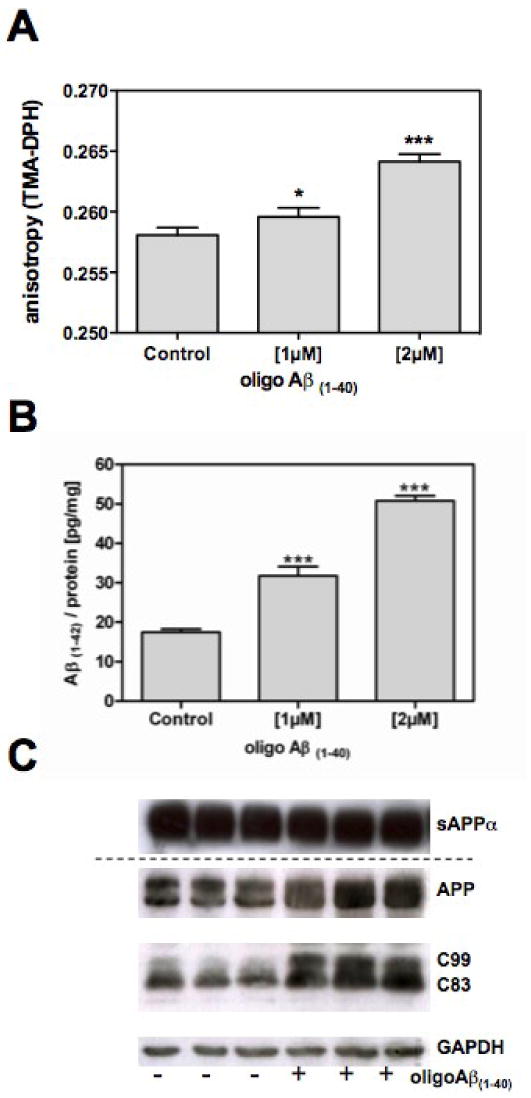
HEK293-APP695 cells were incubated for 24h in the presence or absence of 1μM oligoAβ(1-40); A) Changes in the membrane fluidity, which is inversely correlated to the anisotropy of the fluorescence probe TMA-DPH, were determined using fluorescence polarization spectroscopy in living cells; B) Levels of endogenous Aβ(1-42) were determined by ELISA [pg/mg protein]; C) Representative Western blot analysis of full-length APP and its fragments: The supernatants and cells were collected after incubation; proteins were separated by gel electrophoresis and transferred to PVDF membranes; secreted sAPPα was detected in cell supernatants using antibody 6E10; for APP and CTF analyses, cell homogenates were probed with the antibody C1/6.1; GAPDH served as loading control; Data are means ± SD n=6-9 (p*< 0.05; p***<0.001).
To assess the influence of peptide aggregation on the membrane fluidity of living cells and the production of Aβ(1-42), we compared the effects of monomeric, oligomeric and fibrillar Aβ(1-40), which were characterized by native gel electrophoresis and electron microscopy (supplementary figure 1). Monomeric Aβ(1-40) has no effect on membrane fluidity and Aβ(1-42) production (supplementary figure 2). Effects of oligoAβ(1-40) on membrane fluidity and on production of Aβ(1-42) were much more pronounced compared to fibrillar Aβ(1-40) (supplementary figure 2).
Cellular Aβ levels determine membrane fluidity of living cells
We next investigated, if endogenous, Aβ produced by cells harboring the human APP695 protein was associated with membrane fluidity of living cells. A majority of the mutations in human APP that have been tested, when transfected in cellular models, induce an increase in Aβ(1-40) and Aβ(1-42) levels [31]. Therefore, we examined fluidity in cells, expressing human APP695 (HEK-APP) and human APP695 harboring the Swedish mutation (HEK-APPsw). Expression of human APP is related to enhanced levels of Aβ (Fig. 2A). Cells expressing human APP harboring the Swedish mutation release excessive amounts of Aβ (Fig. 2 A). Enhanced levels of cellular Aβ(1-42) were related to reduced membrane fluidity (fig. 2 B), indicating that endogenous Aβ perturbed the plasma membrane.
Figure 2. Effects of endogenous Aβ levels on membrane fluidity.
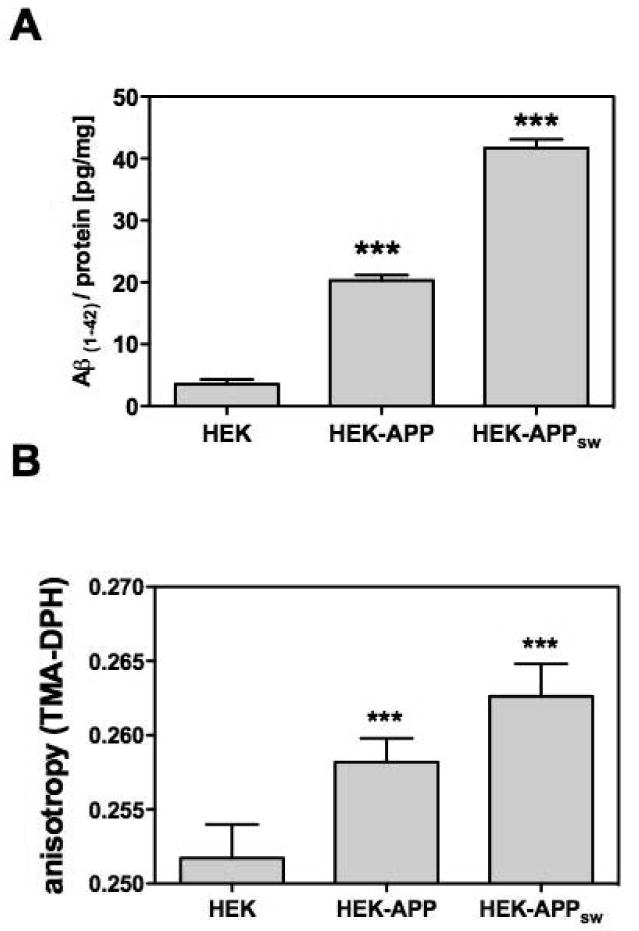
A) Aβ levels were determined by ELISA [pg/mg protein] in HEK293 cells, either tranfected with human AP-P695 (HEK-APP) or human APP695 harboring the Swedish mutation (HEK-APPsw) and in control cells (HEK); B) Changes in the membrane fluidity, which is inversely correlated to the anisotropy of the fluorescence probe TMA-DPH, were determined using fluorescence polarization spectroscopy in living cells; Data are means ± SD; n=6-9 (p***<0.001).
