

Abstract
Pseudoachondroplasia (PSACH) is one of the more common skeletal dysplasias and results from mutations in cartilage oligomeric matrix protein (COMP). Most COMP mutations identified to date cluster in the TSP3 repeat region of COMP and the mutant protein is retained in the rough endoplasmic reticulum (rER) of chondrocytes and may result in increased cell death. In contrast, the pathomolecular mechanism of PSACH resulting from C-terminal domain COMP mutations remain largely unknown. This study describes the generation and analysis of a murine model of mild PSACH resulting from a p.Thr583Met mutation in the C-terminal globular domain (CTD) of COMP. Mutant animals are normal at birth, but grow slower than their wild-type littermates and by 9 weeks of age they have mild short-limb dwarfism. Furthermore, by 16 months of age mutant animals exhibit severe degeneration of articular cartilage, which is consistent with early onset osteoarthritis seen in PSACH patients. In the growth plates of mutant mice the chondrocyte columns are sparser and poorly organized. Mutant COMP is secreted into the extracellular matrix, but its localization is disrupted along with the distribution of several COMP-binding proteins. Although mutant COMP is not retained within the rER there is an unfolded protein/cell stress response and chondrocyte proliferation is significantly reduced, while apoptosis is both increased and spatially dysregulated. Overall, these data suggests a mutation in the CTD of COMP exerts a dominant-negative effect on both intra- and extracellular processes. This ultimately affects the morphology and proliferation of growth plate chondrocytes, eventually leading to chondrodysplasia and reduced long bone growth.
INTRODUCTION
Pseudoachondroplasia (PSACH) (OMIM #177170) is an autosomal dominant skeletal dysplasia characterized by a waddling gait, disproportionate short stature, generalized epimetaphyseal dysplasia and early-onset degenerative joint disease (1,2). The short-limb dwarfism becomes apparent during childhood and most affected individuals develop early-onset osteoarthritis during early adult life, which often requires bilateral hip replacement (2). A mild form of PSACH has also been described in which affected individuals may present with only mild short stature (1).
PSACH results exclusively from mutations in the gene encoding cartilage oligomeric matrix protein (COMP; OMIM *600310) (3,4). COMP is a 550 kDa pentameric glycoprotein found in the extracellular matrix (ECM) of developing and mature cartilage, within and around the tendon (5), in ligament, synovium and the vitreous of the eye (6). It belongs to the thrombospondin family of proteins (7) and consists of a coiled-coil oligomerization domain, four type 2 (EGF-like) domains, eight TSP type 3 (T3) sequence repeats and a large globular C-terminal globular domain (CTD) (8).
The majority of the mutations resulting in PSACH (approximately 85%) cluster in the T3 repeat region of COMP (9) and the cell-matrix pathology of these mutations have been studied in detail. Electron microscopy of cartilage from patients with a T3 mutation consistently shows the retention of mutant COMP within the rough endoplasmic reticulum (rER) of chondrocytes and a general disruption to tissue architecture, including collagen fibril orientation and morphology (10). Furthermore, the primary retention of mutant COMP is associated with the co-retention of other ECM proteins such as matrilin-3 and type IX collagen (11,12). Type II collagen is not co-retained in PSACH chondrocytes, suggesting a different secretory pathway for this molecule (12,13). The retention of protein within the rER results in the formation of large ‘inclusion bodies’, which may in some cases have a distinct lamellar appearance. Eventually, this protein retention is thought to cause rER stress and finally cell death, which ultimately results in an overall reduction in the number of viable cells in the affected tissue (14). However, these studies have mainly used in vitro cell culture systems or articular and iliac crest cartilage samples obtained from surgery and therefore, the mechanistic link between a COMP mutation and disrupted endochondral bone growth has not been determined in vivo. In contrast, the pathomolecular mechanisms of PSACH resulting from CTD–COMP mutations (approximately 15%) remain largely unknown due to the difficulty in obtaining suitable pathological material from the affected patients. Recombinant COMP with mutations engineered into the CTD was shown to be secreted from different cell lines in vitro (15,16), however, the effect of these mutations on endochondral ossification and bone growth have never been studied in vivo.
