

Abstract
Human papillomaviruses (HPV) are the causative agents of cervical cancer and have been shown to increase expression of pro-angiogenic factors from infected cells. Many angiogenic factors are regulated by hypoxia inducible factor 1α (HIF-1α). We investigated whether HPV31 affects the levels of HIF-1α under normal and hypoxic conditions. Our studies indicate that cells containing complete HPV31 genomes showed enhanced levels of HIF-1α upon treatment with the hypoxia mimic DFO, which resulted from protein stabilization and lead to increased expression of some but not all HIF-1α target genes. Both HPV E6 and E7 were able independently to enhance induction of HIF-1α upon DFO treatment. Enhancement of HIF-1α stability was not restricted to high risk HPV types, as HPV11, a low risk HPV type, mediated a similar effect. These findings shed light on mechanisms by which HPV contributes to angiogenesis both in benign cervical lesions and in cervical cancers.
Keywords: Papillomaviruses (HPV), Angiogenesis, Hypoxia, Keratinocytes, Hypoxia inducible factor-1 (HIF-1), Deferoxamine mesylate (DFO), Vascular Endothelial Growth Factor (VEGF), Carbonic Anhydrase (CAIX)
Introduction
Human papillomaviruses (HPVs) are small, non-enveloped DNA viruses that persistently infect the keratinocytes of stratified squamous epithelia (zur Hausen, 1999). HPVs are linked to a variety of malignancies including over 99% of cervical cancers (zur Hausen, 1996). HPVs infect the basal keratinocytes of stratified squamous epithelia (Longworth and Laimins, 2004). As keratinocytes detach from the basement membrane and begin the process of squamous differentiation, HPV maintains the continued expression of cellular DNA replication proteins for use in replicating its own genome, and harnesses cellular regulatory mechanisms to control productive viral replication and expression of its capsid genes (Longworth and Laimins, 2004).
HPV gene products can induce many of the cellular changes characteristic of the tumor phenotype (Hanahan and Weinberg, 2000; Zitvogel, Tesniere, and Kroemer, 2006), including independence from growth control signals (Suprynowicz et al., 2000; Woodworth et al., 1992); bypass of growth inhibitory signals, especially cellular differentiation (Hudson et al., 1990; Ruesch and Laimins, 1998); resistance to apoptosis (Webster et al., 2000; Zhang, Spandau, and Roman, 2002); immune evasion (O’Brien and Saveria Campo, 2002); and angiogenesis (Toussaint-Smith, et al., 2004). The two primary viral oncogenes, E6 and E7, are the only HPV gene products that are consistently retained and expressed in cervical cancers, indicating these two factors are responsible for malignant progression induced by HPV infection (Munger et al., 2004). E6 and E7 function in large part by binding and promoting the degradation of cellular tumor suppressors p53 and pRb, respectively (Munger et al., 2004). Degradation of pRb by E7 results in the activation of E2F transcription factors that drive expression of cellular genes responsible for the S phase of the cell cycle. E7-driven cell cycle progression can result in or potentiate cell death by apoptosis through upregulation of p53. Degradation of p53 mediated by E6 blocks apoptosis to allow continued proliferation. In addition to these well known activities, both oncogenes have a range of other targets (Munger et al., 2004), and the extent to which these additional interactions contribute to HPV associated carcinogenesis is not fully understood.
One important characteristic of tumor development is the promotion of angiogenesis, or the formation of new blood vessels, which allows for access to nutrients and oxygen for growth (Hanahan and Folkman, 1996). Angiogenesis is induced in response to hypoxia, or reduced tissue oxygen levels (Brat, Kaur, and Van Meir, 2003). Under hypoxic conditions, cells secrete a variety of cytokines and growth factors that induce proliferation, migration, and blood vessel formation by endothelial cells (Brat, Kaur, and Van Meir, 2003). The cellular response to hypoxia is primarily regulated through the activity of the transcription factor hypoxia inducible factor-1 (HIF-1)(Bardos and Ashcroft, 2005; Brat, Kaur, and Van Meir, 2003). HIF-1 has two subunits, of which HIF-1α is regulated by oxygen. Under normal oxygen conditions (normoxia), the HIF-1α subunit is hydroxylated, targeting it for rapid degradation through the von Hippel-Lindau (VHL)/proteasome pathway. In hypoxia, reduced oxygen levels block VHL-mediated degradation, resulting in the accumulation of HIF-1α protein, which translocates to the nucleus and activates expression of HIF-1 target genes (Bardos and Ashcroft, 2005). In addition to this hypoxia-dependent stabilization system, a range of other posttranslational modifications and signaling pathways, such as the PI3K/mTOR pathway, also affect HIF-1α synthesis, stability, and activity (Abraham, 2004; Bardos and Ashcroft, 2005; Brat, Kaur, and Van Meir, 2003).
In many cancers, angiogenesis occurs only late in tumor progression, but increased vascular density and production of angiogenic factors is a very early event in the development of HPV-induced pre-malignant lesions and cervical cancers (Smith-McCune et al., 1997; Smith-McCune and Weidner, 1994). Furthermore, several studies have reported that HPV gene products can influence the production of angiogenic factors (Chen et al., 2007; Clere et al., 2007; Tang et al., 2007; Toussaint-Smith, Donner, and Roman, 2004). HIF-1α often continues to be expressed in advanced cervical carcinomas, as well, and is a marker of poor response to therapy (Bachtiary et al., 2003; Ishikawa et al., 2004). Despite these observations, the molecular mechanisms by which HPV proteins induce an angiogenic phenotype remain largely unexplored. Since HIF-1α is a central mediator of the angiogenic response in hypoxia, we investigated whether HPV affects the levels of HIF-1α expressed under normoxic or hypoxic conditions. Our results indicate that the levels of HIF-1α protein are increased in hypoxia when HPV oncogenes are present, and this was true for both high and low risk virus types. Furthermore, this increased HIF-1α induction resulted in increases in some but not all downstream effectors of the hypoxic response, suggesting that HPV specifically manipulates aspects of the cellular hypoxic response.
Materials and Methods
Cell Culture and creation of cell lines
Keratinocyte-derived cell lines were cultured in E-medium containing mouse epidermal growth factor (5 ng/ml; BD Biosciences) with mitomycin C (Medac)-treated NIH 3T3 J2 fibroblast feeders as described previously (Meyers and Laimins, 1994). Prior to harvesting keratinocytes for analysis, feeders were removed via treatment with versene. To create cell lines containing HPV31 episomes, cloned viral DNA was freed from its plasmid vector by digestion with HindIII, unimolecularly ligated with T4 DNA ligase (New England Biolabs) overnight at 16°C, and precipitated with isopropyl alcohol. Human foreskin keratinocytes (HFKs) were transfected with HPV genomes, selected with G418, and pooled cell populations were expanded and analyzed for episomal maintenance as described previously (Wilson and Laimins, 2005). Creation of retroviral constructs has been described previously (Halbert, Demers, and Galloway, 1991; Moody et al., 2007). Hypoxia was mimicked by treatment with 100 μM deferoxamine mesylate (DFO, Sigma). Cycloheximide and rapamycin were obtained from Sigma.