Incubation of HEK-APP cells with the γ-secretase inhibitor DAPT abolished production of endogenous Aβ(1-42) (Fig.3 A) and normalized membrane fluidity in HEK293-APP695 cells (Fig. 3 B). These results indicate that the observed changes in membrane fluidity are due to the endogenous production of Aβ.
Figure 3. Effects of the γ-secretase inhibitor DAPT on the Aβ production and membrane fluidity.
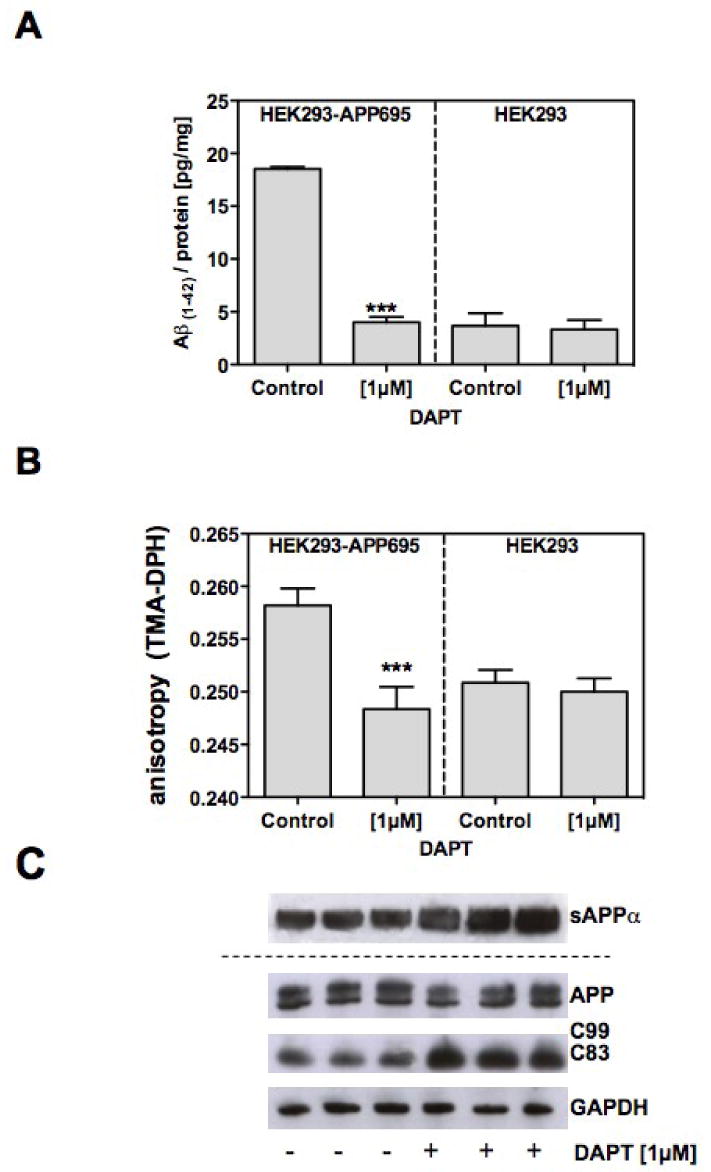
HEK293-AβPP695 cells were treated with 1μM DAPT for at least 10 passages; A) Changes of cellular Aβ(1-42) – levels were determined using ELISA at the end of the incubation period [pg/mg protein]; B) Changes of the membrane fluidity in living cells were measured using TMA-DPH anisotropy as described in the Materials and Methods; C) APP processing fragments were analyzed by Western blot analysis (please refer to Figure 1). Data are means ± SD; n=6-9 (p***<0.001).
Membrane fluidity determines the processing of APP
To confirm that oligoAβ(1-40) stimulates β- and γ-secretase cleavage of APP by altering membrane fluidity (Fig.1), we used compounds that increase (benzyl alcohol) or decrease membrane fluidity (Pluronic F68; PF68) [32, 33]. Treatment with benzyl alcohol increases membrane fluidity (Fig. 4A) and reduces cellular Aβ(1-42) levels in HEK293-APP cells (Fig. 4B). Elevated sAPPα levels induced by benzyl alcohol were associated with reduced CTF C99 levels and elevated CTF C83 levels (Fig. 4C), indicating enhanced α-secretase cleavage of APP. Pluronic PF68 reduced membrane fluidity (Fig. 4D) and increased levels of endogenous Aβ(1-42) (Fig. 4E). Secretion of sAPPα in living HEK293-APP695 cells was reduced (Fig. 4 F). CTF C99 levels were increased and CTF C83 were decreased after PF68 treatment, indicating enhanced β-secretase cleavage of APP (Fig. 4 F).
Figure 4. Impact of membrane fluidity on the cleavage of APP.