Over 15 unique CTD–COMP mutations have now been identified, which can cause phenotypes ranging from mild multiple epiphyseal dysplasia (MED) type ribbing/osteoarthritis through severe PSACH (17). The p.Thr585Met mutation (the equivalent of p.Thr583Met in the mouse) is a single nucleotide substitution in the C-terminal domain of COMP first reported by Briggs et al. in 1998 (18). The family described in this study had a mild form of PSACH with typical radiographic features and waddling gait, but normal or only mild short stature (18). Interestingly, the p.Thr585Met mutation is located in a potential collagen binding site (17,19); it is spatially associated with a cluster of other CTD mutations (most notably p.E583K, p.S681C, p.H587R) (17) and lies 21 amino acids away from a potential integrin-binding sequence. Furthermore, recent crystallography studies of full-length thrombospondin 2 (20) and a C-terminal fragment of thrombospondin 1 (21) (containing the CTD and four T3 repeats) have highlighted the interdependency between these two domains in the overall structure of TSP monomers and oligomers. It is now clear that there are intramolecular interactions between the CTD and T3 domains of individual thrombospondin monomers, in addition to the intermolecular interactions with other ECM components and cells. Mutations in the CTD of COMP are therefore likely to have a profound effect on the structure and function of the protein.
In this paper we describe the generation and comprehensive analysis of the first knock-in mouse model of a CTD–COMP mutation (p.Thr583Met). This is the first model of PSACH to be reported in which the murine phenotype closely resembles the human disease. Furthermore, we have determined that the disease results from a combination of both intra- and extracellular mechanisms that have a profound effect on chondrocyte proliferation and apoptosis within the cartilage growth plate.
RESULTS
Generation of the p.Thr583Met COMP knock-in line
Mice harbouring the equivalent of the human p.Thr585Met mutation in the C-terminal domain of COMP were generated by homologous recombination in R1 embryonic stem (ES) cells using an appropriate targeting strategy (Fig. 1A-D). The ‘long arm’ of the targeting construct (8 kb) contained exons 1–16 of comp and the ‘short arm’ of the construct (2 kb) contained exons 17–19 of comp (Fig. 1B). The required nucleotide substitutions to produce the murine p.Thr583Met COMP mutation (equivalent to human p.Thr585Met) were introduced into exon 16 by site-directed mutagenesis (ACA → ATG). Three hundred and sixty ES cell clones that had been electroporated with the targeting construct and selected by antibiotic resistance were analysed for homologous recombination using Southern blotting with the external probe (Fig. 1C and E). The presence of the mutation was confirmed by direct sequencing of DNA from nine ES clones that had successfully undergone homologous recombination (Fig. 1F). Positive clones resulting from the targeting procedure were microinjected into C57BL/6 blastocysts and then implanted into pseudopregnant foster mothers. The resultant chimeric mice were used for the foundation of a transgenic line and a breeding strategy was used to obtain wild-type mice and littermates that were either heterozygous or homozygous for the mutation.
Figure 1.

Targeting strategy to generate the PSACH mouse. (A) The domain structure of a COMP monomer and the genomic organization of comp showing the location of key restriction sites and the external probe (red bar). (B) The targeting construct with loxP (arrowheads) and flanked Neo cassette in intron 16. An asterisk marks the site of the introduced mutation in exon 16. (C) Modified comp locus following homologous recombination. The external probe detects two HindIII restriction fragments (blue arrows). (D) The recombinant comp allele following Cre-mediated recombination. (E) Southern blot showing wild-type (WT) and recombinant (HR) clones following HindIII digestion. (F) DNA sequencing confirming that the mutation is present in the recombinant alleles (ACA → ATG). Restriction enzyme sites: E, EcoRI; H, HindIII; K, KpnI; M, MluI; C, ClaI. (G) Quantitative real-time PCR (qRT-PCR) analysis showing comparable comp expression in WT and mutant cartilage at 3 weeks of age (n ≥ 5, independent samples t-test). (H) Western blot analysis showing comparable levels of COMP protein in WT and mutant cartilage at 3 weeks of age. Key: +/+ (WT); m/m (mice homozygous for the mutation).
In order to establish the expression levels of the wild-type and mutant comp alleles, quantitative real-time PCR (qRT-PCR) was performed on the mRNA extracted from chondrocytes of 3-week-old growth plate cartilage. The expression levels of comp mRNA from the wild-type and/or mutant alleles was comparable in wild-type, heterozygous and homozygous mice (Fig. 1G). This was confirmed by western blot analysis of protein extracts from the same cartilage (Fig. 1H).