Western blot analysis
Whole cell extracts were prepared using 1x lysis buffer (Cell Signaling) and concentrations were determined using the Bio-Rad (Bradford) protein assay. SDS-PAGE and Western blotting were performed as described previously using 50–100 micrograms of protein (Hebner et al., 2006). HIF-1α antbody (BD Biosciences) was used at 1:500, and GAPDH (Abcam) 1:20,000. Densitometry was performed using Scion Image for Windows (Scion Corp.)
RNA analyses
Total RNA was isolated using RNA STAT-60 (Tel-Test) according to the manufacturer’s directions, and Northern analysis was performed on total RNAs as previously described (Wilson, Fehrmann, and Laimins, 2005). Total RNAs were reverse transcribed using the Omniscript RT kit (Qiagen) and subject to real time PCR analysis using primers specific for human VEGF (5′-TCTACCTCCACCATGCCAAGT-3′, and 5′-GATGATTCTGCCCTCCTCCTT-3′), SYBR green PCR master mix (Applied Biosystems), and an ABI 7900 HT sequence detection system following the manufacturer’s directions. Ribonuclease protection assays were performed using the RPA III kit from Ambion as described previously (Bodily and Meyers, 2005). The probe for Interleukin-8 (IL8) was cloned by PCR into the KpnI/HindIII sites of pGEM 7Zf(+) using Vent polymerase (New England Biolabs) using primers 5′-GCCGGTACCATGACTTCCAAGCTGGCCG-3′ and 5′-GCCAAGCTTCTTTGATAAATTTGGGGTGG-3′. The template (BCMGS neo IL8) was a gift from Steven Polyak at the University of Washington, Seattle, WA and was originally constructed by Naofumi Mukaida, Kanazawa University, Kanazawa, Japan. The internal control for cyclophilin was purchased from Ambion. The CAIX and GLUT1 cDNAs were cloned into pcDNA 3.1(−) by RT-PCR using primers 5′-CGCCTCGAGGCATGGCTCCCCTGTGCCCC-3′ and 5′-CGCGGTACCTCCAGCCTCTAGGCTCCAGT-3′ for CAIX, and 5′-CGCCTCGAG CTGCCATGGAGCCCAGCAGC-3′ and 5′-CGCGGTACCGGCGACTCACACTTGGGAAT-3′ for GLUT1. A cDNA encoding HIF-1α cloned into pcDNA was a gift of Eric Huang (NIH). Northern probe templates were generated from pcDNA-derived plasmids by PCR using the T7 and SP6 primers, and Northern analysis was performed using 10–15 micrograms total RNA as described previously (Fehrmann, Klumpp, and Laimins, 2003).
Luciferase assays
A luciferase reporter consisting of a trimerized 24-mer containing 18 base pairs from the hypoxia response element (HRE) of the phosphoglycerate kinase promoter, HRE-TK-Luc, was a gift from Navdeep Chandel, Department of Medicine, Northwestern University, Chicago, IL. Cells were plated in a six-well dish the day prior to transfection and cotransfected with 1 microgram of a luciferase reporter and 50 nanograms of the Renilla control vector pRL-TK (Promega) using FuGene (Roche Diagnostics) according to manufacturer’s instructions. After overnight incubation, cells were treated with or without DFO for 8 hrs and then harvested for luciferase assay using the Dual Luciferase kit (Promega) according to manufacturer’s instructions. Transfection efficiencies were normalized for Renilla luciferase activity. HIF-1 dependent activity was calculated as the ratio of the activity in cells treated with DFO to that without DFO.
Results
HPV31 and 16 proteins enhance activation of HIF-1α under hypoxic conditions
To determine whether expression of HPV genes had any influence upon HIF-1α activation or levels in normoxia and hypoxia, we first examined whether HIF-1α levels were induced in hypoxia using an HPV31-containing cell line (CIN 612) derived from a premalignant cervical biopsy (Rader et al., 1990). To facilitate analysis we used the hypoxic mimic drug, deferoxamine mesylate (DFO) which has been previously reported to increase the levels of HIF-1α in a manner similar to that seen following growth in 1.5% oxygen (An et al., 1998). As a control we examined the levels of induction with cobalt chloride, a second hypoxic mimic drug. As seen in Figure 1a, treatment of CIN 612 cells with DFO or cobalt chloride induced similar levels of HIF-1α protein as seen following growth for 12 hours in 1.5% oxygen. For ease of use we used DFO treatment in subsequent experiments.
Figure 1. Over-induction of HIF-1α in HPV31 containing cells.
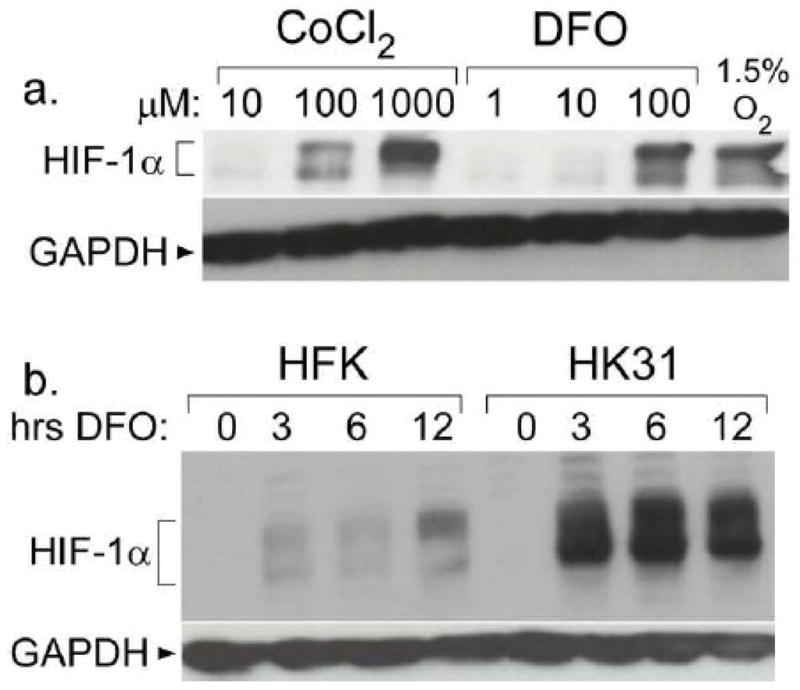
a. CIN 612 cells were treated with the indicated compound for 12 hours or 1.5% oxygen for 48 hours. Total cell extracts were examined for HIF-1α levels by Western blotting. b. HFK-1 and HK31-1 cells were treated with 100 μM DFO for the indicated times, and lysates were analyzed by Western blotting for HIF-1α expression.