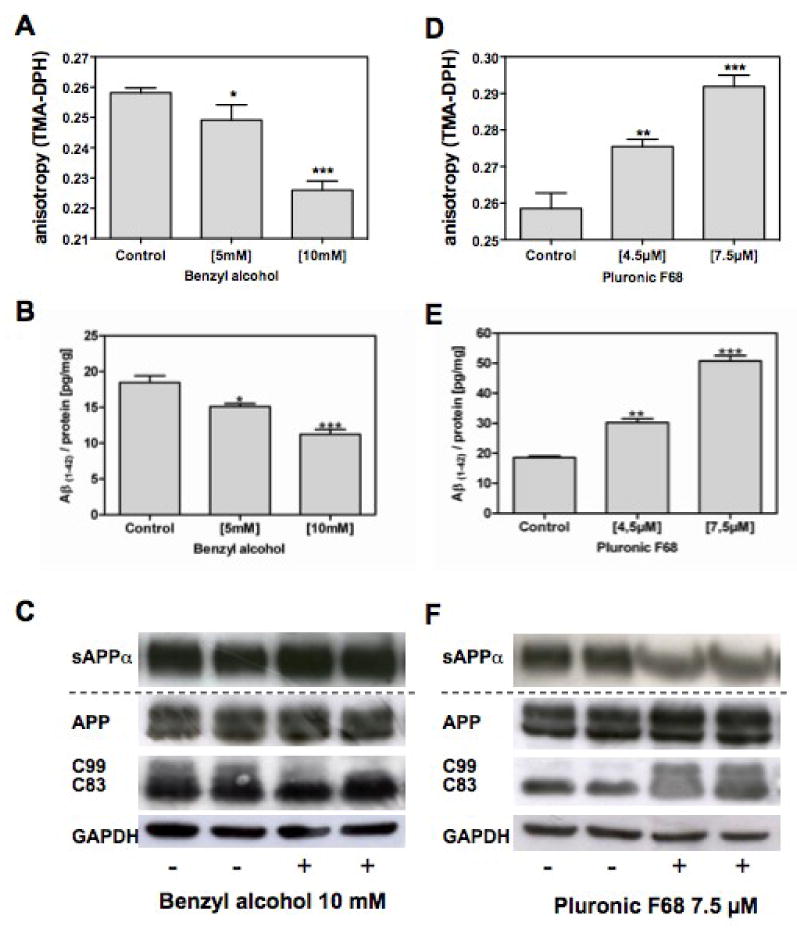
A-C) HEK293-APP695 cells were treated with benzyl alcohol or D-F) Pluronic F68; B,E: The levels of endogenous Aβ(1-42) were determined using ELISA [pg/mg protein]; A, D) Changes in the membrane fluidity were measured as described in the Materials and Methods C,F): APP cleavage products were analyzed by Western blot analysis; the secreted sAPPα was detected in the medium using the antibody 6E10; full-length APP and its C-terminal fragments in the cell homogenates were detected with the antibody C1/6.1; GAPDH served as loading control; Band analysis was performed using BioRad's Quantity One; Data are means ± SD; n=6-12 (p*< 0.05; p**<0.01; p***<0.001).
We also used lovastatin, which increases membrane fluidity by lowering cholesterol levels, and has been shown to stimulate α-secretase-related processing of APP and reducing Aβ production in cells [17, 34, 35]. We confirmed that lovastatin reduced both cellular cholesterol and Aβ(1-42) levels and increased secreted levels of sAPPα and membrane fluidity. β-secretase-related CTF C99 was significantly reduced (data not shown). In contrast to lovastatin, benzyl alcohol and Pluronic F68 did not alter membrane cholesterol levels (data not shown), suggesting that the observed effects of membrane fluidity on APP processing were cholesterol independent.
OligoAβ(1-40) binds to GM-1 ganglioside in living cells
Aβ binds to membrane-bound GM-1 ganglioside and it has been proposed that this binding acts as an endogenous seed for amyloid fibrillization [21]. To test whether the oligoAβ(1-40) induced reduction of membrane fluidity in HEK293-APP695 cells (Figure 1B) involves GM-1 ganglioside, we incubated cells with fluorescein-labeled oligoAβ(1-40) (fluoro-oligoAβ(1-40)). The cells were also stained for GM-1 ganglioside using Alexa555-labelled cholera toxin subunit B and cholesterol using filipin (Figure 5). Blue fluorescence of filipin indicated plasma membrane bound cholesterol (Figure 5). Confocal laser scanning fluorescence microscopy revealed co-localization of Alexa555-labelled cholera toxin subunit B and fluoro-oligoAβ(1-40), which was indicative of oligoAβ(1-40) binding to GM-1 ganglioside (Figure 5).
Figure 5. Binding of Aβ to GM-1 ganglioside.
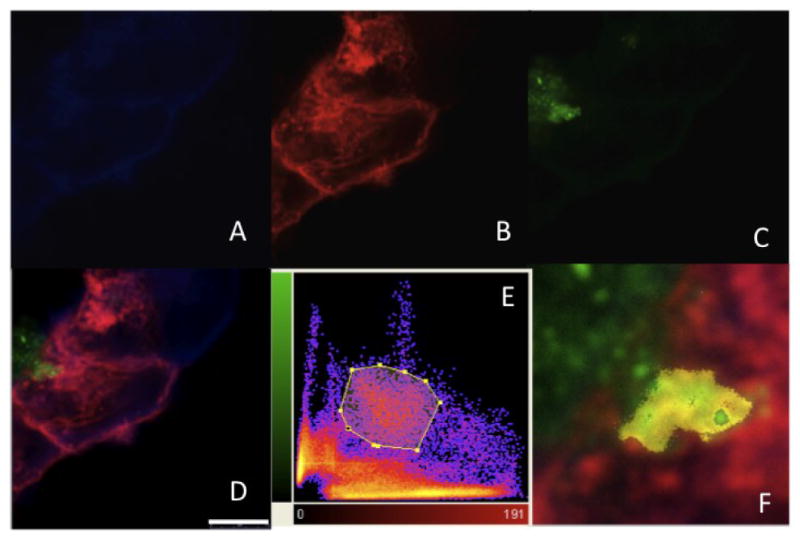
confocal laser-scanning fluorescence microscopy was used to visualize the cellular localization of Aβ and GM-1 ganglioside. A-D) Representative images of the HEK293-APP695 cells; Fluoro-oligoAβ(1-40) was added to the medium and cells were stained for A) cholesterol using filipin, B) GM-1 ganglioside using Alexa 555-conjugated cholera toxin subunit B (CTX-Aexa555) and for C) Aβ using Fluoro-oligoAβ(1-40). D) Merged pictures reveal that oligoAβ(1-40) attaches to the cell membrane (Scale bar = 7.5 μm). Co-localization of oligoAβ(1-40) and GM-1 was described by the presence of the two fluorochromes at the same physical location. E) The 2D-histogramm displays the degree of overlap of the green (Fluor-oligoAβ(1-40)) and red (CTX-Alexa555) fluorescence and reflects the distribution of pairs of voxel intensities occurring in the two selected channels. The range of intensity pairs considered as co-localized was defined after definition of the region of interest on the 2D histogram, its F) result is indicated as yellow color in the picture detail.