Mutant mice are normal at birth
Breeding pairs of mice heterozygous for the p.Thr583Met COMP mutation produced pups in a normal Mendelian distribution of 1:2:1 (+/+:m/+:m/m) and no overt skeletal defects were seen at birth (Fig. 2A). These observations were as expected since PSACH patients have normal stature at birth. Furthermore, there were no differences in the lengths and organization of the skeletal elements between wild-type, heterozygous and homozygous littermates (Fig. 2A) while bone mineralization, as assessed by von Kossa staining, was not altered in the mutant mice (Fig. 2B). At birth, histological analysis of tibial growth plates revealed no gross morphological differences when visualized by H&E staining. The columnar organization of the cells in the growth plate and the demarcation of individual zones were unaffected in animals heterozygous or homozygous for the mutation (Fig. 2C). Immunohistochemistry (IHC) was used to determine the localization of wild-type and mutant COMP in the new born growth plates. At this age COMP had a predominantly pericellular localization and this was comparable in the growth plates of mice of all three genotypes (Fig. 2D).
Figure 2.

COMP mice are normal at birth. (A) Skeletal preparations of new born mice (cartilage stained with Alcian Blue and bone with Alizarin Red). There is no overt phenotype in new born mice homozygous or heterozygous for p.Thr583Met and the skeletal elements appeared normal and in proportion for all three genotypes. (B) Von Kossa staining of mineralized bone showed no differences in bone mineralization and morphology between the new born wild-type and p.Thr583Met COMP mice. (C) H&E staining of the growth plate showed no differences in the morphology between the littermates of all three genotypes. (D) IHC using a COMP antibody revealed no differences in the distribution of COMP in the new born WT and mutant growth plates. Scale bar 100 μm. Key: +/+ (WT); m/+ (mice heterozygous for the mutation); m/m (mice homozygous for the mutation); PZ (proliferative zone); PHZ (prehypertrophic zone); HZ (hypertrophic zone).
Mutant animals develop short limb dwarfism by 9 weeks of age
In order to quantify any changes in growth that may have been caused by the mutation littermates of all three genotypes (i.e. +/+; m/+; m/m) were weighed at 3, 6 and 9 weeks of age and these measurements were used to prepare growth curves (Fig. 3A; ≥15 mice per sex per genotype per time point). At 3 weeks of age the body weights of all the mice were comparable, but by 6 weeks of age the body weights were different according to the genotype and sex of the animal. By 9 weeks of age, when long bone growth had ceased, the differences in weight were statistically significant and males homozygous for the mutation were 6% lighter than their wild-type littermates. A similar difference was noted for female mice. Bone length measurements were performed on whole mouse radiographs taken at 3, 6 and 9 weeks of age to determine the effect of the mutation on endochondral and intramembranous ossification (Fig. 3B; ≥10 mice per sex per genotype per time point). The inner-canthal distance (ICD) was used as a measure of intramembranous ossification and these measurements were comparable in mice of all three genotypes. However, long bone growth was significantly affected and by 9 weeks of age the tibia of male mice homozygous for the mutation were 4% shorter than those of their wild-type littermates. An intermediate phenotype was observed in males heterozygous for the mutation, which was characterized by a 2% decrease in tibia length when compared with wild-type littermates. In addition to reduced long bone length, mice that were homozygous for the p.Thr583Met COMP mutation also exhibited a mild, but significant, hip dysplasia that was characterized by the tuberosity of the ischium protruding from the pelvic region at an angle of >15° (Fig. 4A and B). Mice that were heterozygous for the mutation exhibited a milder form of pelvic deformation.
Figure 3.

Mutant mice develop a mild short limb dwarfism with increasing age. (A) Mice were weighed at 3, 6 and 9 weeks of age and the weights used to construct the growth curves. Mice of both sexes and of all genotypes had comparable weights early in development but by 9 weeks of age mutant animals were significantly lighter with a >6% decrease in male body weight and >7% decrease in female body weight for mice homozygous for the mutation (n ≥ 15; one-way ANOVA). (B) Bone measurements of male littermates with inner canthal distance (ICD) used as a marker of intramembranous ossification and the tibia length as a marker of endochondral ossification. The difference in tibia length between the WT males and males homozygous for the mutation was >4% at 9 weeks of age and the difference between WT males and males heterozygous for the mutation was >2% (n ≥ 10; one-way ANOVA). Key: +/+ (WT); m/+ (mice heterozygous for the mutation); m/m (mice homozygous for the mutation); *P > 0.05; **P > 0.01.