We next investigated whether HPV proteins enhanced the levels of HIF-1α in normoxia or hypoxia. Additional HPV31 positive cell lines were generated by transfection of HPV31 DNA into human foreskin keratinocytes (HFK) followed by drug selection and expansion (Wilson and Laimins, 2005). This allowed us to make direct comparisons between normal keratinocytes and keratinocytes containing HPV genomes in the same genetic background. Southern blotting confirmed that these cell lines maintained HPV episomes at similar copy number (not shown). To ensure that any differences we observed were not a result of genetic factors, we performed all experiments multiple times using different HFK donors and compared HPV31-positive cells with HFKs from the same donor. Cell lines were designated HK31 cells, and distinguished from each other according to the HFK donor (i.e. the HK31-1 line was derived from HFK-1, etc.) HFKs and HK31 cells were treated with DFO for various lengths of time and compared the levels of HIF-1α by Western blot analysis. As seen in Figure 1b, HIF-1α was not detected above background in either HFK cells or HK31 cells in the absence of DFO treatment. This indicates that the presence of HPV genomes is not sufficient by itself to induce HIF-1α expression under conditions of normoxia. Failure to detect HIF-1α in normoxia was also seen in cells containing only the HPV E6 and E7 genes, and in cervical cancer cell lines HeLa and C33A (not shown). After incubation with DFO, HIF-1α levels were increased in both cell types within 3 hours after the initiation of treatment. This indicates HIF-1α levels can be increased in response to hypoxia in cells containing HPV. However, HIF-1α levels were consistently increased to a higher degree in HK31 cells as compared to HFK cells, indicating that HPV31 proteins act to enhance induction of HIF-1α in hypoxia. Quantitative analysis of eight experiments indicated that HIF-1α levels were approximately 2.5 fold higher on average in HPV31 positive cells as compared to HFK with a range of 1.5 to 7 fold (data not shown). Similar enhancement of HIF-1α levels was also seen in cell lines harboring episomal forms of HPV16 (data not shown). The multiple bands occasionally seen in HIF-1α Western blots (see also Fig. 5) are of unknown significance, but have also been seen in other published reports (Schmid et al., 2004; Tang et al., 2007) and may represent post-translational modifications such as phosphorylation or ubiquitination.
Figure 5. Enhanced induction of HIF-1α in cells expressing either E6 or E7 from HPV16 or 31.

HFKs or HFKs transduced with either HPV31 E7 (a) or HPV31 E6 (b) were treated with 100 μM DFO for the indicated times, and analyzed by Western blot for HIF-1α expression. c. HFK cells or HFKs expressing the indicated HPV16 oncogenes were treated with 100 μM DFO for the indicated times and analyzed by Western blot for HIF-1α expression.
The oncogenic herpesvirus, HHV8, increases the levels of HIF-1α in infected cells by increasing transcription of the HIF1A gene (Carroll et al., 2006), whereas regulation of HIF-1α usually occurs through post-translational mechanisms in other systems (Bardos and Ashcroft, 2005). To determine if increased levels of HIF-1α seen in HK31 cells was due to increased transcription of the HIF1A gene, Northern blot analysis was performed on RNAs from DFO-treated HFKs and HK31 cells. No differences in HIF-1α mRNA levels were found (Fig. 2a), indicating that the upregulation of HIF-1α is due to post-transcriptional events. We next investigated whether the increase in HIF-1α protein levels was due to changes in protein stability. HFKs or HK31 cells were treated with DFO for 6 hours, with the addition of cycloheximide for various lengths of time before harvesting (Fig. 2b). Levels of HIF-1α were determined by Western blotting, and the half lives of HIF-1α in both cell types were calculated, taking into account the difference in total expression levels between HFKs and HK31 cells. Our studies found that the half life of HIF-1α was extended from approximately 39 minutes in HFKs to 59 minutes in HK31 cells (Fig. 2c). These data indicate that enhanced induction of HIF-1α by HPV31 is due to increased stability of the HIF-1α protein.
Figure 2. HIF-1α regulation by increased protein stability.
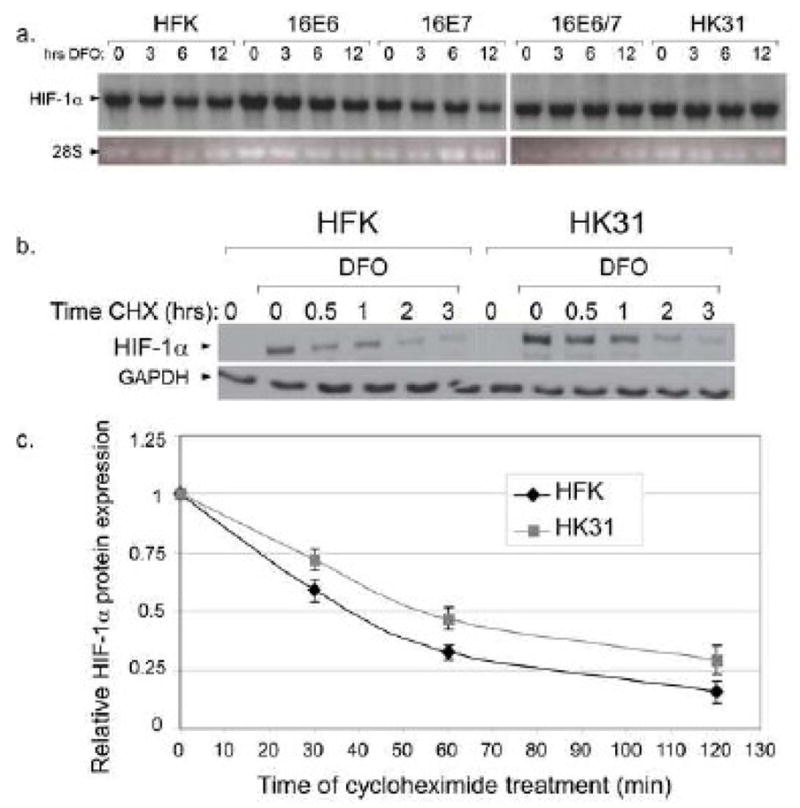
a. HFK-1 cells, HK31-1 cells, or HFKs transduced with the indicated HPV16 oncogene(s) were treated with 100 μM DFO for the indicated times. Total RNA was isolated and subjected to Northern analysis using a probe specific for HIF-1α. b. HFK cells or HK31 cells were treated with DFO for 6 hours. Cycloheximide (50 μg/ml final) was added at the indicated times before cells were harvested. Lysates were analyzed by SDS-PAGE/Western blot for HIF-1α. c. Band densities from three independent experiments were normalized first to the level of GAPDH in each sample and then to the total level of HIF-1α without cycloheximide treatment for each cell line. Bars represent ± one standard error of the mean.
The PI3K/mTOR pathway has also been reported to result in hypoxia-specific upregulation of HIF-1α protein synthesis, an effect that can be inhibited by treatment with rapamycin (Abraham, 2004; Bardos, Chau, and Ashcroft, 2004). To investigate whether the mTOR pathway contributed to increased HIF-1α expression in HK31 cells, we treated cells with both DFO and rapamycin, and then examined HIF-1α levels by Western blot analysis. Rapamycin had a minimal effect on induction of HIF-1α in either HK31 cells or HFKs, regardless of DFO treatment (Fig. 3). We conclude that HPV proteins do not modulate HIF-1α protein levels through the PI3K/mTOR pathway under our culture conditions.
Figure 3. Effect of rapamycin on HIF-1α protein levels.