Choleratoxin reduces membrane fluidity and enhances cellular Aβ production
If GM-1 ganglioside contributes to the oligoAβ(1-40) induced reduction of membrane fluidity and altered processing of APP, then binding of GM-1 ganglioside to other compounds should mimic effects of Aβ. HEK293-APP695 cells were incubated with choleratoxin subunit B (CTX), which specifically binds to its cell surface receptor GM-1 [36] and it was determined if CTX also reduces membrane fluidity and increases endogenous Aβ levels in cells. Binding of CTX to GM-1 ganglioside significantly reduced membrane fluidity and increased cellular Aβ(1-42) levels (Figure 6 A & B) findings, which were similar to effects of oligoAβ(1-40) as described above. These findings suggest that GM-1 gangliosides play a role in Aβ effects on membrane fluidity and APP cleavage.
Figure 6. Impact of GM-1 ganglioside on the production of cellular Aβ.
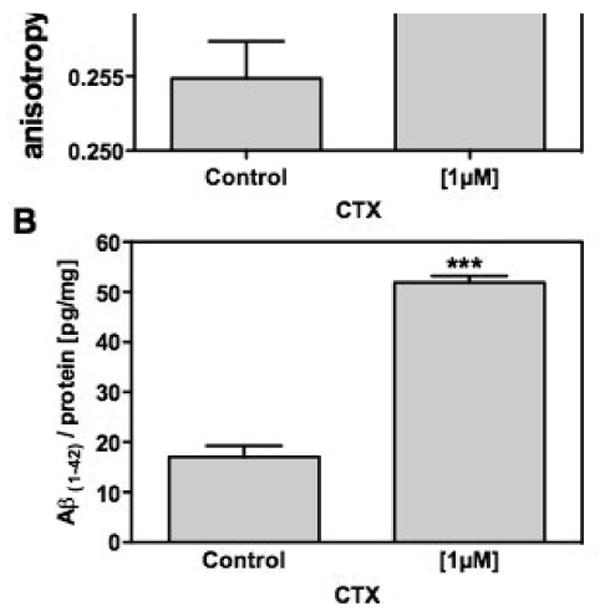
HEK293-APP695 cells were treated for 24h with 1 μM Choleratoxin subunit B (CTX), which specifically binds to GM-1 ganglioside. A) Changes in the membrane fluidity after incubation with CTX were measured as described in the Materials and Methods B) The levels of cellular Aβ(1-42) after incubation CTX were determined using ELISA [pg/mg protein]; Data are means ± SD; n=6-12 (p***<0.001).
In order to investigate possible effects of Aβ on its own production, we studied the influence of oligoAβ(1-40) on the release of cellular Aβ(1-42) in HEK293-APP695 cells. Aβ(1-42) could be determined by a specific and very sensitive ELISA. In order to prove that Aβ also regulates ectodomain shedding of APP in neuronal cells, we used the SH-SY5Y cell line.
Again, a very good correlation was found between membrane fluidity (modulated by Pluronic F68, benzyl alcohol and lovastatin) and cellular Aβ(1-40) production (Figure 7A). OligoAβ(1-40) modulation of Aβ(1-42) release could not be investigated, since levels of Aβ(1-42) were below the detection limit.
Figure 7. Membrane fluidity and cleavage of APP in neuronal cells.
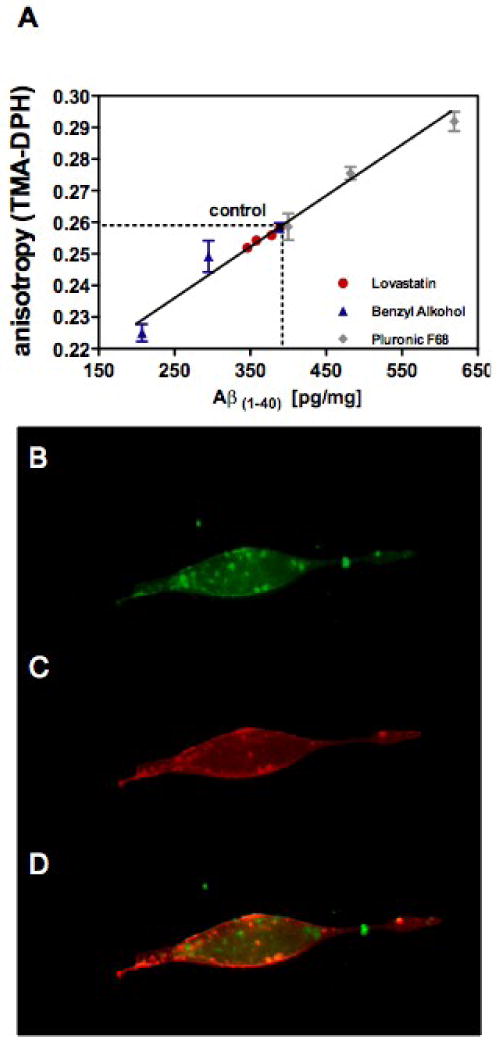
SH-SY5Y-APP695 cells were treated as described in Materials and Methods; A) Membrane fluidity of living cells was correlated to endogenous Aβ(1-40) levels [pg/mg protein]; cells were treated for 24 h with Lovastatin (1, 2 & 4 μM), Benzyl alcohol (5 & 10 mM) and Pluronic F68 (4.5 & 7.5 μM); B-D) Representative fluorescence microscopy images of the SH-SY5Y-AβPP695 cells; B) Fluoro-oligoAβ(1-40) was added to the medium and cells were incubated for 24h; C) Cells were stained for GM-1 ganglioside using Alexa 555-conjugated cholera toxin subunit B; D) Co-localization of Fluoro-oligoAβ(1-40) and Alexa 555-conjugated cholera toxin sub-unit B. Data are means ± SD; n=6-12.