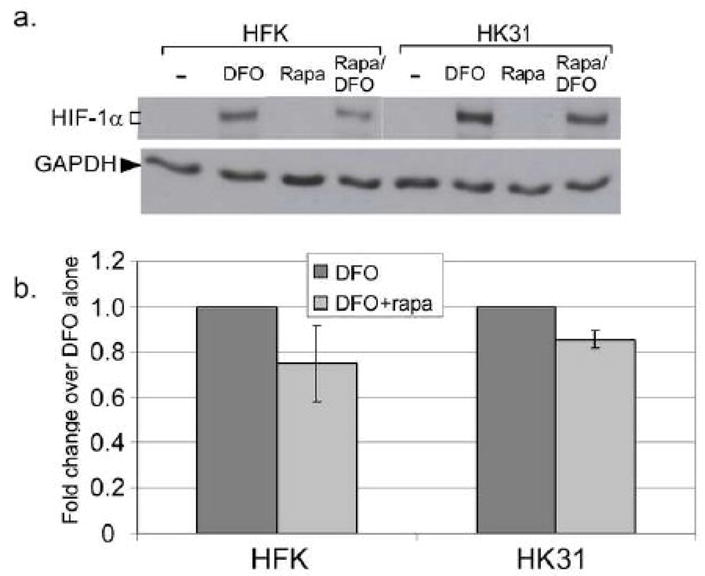
a. HFKs or HK31-2 cells were treated with the indicted drug for 6 hours. Total cell lysates were analyzed for HIF-1α expression by Western blotting. b. Band densities from three independent experiments were normalized first to the level of GAPDH in each sample and then to the total level of HIF-1α without treatment for each cell line. Bars represent ± one standard error of the mean
Activation of HIF-1α target genes by HPV31
HIF-1 regulates the transcription of dozens of genes associated with angiogenesis, tumorigenesis, and glycolytic metabolism, many of which contain hypoxia response elements (HRE) in their promoters (Bardos and Ashcroft, 2005; Brat, Kaur, and Van Meir, 2003). To investigate whether the ability of HIF-1 to act as a transactivator is altered in HK31 cells, we first used reporter plasmids containing several HREs located upstream of luciferase under the control of the tk promoter (Alam et al., 2004). This HIF-1 responsive reporter was transfected into HK31 cells or HFKs that had been infected as an LXSN drug resistance-encoding retrovirus as a control. After overnight incubation, cells were treated with DFO for 8 hours or left untreated. HIF-1 dependent luciferase activity in cell lysates is shown in Figure 4a as a ratio of luciferase activity in cells treated with DFO versus untreated cells. While both HK31 cells and controls showed an increased level of HRE-dependent luciferase activity following treatment with DFO, HK31 cells showed a greater than two-fold increase in activity as compared to controls, in line with the enhanced HIF-1α levels we observed in HK31 cells.
Figure 4. Regulation of HIF-1 targets by HPV31.

a. HK31-1 or HFK-1 cells transduced with a LXSN retroviral vector were transfected with the HRE-TK-Luc reporter plasmid. The next day, cells were treated or untreated with 100 μM DFO for 8 hours and assayed for luciferase activity. The data points represent a ratio of the activity with DFO treatment to the activity without for three experiments and bars represent ± one standard error of the mean. b. Total RNAs from HK31-1 or HFK-1 cells treated with DFO for the indicated times were subjected to reverse transcription followed by real-time PCR using primers specific for VEGF. Bars represent ± one standard error of the mean. Total RNAs from the HFK-3 or HK31-3 cells were examined for expression of (c) CAIX or GLUT1 by Northern analysis or (d) IL8 expression by ribonuclease protection assay.
We next investigated whether expression of endogenous HIF-1 targets would be similarly enhanced in HPV-containing cells. Vascular endothelial growth factor (VEGF) is an important pro-angiogenic HIF-1 target gene and a central player in angiogenic signaling (Rankin and Giaccia, 2008). The levels of VEGF transcripts were examined by quantitative RT-PCR in normoxia and following treatment with DFO. VEGF transcripts were induced to a higher level in HK31 cells than in HFKs following treatment with DFO, consistent with higher levels of HIF-1α in these cells (Fig. 4b). We next sought to determine whether HPV would enhance the expression of all HIF-1 targets or only those associated with angiogenesis. Among the many HIF-1 targets, carbonic anhydrase IX (CAIX), which helps control cellular pH, and GLUT1, a glucose transporter, are not 11 associated directly with angiogenesis but are upregulated during carcinogenesis (Lee et al., 2007). Northern blot analysis indicated CAIX transcripts were increased to a greater degree (mean of about 1.8 fold) in HK31 cells treated with DFO as compared to similarly treated HFKs. In contrast, GLUT1 transcripts were expressed at similar basal levels in untreated HFK and HK31 cells, and induced to comparable levels following DFO treatment.
HIF-1 regulates some genes which could be detrimental to the persistence of virally infected cells. One of these is interleukin-8 (IL8), which, in addition to angiogenic activity also promotes inflammatory responses (Brat, Kaur, and Van Meir, 2003). To test whether HPV31 induces IL8 in hypoxic conditions, we performed ribonuclease protection assays on RNA from cells treated or untreated with DFO. As shown in Figure 4d, not only were transcripts for IL8 not induced by DFO in HFK-derived cells, expression of this gene was dramatically reduced by HPV31, in contrast to the expression profile of HIF-1α protein (Fig. 1), but in agreement with the downregulation of IL8 by HPV oncogenes observed by others in normoxia (Huang and McCance, 2002). Together these experiments indicate that enhanced HIF-1α induction by HPV results in activation of a subset of HIF-1 target genes, and that HPV may alter the spectrum of genes activated by HIF-1α.
Both E6 and E7 can enhance induction of HIF-1α
HK31 cells maintain the entire HPV31 episomal genome, and so any of the HPV31 gene products could potentially be involved in regulating HIF-1α. We investigated whether the oncogenes E6 and E7 were sufficient to enhance HIF-1α levels since these proteins are selectively retained in cancers and previous reports indicate that they are capable of regulating some angiogenic genes (Baker et al., 1987; Tang et al., 2007; Toussaint-Smith, Donner, and Roman, 2004). Furthermore, E6 promotes the degradation of p53 (Scheffner et al., 1990), which is reported to negatively regulate HIF-1α stability in hypoxia (Ravi et al., 2000). HFKs stably expressing HPV31 E6 or E7 were generated following infection with LXSN-based retroviral vectors, selection for G418 resistance, and expansion in culture (Thomas and Laimins, 1998). Following treatment with DFO, the levels of HIF-1α were examined. As seen in Figure 5a and b, HPV31 E6 and E7 were each sufficient on their own to cause enhanced induction of HIF-1α upon DFO treatment. Similar effects were seen in cell lines expressing E6 or E7 from HPV16 (Fig. 5c). In none of these cell lines were the levels HIF-1α transcripts altered (Fig. 2a). We confirmed that p53 levels were reduced in E6-expressing cells and enhanced in E7 expressing cells, in line with previous observations (Eichten et al., 2002; Scheffner et al., 1990) (data not shown). This indicates that the mechanism(s) regulating enhanced stability of HIF-1α in HPV positive cells does not depend exclusively on p53 protein levels.
Low risk HPV11 also enhances HIF-1α levels upon DFO treatment
Infection by low-risk HPV types induces benign hyperproliferations that rarely progress to cancers. Stabilization of HIF-1α as a mechanism of inducing angiogenesis during the development of cancers has been well documented (Zhou et al., 2006), but benign HPV-induced lesions likely also require the expression of angiogenic factors to ensure adequate oxygen and nutrient supply for growth. We therefore sought to determine whether cells containing the low risk type HPV11 would also show enhanced induction of HIF-1α under hypoxic conditions. For these studies, we transfected HFKs with cloned and recircularized HPV11 genomes (Oh, Longworth, and Laimins, 2004). Following drug selection and expansion, we determined by Southern analysis that viral DNA was maintained as episomes (not shown). We then treated HPV11 positive cells with DFO and examined the levels of HIF-1α by Western blot analysis. As shown in Figure 6, HIF-1α expression in hypoxia was enhanced in HFKs containing HPV11 episomes, similar to the effect seen with HK31 cells, with a mean enhancement of about 2.5 fold. Thus, the increased levels of HIF-1α proteins induced during hypoxia appear to be a property of both high and low risk papillomaviruses.
Figure 6. Effect of HPV11 on HIF-1α induction.
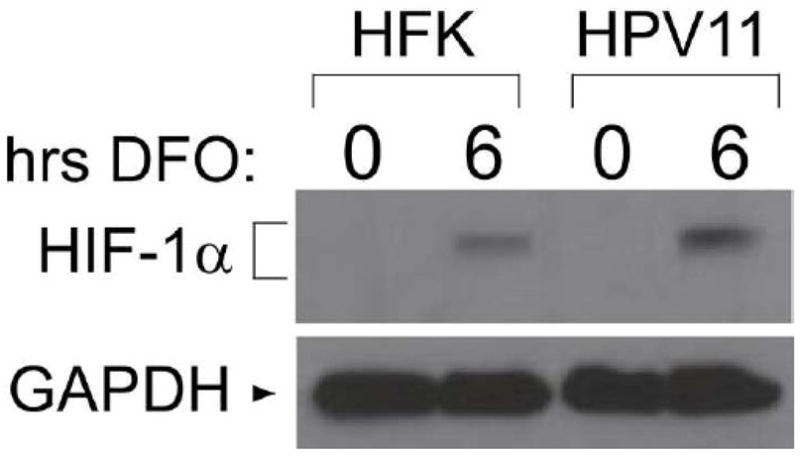
HFKs or HFKs containing HPV11 episomes were treated with 100 μM DFO for the indicated time and analyzed by Western blotting for the expression of HIF-1α.
Discussion
The development of cervical cancers requires angiogenesis, but exhibits some unique differences from other solid tumors. Normal stratified epithelia, including cervix, have characteristics of chronically hypoxic tissue, such as HIF-1α stabilization and VEGF expression, which are further increased during progression to malignancy (Evans et al., 2006; Lee et al., 2007; Mayer et al., 2004; Mazibrada et al., 2008; Smith-McCune et al., 1997). Increased vascularity is observed in low grade HPV infections of the cervix, while this is usually a late event in the progression of many other cancers (Mazibrada et al., 2008; Smith-McCune et al., 1997; Smith-McCune and Weidner, 1994). From the early stages of infection to lesion development and eventually progression to cancer, HPV modulates the cellular response to hypoxia. Our results indicate that HPV enhances the levels of HIF-1α through protein stabilization, resulting in the increased expression of a subset of HIF-1 target genes. This enhanced expression likely plays a central role in the progression of HPV-induced disease.
Our studies provide insight into how HIF-1α levels are manipulated by HPV proteins. We found that upregulation of HIF-1α by HPV proteins was not limited to cervical cancers: keratinocytes stably maintaining HPV genomes, which model cells of early, precancerous lesions, also upregulated HIF-1α. Furthermore, both high-risk HPV16 and 31 as well as the low risk HPV11 were found to be equally capable of enhancing HIF-1α levels, indicating that manipulation of the hypoxic response is important for many papillomavirus types. This may not be surprising, given that nutrient and oxygen delivery are required in proliferating lesions regardless of whether they are induced by high or low risk viruses. Our studies also indicate that the effects of HPV proteins on HIF-1α levels occur specifically in hypoxia, with no detectible effect in normoxia. Since cervical tissues have characteristics of chronic hypoxia even under normal conditions (Lee et al., 2007; Mayer et al., 2004; Smith-McCune et al., 1997), HPV-infected cervical cells in vivo may activate angiogenic factors before the development of high-grade cancers.
A wide range of cellular factors and pathways have been reported to affect HIF-1α protein levels (Bardos and Ashcroft, 2005; Brat, Kaur, and Van Meir, 2003). A previous report (Huh et al., 2007) has shown that E7 can interact with a cullin-2 ubiquitin ligase complex that has been reported to mediate VHL-dependent HIF-1α degradation (Brat, Kaur, and Van Meir, 2003). Alteration of the activity of this complex by E7 should affect the level of HIF-1α in normoxia. However, we failed to detect HIF-1α in normoxia, even in cells expressing E7. This suggests that the normal VHL-mediated HIF-1α degradation pathway still functions in HPV-infected cells regardless of any effects E7 may have on other aspects the VHL pathway. Furthermore, there was no strong effect of rapamycin on HIF-1α levels, either with or without treatment with DFO, suggesting that the mTOR pathway (Abraham, 2004) is also not a major player. The loss of PML has been reported to result in hypoxia-specific enhancement of HIF-1α induction (Bernardi et al., 2006), and PML can be targeted by E7 (Bischof, Nacerddine, and Dejean, 2005). Loss of p53 has also been reported to enhance HIF-1α induction in hypoxia (Ravi et al., 2000). Although E6 and E7 affect the absolute levels of p53 protein in opposite ways, both oncogenes have been reported to affect the function of p53 (Munger et al., 2004). Both E6 and E7 were independently sufficient to alter HIF-1α levels, indicating that activation of HIF-1α in HPV positive cells cannot be strictly dependent on p53 protein levels, but whether E6 and E7 alter HIF-1α through alteration of p53 function remains to be determined.
Several studies have shown that high-risk E6 and E7 can activate expression or enhance levels of a number of angiogenic factors, but it was not established that HIF-1α activation was responsible (Toussaint-Smith, Donner, and Roman, 2004). Transfection of siRNAs against HPV18 E6 was found to reduce VEGF expression in HeLa cells (Clere et al., 2007). In addition, conditioned supernatants from HPV16 E6 or E7 expressing cells were able to induce tube formation and proliferation of endothelial cells consistent with the activation of angiogenic factors (Bequet-Romero and Lopez-Ocejo, 2000; Chen et al., 2007; Lopez-Ocejo et al., 2000; Tang et al., 2007). Using transient transfections of cervical cancer cells, E6 and E7 were able to increase expression of genes associated with angiogenesis which are also responsive to HIF-1 (Clere et al., 2007; Lopez-Ocejo et al., 2000; Tang et al., 2007). Finally, transient transfection of E6 and E7 expression vectors into cervical cancer cell lines was reported to induce HIF-1α levels under normoxic conditions but without altering protein stability (Tang et al., 2007). In contrast, our studies using stable cell lines show that HIF-1α can be upregulated by E6 and E7 but only in hypoxia and through protein stabilization. We suggest that the hypoxia-specific stabilization effect that we report is more likely to reflect conditions in vivo, given that our cells stably express HPV proteins.