SH-SY5Y-APP695 cells were also incubated with fluoro-oligoAβ(1-40) (Figure 7 B) and stained for GM-1 ganglioside using Alexa555-labelled cholera toxin subunit B (Figure 7 C). Co-localization of Alexa555-labeled cholera toxin subunit B and fluoro-oligoAβ(1-40) is indicative of oligoAβ(1-40) binding to GM-1 ganglioside (Figure 7 D), also in neuronal cells.
In summary, manipulation of the membrane fluidity using different compounds also modulates cellular Aβ levels and oligoAβ(1-40) binds to GM-1 ganglioside in SH-SY5Y-APP695 cells, indicating that theses data are confirmatory of the findings we obtained using non-neuronal cells.
Discussion
There is evidence that the membrane lipid environment modulates secretase activity and potentially affects the function and conformation of the enzymes, influencing substrate selection and the distribution of APP cleavage sites [37]. In the present study we showed that Aβ triggers the amyloidogenic processing of APP by decreasing membrane fluidity and stimulated its own production in living cells. The Aβ perturbation of the membrane was associated with Aβ complexing with GM-1 ganglioside. Our data showed that inhibition of γ-secretase inhibited Aβ production, increased membrane fluidity and stimulated α-secretase cleavage of APP in APP695 over-expressing cells underlines this notion. In vivo support for the potential importance of membrane fluidity in APP processing are reports that the polyunsaturated fatty acid docosahexaenoic acid enhanced synaptic membrane fluidity in aged mice [38] and decreased Aβ levels in brains of murine AD models [39, 40].
The Aβ-induced reduction in fluidity may involve GM-1 ganglioside. This idea is based on several different lines of evidence. The cell surface association of fluoro-oligoAβ(1-40) with GM-1-ganglioside was observed in plasma membranes of intact cells, which confirms GM-1 ganglioside as a potential target molecule of Aβ in plasma membranes [21]. These results are in agreement with recent findings showing that GM-1 ganglioside induces Aβ assembly on the cell surface[41]. Binding of oligoAβ to GM-1 ganglioside may induce changes in membrane fluidity, which in turn provides an energy favorable environment for β- and γ-secretase within the plasma membrane. GM-1 ganglioside regulates membrane structure, increases the order of hydrocarbon chains and decreases fluidity in sphingolipid-enriched membranes [42]. GM-1 ganglioside reduces the membrane fluidity of PC12 cells and alters the localization of receptors within the plasma membrane [43]. Binding of cholera toxin protein to membrane-incorporated GM-1 ganglioside alters the long-range lateral diffusion of fluorescently labeled lipids [44]. Importantly, we demonstrate that binding of cholera toxin to GM-1 reduces membrane fluidity and enhances the production of cellular Aβ(1-42), which are similar to effects of oligoAβ(1-40). Moreover, previous reports show that GM-1 ganglioside regulates APP cleavage [45-47]. These findings further support the notion that Aβ affects the processing of APP in part by binding to GM-1. While Aβ targeting of GM-1 ganglioside may reduce fluidity, it is worth noting that we have been reported previously that aggregated Aβ has a high binding affinity for cholesterol as compared with phosphatidylcholine and saturated fatty acids [25]. It is well-established that membrane fluidity is reduced when cholesterol levels are increased and that APP processing is influenced by cholesterol abundance [17, 34, 35]. However, our findings with lovastatin indicate that membrane fluidity determines the processing of APP independent of cholesterol levels.
Sporadic AD is related to advancing age far more than any other risk factor and there is evidence that sporadic AD overlaps with normal aging in many clinical and pathologic features [48]. Changes in membrane properties are distinguishing markers of brain aging [49, 50]. In particular, membrane fluidity is less in synaptosomal plasma membranes isolated from brains of aged versus young mice [29]. Hippocampal membranes, isolated from brains of AD patients were significantly less fluid compared to membranes of age-matched controls [51]. Thus, age-related reduction of synaptosomal membrane fluidity may provide the optimal environment for β- and γ-secretase cleavage of APP.
Based on our novel findings, we propose that oligoAβ interacts with neuronal membranes by decreasing membrane fluidity and binding to GM-1 ganglioside, which accelerates the proteolytic cleavage of APP and starting a vicious circle in which endogenous Aβ stimulates its own production. This process may be further be enhanced by reduced membrane fluidity which occurs during brain aging.