The enhanced levels of HIF-1α in HPV positive cells were found to activate some downstream genes such as VEGF and CAIX. On the other hand, upregulation of IL8 and GLUT1 were either not seen or not enhanced in HPV positive cells. This indicates that HPV proteins have a mechanism to modulate the spectra of HIF-1 target genes that are activated upon DFO treatment. Studies from the Roman laboratory (Toussaint-Smith, Donner, and Roman, 2004) have shown that cells expressing HPV oncogenes can induce the expression of angiogenic genes such as VEGF even in non-hypoxic conditions. The state of HIF-1α was, however, not examined and it is possible that there was low level activation in their cells that was not observed in our assays. Alternatively, it is possible that HIF-1 independent mechanisms are responsible for activation of these genes. Our studies examining the levels of VEGF in normoxia showed low levels of expression in HPV positive cells in the absence of DFO, consistent with HIF-1 independent mechanisms of activation. It is also possible that different culture methods could be responsible for the effects observed. Consistent with published studies, we have observed that HIF-1α can be induced at low levels in normoxia by growth at high confluency (data not shown, see also (Rempe et al., 2007)) which could lead to activation of angiogenic genes. Our survey of angiogenesis mediators produced in HPV positive cells was not as complete as Toussaint-Smith et al., but we note that there appears to have been a somewhat higher expression of VEGF in HK31 cells and in some experiments CAIX (not shown) in the absence of DFO treatment. In any case, more detailed analysis of HIF-1 downstream mediators will be needed to understand the role of hypoxia and HIF-1α stabilization in the angiogenic phenotype in HPV infection.
The ability of HPV to inhibit the expression of IL8 transcripts, as seen in Figure 4d, was striking, but is consistent with the results of Huang et al, who observed that E6 and E7 could inhibit IL8 transcription by binding to coactivators (Huang and McCance, 2002). Future studies will further clarify this effect.
Finally, it was possible that the replication or transcription of the HPV31 genomes was sensitive to hypoxia, but we found that there was no effect of 12 hours of DFO treatment on viral copy number, and only a slight increase in viral transcripts and luciferase activity from early and late promoter reporters (unpublished). Whether viral replication and gene expression are impacted by more chronic hypoxia remains to be determined. In summary, we have found that several HPV types cause an enhancement of HIF-1α expression in hypoxia, an effect mediated by increasing stability of the HIF-1α protein. Furthermore, this increased HIF-1α expression is reflected in increases in some but not all downstream effectors of the hypoxic response, and is seen upon the expression of either E6 or E7. These results shed light on the relationship between HPV infection and hypoxia in the development of cervical cancer and may help us better understand how to manage HPV-induced disease.
Acknowledgments
We thank Eric Huang, Navdeep Chandel, Stephen Polyak, and Naofumi Mukaida for reagents, and Hiroshi Ishikawa for assistance performing real time PCR analysis. This study was supported by a STD center grant from the NIAID to LAL. JMB was supported by fellowships from NIH (NRSA) and Illinois Department of Public Health.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abraham RT. mTOR as a positive regulator of tumor cell responses to hypoxia. Curr Top Microbiol Immunol. 2004;279:299–319. doi: 10.1007/978-3-642-18930-2_18. [DOI] [PubMed] [Google Scholar]
- Alam H, Maizels ET, Park Y, Ghaey S, Feiger ZJ, Chandel NS, Hunzicker-Dunn M. Follicle-stimulating hormone activation of hypoxia-inducible factor-1 by the phosphatidylinositol 3-kinase/AKT/Ras homolog enriched in brain (Rheb)/mammalian target of rapamycin (mTOR) pathway is necessary for induction of select protein markers of follicular differentiation. J Biol Chem. 2004;279 (19):19431–40. doi: 10.1074/jbc.M401235200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- An WG, Kanekal M, Simon MC, Maltepe E, Blagosklonny MV, Neckers LM. Stabilization of wild-type p53 by hypoxia-inducible factor 1alpha. Nature. 1998;392 (6674):405–8. doi: 10.1038/32925. [DOI] [PubMed] [Google Scholar]
- Bachtiary B, Schindl M, Potter R, Dreier B, Knocke TH, Hainfellner JA, Horvat R, Birner P. Overexpression of hypoxia-inducible factor 1alpha indicates diminished response to radiotherapy and unfavorable prognosis in patients receiving radical radiotherapy for cervical cancer. Clin Cancer Res. 2003;9(6):2234–40. [PubMed] [Google Scholar]
- Baker CC, Phelps WC, Lindgren V, Braun MJ, Gonda MA, Howley PM. Structural and transcriptional analysis of human papillomavirus type 16 sequences in cervical carcinoma cell lines. J Virol. 1987;61 (4):962–71. doi: 10.1128/jvi.61.4.962-971.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bardos JI, Ashcroft M. Negative and positive regulation of HIF-1: a complex network. Biochim Biophys Acta. 2005;1755 (2):107–20. doi: 10.1016/j.bbcan.2005.05.001. [DOI] [PubMed] [Google Scholar]
- Bardos JI, Chau NM, Ashcroft M. Growth factor-mediated induction of HDM2 positively regulates hypoxia-inducible factor 1alpha expression. Mol Cell Biol. 2004;24 (7):2905–14. doi: 10.1128/MCB.24.7.2905-2914.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bequet-Romero M, Lopez-Ocejo O. Angiogenesis modulators expression in culture cell lines positives for HPV-16 oncoproteins. Biochem Biophys Res Commun. 2000;277(1):55–61. doi: 10.1006/bbrc.2000.3628. [DOI] [PubMed] [Google Scholar]
- Bernardi R, Guernah I, Jin D, Grisendi S, Alimonti A, Teruya-Feldstein J, Cordon-Cardo C, Simon MC, Rafii S, Pandolfi PP. PML inhibits HIF-1alpha translation and neoangiogenesis through repression of mTOR. Nature. 2006;442 (7104):779–85. doi: 10.1038/nature05029. [DOI] [PubMed] [Google Scholar]