Supplementary Material
Acknowledgments
This work was supported in part by grants from the Hanna Bragard-Apfel Foundation, Alzheimer Forschung Initiative e.V. (AFI #07821 to D.K. & G.P.E. and #08823 to G.P.E.) and the National Institutes of Health AG-23524, AG-18357 and the Department of Veterans Affairs to W.G.W. We thank Dr. Tobias Hartmann, Homburg, Germany for providing us with SH-SY5YAβPP695 cells and Dr. Paul M. Mathews, Nathan Kline Institute, Orangeburg, NY, for providing us with the mouse IgG C1/6.1 antibody. We also acknowledge the technical help of Claudia Jourdan and Anke Biczysko.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Mattson MP. Pathways towards and away from Alzheimer's disease. Nature. 2004;430:631–639. doi: 10.1038/nature02621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science. 2002;297:353–356. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- 3.Golde TE, Dickson D, Hutton M. Filling the gaps in the AB cascade hypothesis of Alzheimer's disease. Curr Alzheimer Res. 2006;3:421–430. doi: 10.2174/156720506779025189. [DOI] [PubMed] [Google Scholar]
- 4.Haass C, Selkoe DJ. Soluble protein oligomers in neurodegeneration: lessons from the Alzheimer's amyloid beta-peptide. Nat Rev Mol Cell Biol. 2007;8:101–112. doi: 10.1038/nrm2101. [DOI] [PubMed] [Google Scholar]
- 5.Caughey B, Lansbury PT. Protofibrils, pores, fibrils, and neurodegeneration: separating the responsible protein aggregates from the innocent bystanders. Annu Rev Neurosci. 2003;26:267–298. doi: 10.1146/annurev.neuro.26.010302.081142. [DOI] [PubMed] [Google Scholar]
- 6.Lacor PN, Buniel MC, Furlow PW, Clemente AS, Velasco PT, Wood M, Viola KL, Klein WL. Abeta oligomer-induced aberrations in synapse composition, shape, and density provide a molecular basis for loss of connectivity in Alzheimer's disease. J Neurosci. 2007;27:796–807. doi: 10.1523/JNEUROSCI.3501-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Walsh DM, Klyubin I, Fadeeva JV, Cullen WK, Anwyl R, Wolfe MS, Rowan MJ, Selkoe DJ. Naturally secreted oligomers of amyloid beta protein potently inhibit hippocampal long-term potentiation in vivo. Nature. 2002;416:535–539. doi: 10.1038/416535a. [DOI] [PubMed] [Google Scholar]
- 8.Postina R. A Closer Look at alpha-Secretase. Curr Alzheimer Res. 2008;5:179–186. doi: 10.2174/156720508783954668. [DOI] [PubMed] [Google Scholar]
- 9.Deuss M, Reiss K, Hartmann D. Part-Time alpha-Secretases: The Functional Biology of ADAM 9, 10 and 17. Curr Alzheimer Res. 2008;5:187–201. doi: 10.2174/156720508783954686. [DOI] [PubMed] [Google Scholar]
- 10.Kogel D, Schomburg R, Schurmann T, Reimertz C, Konig HG, Poppe M, Eckert A, Muller WE, Prehn JH. The amyloid precursor protein protects PC12 cells against endoplasmic reticulum stress-induced apoptosis. J Neurochem. 2003;87:248–256. doi: 10.1046/j.1471-4159.2003.02000.x. [DOI] [PubMed] [Google Scholar]
- 11.Ziani-Cherif C, Mostefa-Kara B, Brixi-Gormat FZ. Gamma-Secretase as a Pharmacological Target in Alzheimer Disease Research: When, Why and How? Curr Pharm Des. 2006;12:4313–4335. doi: 10.2174/138161206778792994. [DOI] [PubMed] [Google Scholar]
- 12.Vassar R, Bennett BD, Babu-Khan S, Kahn S, Mendiaz EA, Denis P, Teplow DB, Ross S, Amarante P, Loeloff R, Luo Y, Fisher S, Fuller J, Edenson S, Lile J, Jarosinski MA, Biere AL, Curran E, Burgess T, Louis JC, Collins F, Treanor J, Rogers G, Citron M. Beta-secretase cleavage of Alzheimer's amyloid precursor protein by the transmembrane aspartic protease BACE. Science. 1999;286:735–741. doi: 10.1126/science.286.5440.735. [DOI] [PubMed] [Google Scholar]
- 13.Maltese WA, Wilson S, Tan Y, Suomensaari S, Sinha S, Barbour R, McConlogue L. Retention of the Alzheimer's amyloid precursor fragment C99 in the endoplasmic reticulum prevents formation of amyloid beta-peptide. J Biol Chem. 2001;276:20267–20279. doi: 10.1074/jbc.M007238200. [DOI] [PubMed] [Google Scholar]
- 14.Kim SH, Leem JY, Lah JJ, Slunt HH, Levey AI, Thinakaran G, Sisodia SS. Multiple effects of aspartate mutant presenilin 1 on the processing and trafficking of amyloid precursor protein. J Biol Chem. 2001;276:43343–43350. doi: 10.1074/jbc.M108245200. [DOI] [PubMed] [Google Scholar]
- 15.Cuello AC. Intracellular and extracellular Abeta, a tale of two neuropathologies. Brain Pathol. 2005;15:66–71. doi: 10.1111/j.1750-3639.2005.tb00101.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bodovitz S, Klein WL. Cholesterol modulates alpha-secretase cleavage of amyloid precursor protein. J Biol Chem. 1996;271:4436–4440. doi: 10.1074/jbc.271.8.4436. [DOI] [PubMed] [Google Scholar]
- 17.Kojro E, Gimpl G, Lammich S, Marz W, Fahrenholz F. Low cholesterol stimulates the nonamyloidogenic pathway by its effect on the alpha -secretase ADAM 10. Proc Natl Acad Sci USA. 2001;98:5815–5820. doi: 10.1073/pnas.081612998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gamerdinger M, Clement AB, Behl C. Effects of sulindac sulfide on the membrane architecture and the activity of gamma-secretase. Neuropharmacology. 2008;54:998–1005. doi: 10.1016/j.neuropharm.2008.02.009. [DOI] [PubMed] [Google Scholar]
- 19.von Arnim CA, von Einem B, Weber P, Wagner M, Schwanzar D, Spoelgen R, Strauss WL, Schneckenburger H. Impact of cholesterol level upon APP and BACE proximity and APP cleavage. Biochem Biophys Res Commun. 2008;370:207–212. doi: 10.1016/j.bbrc.2008.03.047. [DOI] [PubMed] [Google Scholar]
- 20.Wood WG, Eckert GP, Igbavboa U, Müller WE. Amyloid beta-protein interactions with membranes and cholesterol: causes or casualties of Alzheimer's disease. Biochim Biophys Acta. 2003;1610:281–290. doi: 10.1016/s0005-2736(03)00025-7. [DOI] [PubMed] [Google Scholar]
- 21.Yanagisawa K. Role of gangliosides in Alzheimer's disease. Biochim Biophys Acta. 2007;1768:1943–1951. doi: 10.1016/j.bbamem.2007.01.018. [DOI] [PubMed] [Google Scholar]
- 22.Mathews PM, Jiang Y, Schmidt SD, Grbovic OM, Mercken M, Nixon RA. Calpain activity regulates the cell surface distribution of amyloid precursor protein. Inhibition of clapains enhances endosomal generation of beta-cleaved C-terminal APP fragments. J Biol Chem. 2002;277:36415–36424. doi: 10.1074/jbc.M205208200. [DOI] [PubMed] [Google Scholar]
- 23.Stine WB, Jr, Dahlgren KN, Krafft GA, LaDu MJ. In vitro characterization of conditions for amyloid-beta peptide oligomerization and fibrillogenesis. J Biol Chem. 2003;278:11612–11622. doi: 10.1074/jbc.M210207200. [DOI] [PubMed] [Google Scholar]
- 24.Johnson-Anuna LN, Eckert GP, Franke C, Igbavboa U, Muller WE, Wood WG. Simvastatin protects neurons from cytotoxicity by up-regulating Bcl-2 mRNA and protein. J Neurochem. 2007;101:77–86. doi: 10.1111/j.1471-4159.2006.04375.x. [DOI] [PubMed] [Google Scholar]
- 25.Avdulov NA, Chochina SV, Igbavboa U, Warden CS, Vassiliev AV, Wood WG. Lipid binding to amyloid beta-peptide aggregates: Preferential binding of cholesterol as compared with phosphatidylcholine and fatty acids. J Neurochem. 1997;69:1746–1752. doi: 10.1046/j.1471-4159.1997.69041746.x. [DOI] [PubMed] [Google Scholar]
- 26.Ward RV, Jennings KH, Jepras R, Neville W, Owen DE, Hawkins J, Christie G, Davis JB, George A, Karran EH, Howlett DR. Fractionation and characterization of oligomeric, protofibrillar and fibrillar forms of beta-amyloid peptide. Biochem J. 2000;348:137–144. [PMC free article] [PubMed] [Google Scholar]
- 27.Albers DS, Beal MF. Mitochondrial dysfunction and oxidative stress in aging and neurodegenerative disease. J Neural Transm Suppl. 2000;5959:133–54. 133–54, 133–54. doi: 10.1007/978-3-7091-6781-6_16. [DOI] [PubMed] [Google Scholar]
- 28.Kuhry JG, Duportail G, Bronner C, Laustriat G. Plasma membrane fluidity measurements on whole living cells by fluorescence anisotropy of trimethylammoniumdiphenylhexatriene. Biochim Biophys Acta. 1985;845:60–67. doi: 10.1016/0167-4889(85)90055-2. [DOI] [PubMed] [Google Scholar]
- 29.Eckert GP, Wood WG, Müller WE. Effects of aging and beta-amyloid on the properties of brain synaptic and mitochondrial membranes. J Neural Transm. 2001;108:1051–1064. doi: 10.1007/s007020170024. [DOI] [PubMed] [Google Scholar]
- 30.Leutz S, Steiner B, Marques CA, Haass C, Müller WE, Eckert A. Reduction of trophic support enhances apoptosis in PC12 cells expressing Alzheimer's APP mutation and sensitizes cells to staurosporine-induced cell death. J Mol Neurosci. 2002;18:189–201. doi: 10.1385/JMN:18:3:189. [DOI] [PubMed] [Google Scholar]
- 31.Duyckaerts C, Potier MC, Delatour B. Alzheimer disease models and human neuropathology: similarities and differences. Acta Neuropathol. 2008;115:5–38. doi: 10.1007/s00401-007-0312-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.de Foresta B, Rogard M, le Maire M, Gallay J. Effects of temperature and benzyl alcohol on the structure and adenylate cyclase activity of plasma membranes from bovine adrenal cortex. Biochim Biophys Acta. 1987;905:240–256. doi: 10.1016/0005-2736(87)90452-4. [DOI] [PubMed] [Google Scholar]
- 33.Clarke MS, Prendergast MA, Terry AV., Jr Plasma membrane ordering agent pluronic F-68 (PF-68) reduces neurotransmitter uptake and release and produces learning and memory deficits in rats. Learn Mem. 1999;6:634–649. doi: 10.1101/lm.6.6.634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fassbender K, Simons M, Bergmann C, Stroick M, Lutjohann D, Keller P, Runz H, Kuhl S, Bertsch T, von Bergmann K, Hennerici M, Beyreuther K, Hartmann T. Simvastatin strongly reduces levels of Alzheimer's disease beta -amyloid peptides Abeta 42 and Abeta 40 in vitro and in vivo. Proc Natl Acad Sci USA. 2001;98:5856–5861. doi: 10.1073/pnas.081620098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Frears ER, Stephens DJ, Walters CE, Davies H, Austen BM. The role of cholesterol in the biosynthesis of beta-amyloid. Neuroreport. 1999;10:1699–1705. doi: 10.1097/00001756-199906030-00014. [DOI] [PubMed] [Google Scholar]