- Bischof O, Nacerddine K, Dejean A. Human Papillomavirus Oncoprotein E7 Targets the Promyelocytic Leukemia Protein and Circumvents Cellular Senescence via the Rb and p53 Tumor Suppressor Pathways. Mol Cell Biol. 2005;25(3):1013–24. doi: 10.1128/MCB.25.3.1013-1024.2005. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Bodily JM, Meyers C. Analysis of HPV transcription by RPA. Methods Mol Med. 2005;119:279–90. doi: 10.1385/1-59259-982-6:279. [DOI] [PubMed] [Google Scholar]
- Brat DJ, Kaur B, Van Meir EG. Genetic modulation of hypoxia induced gene expression and angiogenesis: relevance to brain tumors. Front Biosci. 2003;8:d 100–16. doi: 10.2741/942. [DOI] [PubMed] [Google Scholar]
- Carroll PA, Kenerson HL, Yeung RS, Lagunoff M. Latent Kaposi’s sarcoma-associated herpesvirus infection of endothelial cells activates hypoxia-induced factors. J Virol. 2006;80 (21):10802–12. doi: 10.1128/JVI.00673-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen W, Li F, Mead L, White H, Walker J, Ingram DA, Roman A. Human papillomavirus causes an angiogenic switch in keratinocytes which is sufficient to alter endothelial cell behavior. Virology. 2007;367 (1):168–74. doi: 10.1016/j.virol.2007.05.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clere N, Bermont L, Fauconnet S, Lascombe I, Saunier M, Vettoretti L, Plissonnier ML, Mougin C. The human papillomavirus type 18 E6 oncoprotein induces Vascular Endothelial Growth Factor 121 (VEGF121) transcription from the promoter through a p53-independent mechanism. Exp Cell Res. 2007;313 (15):3239–50. doi: 10.1016/j.yexcr.2007.06.029. [DOI] [PubMed] [Google Scholar]
- Eichten A, Westfall M, Pietenpol JA, Munger K. Stabilization and functional impairment of the tumor suppressor p53 by the human papillomavirus type 16 E7 oncoprotein. Virology. 2002;295 (1):74–85. doi: 10.1006/viro.2002.1375. [DOI] [PubMed] [Google Scholar]
- Evans SM, Schrlau AE, Chalian AA, Zhang P, Koch CJ. Oxygen levels in normal and previously irradiated human skin as assessed by EF5 binding. J Invest Dermatol. 2006;126 (12):2596–606. doi: 10.1038/sj.jid.5700451. [DOI] [PubMed] [Google Scholar]
- Fehrmann F, Klumpp DJ, Laimins LA. Human papillomavirus type 31 E5 protein supports cell cycle progression and activates late viral functions upon epithelial differentiation. J Virol. 2003;77 (5):2819–31. doi: 10.1128/JVI.77.5.2819-2831.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halbert CL, Demers GW, Galloway DA. The E7 gene of human papillomavirus type 16 is sufficient for immortalization of human epithelial cells. J Virol. 1991;65 (1):473–8. doi: 10.1128/jvi.65.1.473-478.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanahan D, Folkman J. Patterns and emerging mechanisms of the angiogenic switch during tumorigenesis. Cell. 1996;86 (3):353–64. doi: 10.1016/s0092-8674(00)80108-7. [DOI] [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100 (1):57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- Hebner CM, Wilson R, Rader J, Bidder M, Laimins LA. Human papillomaviruses target the double-stranded RNA protein kinase pathway. J Gen Virol. 2006;87 (Pt 11):3183–93. doi: 10.1099/vir.0.82098-0. [DOI] [PubMed] [Google Scholar]
- Huang SM, McCance DJ. Down regulation of the interleukin-8 promoter by human papillomavirus type 16 E6 and E7 through effects on CREB binding protein/p300 and P/CAF. J Virol. 2002;76 (17):8710–21. doi: 10.1128/JVI.76.17.8710-8721.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudson JB, Bedell MA, McCance DJ, Laiminis LA. Immortalization and altered differentiation of human keratinocytes in vitro by the E6 and E7 open reading frames of human papillomavirus type 18. J Virol. 1990;64(2):519–26. doi: 10.1128/jvi.64.2.519-526.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huh K, Zhou X, Hayakawa H, Cho JY, Libermann TA, Jin J, Harper JW, Munger K. Human papillomavirus type 16 E7 oncoprotein associates with the cullin 2 ubiquitin ligase complex, which contributes to degradation of the retinoblastoma tumor suppressor. J Virol. 2007;81 (18):9737–47. doi: 10.1128/JVI.00881-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishikawa H, Sakurai H, Hasegawa M, Mitsuhashi N, Takahashi M, Masuda N, Nakajima M, Kitamoto Y, Saitoh J, Nakano T. Expression of hypoxic-inducible factor 1alpha predicts metastasis-free survival after radiation therapy alone in stage IIIB cervical squamous cell carcinoma. Int J Radiat Oncol Biol Phys. 2004;60 (2):513–21. doi: 10.1016/j.ijrobp.2004.03.025. [DOI] [PubMed] [Google Scholar]
- Lee WY, Huang SC, Hsu KF, Tzeng CC, Shen WL. Roles for hypoxia-regulated genes during cervical carcinogenesis: Somatic evolution during the hypoxia-glycolysis-acidosis sequence. Gynecol Oncol. 2007;108 (2):377–84. doi: 10.1016/j.ygyno.2007.10.034. [DOI] [PubMed] [Google Scholar]
- Longworth MS, Laimins LA. Pathogenesis of human papillomaviruses in differentiating epithelia. Microbiol Mol Biol Rev. 2004;68 (2):362–72. doi: 10.1128/MMBR.68.2.362-372.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Ocejo O, Viloria-Petit A, Bequet-Romero M, Mukhopadhyay D, Rak J, Kerbel RS. Oncogenes and tumor angiogenesis: the HPV-16 E6 oncoprotein activates the vascular endothelial growth factor (VEGF) gene promoter in a p53 independent manner. Oncogene. 2000;19 (40):4611–20. doi: 10.1038/sj.onc.1203817. [DOI] [PubMed] [Google Scholar]
- Mayer A, Wree A, Hockel M, Leo C, Pilch H, Vaupel P. Lack of correlation between expression of HIF-1alpha protein and oxygenation status in identical tissue areas of squamous cell carcinomas of the uterine cervix. Cancer Res. 2004;64 (16):5876–81. doi: 10.1158/0008-5472.CAN-03-3566. [DOI] [PubMed] [Google Scholar]
- Mazibrada J, Ritta M, Mondini M, De Andrea M, Azzimonti B, Borgogna C, Ciotti M, Orlando A, Surico N, Chiusa L, Landolfo S, Gariglio M. Interaction between inflammation and angiogenesis during different stages of cervical carcinogenesis. Gynecol Oncol. 2008;108 (1):112–20. doi: 10.1016/j.ygyno.2007.08.095. [DOI] [PubMed] [Google Scholar]
- Meyers C, Laimins LA. In vitro systems for the study and propagation of human papillomaviruses. Curr Top Microbiol Immunol. 1994;186:199–215. doi: 10.1007/978-3-642-78487-3_11. [DOI] [PubMed] [Google Scholar]