- 36.Miller CE, Majewski J, Faller R, Satija S, Kuhl TL. Cholera toxin assault on lipid monolayers containing ganglioside GM1 2. Biophys J. 2004;86:3700–3708. doi: 10.1529/biophysj.103.032508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhou S, Zhou H, Walian PJ, Jap BK. Regulation of gamma-secretase activity in Alzheimer's disease. Biochemistry. 2007;46:2553–2563. doi: 10.1021/bi602509c. [DOI] [PubMed] [Google Scholar]
- 38.Suzuki H, Park SJ, Tamura M, Ando S. Effect of the long-term feeding of dietary lipids on the learning ability, fatty acid composition of brain stem phospholipids and synaptic membrane fluidity in adult mice: a comparison of sardine oil diet with palm oil diet. Mech Ageing Dev. 1998;101:119–128. doi: 10.1016/s0047-6374(97)00169-3. [DOI] [PubMed] [Google Scholar]
- 39.Green KN, Martinez-Coria H, Khashwji H, Hall EB, Yurko-Mauro KA, Ellis L, LaFerla FM. Dietary docosahexaenoic acid and docosapentaenoic acid ameliorate amyloid-beta and tau pathology via a mechanism involving presenilin 1 levels. J Neurosci. 2007;27:4385–4395. doi: 10.1523/JNEUROSCI.0055-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Oksman M, Iivonen H, Hogyes E, Amtul Z, Penke B, Leenders I, Broersen L, Lutjohann D, Hartmann T, Tanila H. Impact of different saturated fatty acid, polyunsaturated fatty acid and cholesterol containing diets on beta-amyloid accumulation in APP/PS1 transgenic mice. Neurobiol Dis. 2006;23:563–572. doi: 10.1016/j.nbd.2006.04.013. [DOI] [PubMed] [Google Scholar]
- 41.Wakabayashi M, Okada T, Kozutsumi Y, Matsuzaki K. GM1 ganglioside-mediated accumulation of amyloid beta-protein on cell membranes. Biochem Biophys Res Commun. 2005;328:1019–1023. doi: 10.1016/j.bbrc.2005.01.060. [DOI] [PubMed] [Google Scholar]
- 42.Pei B, Chen JW. More ordered, convex ganglioside-enriched membrane domains: the effects of GM1 on sphingomyelin bilayers containing a low level of cholesterol. J Biochem (Tokyo) 2003;134:575–581. doi: 10.1093/jb/mvg176. [DOI] [PubMed] [Google Scholar]
- 43.Nishio M, Fukumoto S, Furukawa K, Ichimura A, Miyazaki H, Kusunoki S, Urano T. Overexpressed GM1 suppresses nerve growth factor (NGF) signals by modulating the intracellular localization of NGF receptors and membrane fluidity in PC12 cells. J Biol Chem. 2004;279:33368–33378. doi: 10.1074/jbc.M403816200. [DOI] [PubMed] [Google Scholar]
- 44.Forstner MB, Yee CK, Parikh AN, Groves JT. Lipid lateral mobility and membrane phase structure modulation by protein binding. J Am Chem Soc. 2006;128:15221–15227. doi: 10.1021/ja064093h. [DOI] [PubMed] [Google Scholar]
- 45.Tamboli IY, Prager K, Barth E, Heneka M, Sandhoff K, Walter J. Inhibition of Glycosphingolipid Biosynthesis Reduces Secretion of the {beta}-Amyloid Precursor Protein and Amyloid {beta}-Peptide. J Biol Chem. 2005;280:28110–28117. doi: 10.1074/jbc.M414525200. [DOI] [PubMed] [Google Scholar]
- 46.Zha Q, Ruan Y, Hartmann T, Beyreuther K, Zhang D. GM1 ganglioside regulates the proteolysis of amyloid precursor protein. Mol Psychiatry. 2004;9:946–952. doi: 10.1038/sj.mp.4001509. [DOI] [PubMed] [Google Scholar]
- 47.Tashima Y, Oe R, Lee S, Sugihara G, Chambers EJ, Takahashi M, Yamada T. The effect of cholesterol and monosialoganglioside (GM1) on the release and aggregation of amyloid beta-peptide from liposomes prepared from brain membrane-like lipids. J Biol Chem. 2004;279:17587–17595. doi: 10.1074/jbc.M308622200. [DOI] [PubMed] [Google Scholar]
- 48.Drachman DA. Aging of the brain, entropy, and Alzheimer disease. Neurology. 2006;67:1340–1352. doi: 10.1212/01.wnl.0000240127.89601.83. [DOI] [PubMed] [Google Scholar]
- 49.Choi JH, Yu BP. Brain synaptosomal aging: free radicals and membrane fluidity. Free Radic Biol Med. 1995;18:133–139. doi: 10.1016/0891-5849(94)00106-t. [DOI] [PubMed] [Google Scholar]
- 50.Igbavboa U, Avdulov NA, Schroeder F, Wood WG. Increasing Age Alters Transbilayer Fluidity and Cholesterol Asymetry in Synaptic Plasma Membrane of Mice. J Neurochem. 1996;66:1717–1725. doi: 10.1046/j.1471-4159.1996.66041717.x. [DOI] [PubMed] [Google Scholar]
- 51.Eckert GP, Cairns NJ, Maras A, Gattaz WF, Müller WE. Cholesterol modulates the membrane-disordering effects of beta-amyloid peptides in the hippocampus: specific changes in Alzheimer's disease. Dement Geriatr Cogn Disord. 2000;11:181–186. doi: 10.1159/000017234. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.