- Moody CA, Fradet-Turcotte A, Archambault J, Laimins LA. Human papillomaviruses activate caspases upon epithelial differentiation to induce viral genome amplification. Proc Natl Acad Sci U S A. 2007;104 (49):19541–6. doi: 10.1073/pnas.0707947104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munger K, Baldwin A, Edwards KM, Hayakawa H, Nguyen CL, Owens M, Grace M, Huh K. Mechanisms of human papillomavirus-induced oncogenesis. J Virol. 2004;78 (21):11451–60. doi: 10.1128/JVI.78.21.11451-11460.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Brien PM, Saveria Campo M. Evasion of host immunity directed by papillomavirus-encoded proteins. Virus Res. 2002;88 (1–2):103–17. doi: 10.1016/s0168-1702(02)00123-5. [DOI] [PubMed] [Google Scholar]
- Oh ST, Longworth MS, Laimins LA. Roles of the E6 and E7 proteins in the life cycle of low-risk human papillomavirus type 11. J Virol. 2004;78 (5):2620–6. doi: 10.1128/JVI.78.5.2620-2626.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rader JS, Golub TR, Hudson JB, Patel D, Bedell MA, Laimins LA. In vitro differentiation of epithelial cells from cervical neoplasias resembles in vivo lesions. Oncogene. 1990;5 (4):571–6. [PubMed] [Google Scholar]
- Rankin EB, Giaccia AJ. The role of hypoxia-inducible factors in tumorigenesis. Cell Death Differ. 2008;15 (4):678–85. doi: 10.1038/cdd.2008.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravi R, Mookerjee B, Bhujwalla ZM, Sutter CH, Artemov D, Zeng Q, Dillehay LE, Madan A, Semenza GL, Bedi A. Regulation of tumor angiogenesis by p53-induced degradation of hypoxia-inducible factor 1alpha. Genes Dev. 2000;14 (1):34–44. [PMC free article] [PubMed] [Google Scholar]
- Rempe DA, Lelli KM, Vangeison G, Johnson RS, Federoff HJ. In cultured astrocytes, p53 and MDM2 do not alter hypoxia-inducible factor-1alpha function regardless of the presence of DNA damage. J Biol Chem. 2007;282 (22):16187–201. doi: 10.1074/jbc.M702203200. [DOI] [PubMed] [Google Scholar]
- Ruesch MN, Laimins LA. Human papillomavirus oncoproteins alter differentiation-dependent cell cycle exit on suspension in semisolid medium. Virology. 1998;250 (1):19–29. doi: 10.1006/viro.1998.9359. [DOI] [PubMed] [Google Scholar]
- Scheffner M, Werness BA, Huibregtse JM, Levine AJ, Howley PM. The E6 oncoprotein encoded by human papillomavirus types 16 and 18 promotes the degradation of p53. Cell. 1990;63 (6):1129–36. doi: 10.1016/0092-8674(90)90409-8. [DOI] [PubMed] [Google Scholar]
- Schmid T, Zhou J, Kohl R, Brune B. p300 relieves p53-evoked transcriptional repression of hypoxia-inducible factor-1 (HIF-1) Biochem J. 2004;380(Pt 1):289–95. doi: 10.1042/BJ20031299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith-McCune K, Zhu YH, Hanahan D, Arbeit J. Cross-species comparison of angiogenesis during the premalignant stages of squamous carcinogenesis in the human cervix and K14-HPV16 transgenic mice. Cancer Res. 1997;57 (7):1294–300. [PubMed] [Google Scholar]
- Smith-McCune KK, Weidner N. Demonstration and characterization of the angiogenic properties of cervical dysplasia. Cancer Res. 1994;54 (3):800–4. [PubMed] [Google Scholar]
- Suprynowicz FA, Sparkowski J, Baege A, Schlegel R. E5 oncoprotein mutants activate phosphoinositide 3-kinase independently of platelet-derived growth factor receptor activation. Journal of Biological Chemistry. 2000;275(7):5111–5119. doi: 10.1074/jbc.275.7.5111. [DOI] [PubMed] [Google Scholar]
- Tang X, Zhang Q, Nishitani J, Brown J, Shi S, Le AD. Overexpression of human papillomavirus type 16 oncoproteins enhances hypoxia-inducible factor 1 alpha protein accumulation and vascular endothelial growth factor expression in human cervical carcinoma cells. Clin Cancer Res. 2007;13(9):2568–76. doi: 10.1158/1078-0432.CCR-06-2704. [DOI] [PubMed] [Google Scholar]
- Thomas JT, Laimins LA. Human papillomavirus oncoproteins E6 and E7 independently abrogate the mitotic spindle checkpoint. J Virol. 1998;72 (2):1131–7. doi: 10.1128/jvi.72.2.1131-1137.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toussaint-Smith E, Donner DB, Roman A. Expression of human papillomavirus type 16 E6 and E7 oncoproteins in primary foreskin keratinocytes is sufficient to alter the expression of angiogenic factors. Oncogene. 2004;23(17):2988–95. doi: 10.1038/sj.onc.1207442. [DOI] [PubMed] [Google Scholar]
- Webster K, Parish J, Pandya M, Stern PL, Clarke AR, Gaston K. The human papillomavirus (HPV) 16 E2 protein induces apoptosis in the absence of other HPV proteins and via a p53-dependent pathway. J Biol Chem. 2000;275(1):87–94. doi: 10.1074/jbc.275.1.87. [DOI] [PubMed] [Google Scholar]
- Wilson R, Fehrmann F, Laimins LA. Role of the E1^E4 Protein in the Differentiation-Dependent Life Cycle of Human Papillomavirus Type 31. J Virol. 2005;79 (11):6732–40. doi: 10.1128/JVI.79.11.6732-6740.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson R, Laimins LA. Differentiation of HPV-containing cells using organotypic “raft” culture or methylcellulose. Methods Mol Med. 2005;119:157–69. doi: 10.1385/1-59259-982-6:157. [DOI] [PubMed] [Google Scholar]
- Woodworth CD, Cheng S, Simpson S, Hamacher L, Chow LT, Broker TR, DiPaolo JA. Recombinant retroviruses encoding human papillomavirus type 18 E6 and E7 genes stimulate proliferation and delay differentiation of human keratinocytes early after infection. Oncogene. 1992;7(4):619–26. [PubMed] [Google Scholar]
- Zhang B, Spandau DF, Roman A. E5 protein of human papillomavirus type 16 protects human foreskin keratinocytes from UV B-irradiation-induced apoptosis. J Virol. 2002;76 (1):220–31. doi: 10.1128/JVI.76.1.220-231.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J, Schmid T, Schnitzer S, Brune B. Tumor hypoxia and cancer progression. Cancer Lett. 2006;237 (1):10–21. doi: 10.1016/j.canlet.2005.05.028. [DOI] [PubMed] [Google Scholar]
- Zitvogel L, Tesniere A, Kroemer G. Cancer despite immunosurveillance: immunoselection and immunosubversion. Nat Rev Immunol. 2006;6 (10):715–27. doi: 10.1038/nri1936. [DOI] [PubMed] [Google Scholar]
- zur Hausen H. Papillomavirus infections--a major cause of human cancers. Biochim Biophys Acta. 1996;1288 (2):F55–78. doi: 10.1016/0304-419x(96)00020-0. [DOI] [PubMed] [Google Scholar]
- zur Hausen H. Papillomaviruses in human cancers. Proc Assoc Am Physicians. 1999;111(6):581–7. doi: 10.1046/j.1525-1381.1999.99723.x. [DOI] [PubMed] [Google Scholar]