Figure 4.

Mutant mice exhibit a mild hip dysplasia and with age develop severe articular cartilage degeneration. (A) The angle between the tuberosity of the ischium and the pelvic region (°) were measured at 3, 6 and 9 weeks of age. (B) The hip angle was approximately 56% greater for males homozygous for the mutation and 44% greater for males heterozygous for the mutation when compared with their wild-type littermates at 9 weeks (n ≥ 10 mice per genotype; one-way ANOVA). (C) H&E (top panel) and Safranin O (bottom panel) staining of the articular surface of the knee joints at 16 months of age showing severe articular cartilage degradation associated with a complete loss of sulphated proteoglycans in mice homozygous for the mutation. Scale bar 100 μm. Key: +/+ (wild-type); m/+ (heterozygous for the mutation); m/m (homozygous for the mutation); ***P < 0.001; **P < 0.01; *P < 0.05.
Mutant mice develop degenerative joint disease in adulthood
Mice homozygous for the p.Thr583Met mutation developed degenerative joint disease by 16 months of age, which was characterized by a loss of Safranin O staining at the articular surface of the knee (Fig. 4C, bottom panel; >15 sections per genotype from two mice per genotype). In contrast, wild-type mice showed uniform Safranin O staining at the articular surfaces of the knee joints indicating the presence of sulphated proteoglycans. The specific loss of cartilage from the articular surface was verified by H&E staining (Fig. 4C, top panel).
Growth plate organization is disrupted in mutant mice
Histological (H&E) analysis was performed in order to determine the morphological changes that had occurred in the growth plates of mutant mice (Fig. 5A). Wild-type mice had well organized growth plates with clearly distinguishable resting, proliferative and hypertrophic zones. In particular, the chondrocytes in the proliferative zones were aligned into well organized columns evenly distributed along the horizontal axis of the growth plate. However, chondrocyte alignment in the proliferative zone of mice homozygous for the p.Thr583Met mutation was disrupted from 2 weeks of age. For example, chondrocyte columns were reduced in number and in some cases they terminated prematurely with a corresponding increase in the amount of space between individual columns. At 3 weeks of age the growth plates of mice homozygous for the p.Thr583Met mutation exhibited a significantly enlarged proliferative zone of ≥15% (Fig. 5A; 10 sections per mouse, three mice per genotype; independent samples t-test), which was possibly the result of chondrocyte misalignment (Fig. 5B). In contrast, the morphology of the hypertrophic zone did not appear to be affected in littermates that were either heterozygous or homozygous for p.Thr583Met.
Figure 5.

Organization of the growth plate in mutant mice is disrupted by 3 weeks of age. (A) H&E staining showed disruption to the growth plate by 3 weeks of age in mice carrying the comp mutation. This disruption was characterized by disorganized and sparse columns of chondrocytes in the proliferating zone. Note the enlarged proliferating zone in the 3 week mutant growth, possibly due to chondrocyte misalignment (n ≥ 8 sections per genotype; independent samples t-test; *P < 0.05). The black line marks the boundary between the hypertrophic zone and proliferative zone in the growth plate. Scale bar 100 μm. (B) TEM of the proliferating zone in the tibial growth plates at 1 week shows misaligned and abnormally shaped chondrocytes in mice homozygous for p.Thr583Met. Scale bar 23 μm. Key: +/+ (wild-type); m/+ (heterozygous for the mutation); m/m (homozygous for the mutation).
The alignment of proliferating chondrocytes is significantly affected in the mutant growth plate
The ultrastructure of 1 week old tibial growth plates was studied by transmission electron microscopy (TEM). In the growth plates of wild-type mice the resting chondrocytes were evenly distributed in the ECM, while the proliferating chondrocytes had a more flattened appearance and were found in the characteristic 2-, 4- and 8-cell chondron arrangement (Fig. 5B). In the prehypertrophic zone of the wild-type growth plate, the cells were larger and assumed a more rounded appearance, whereas in the late hypertrophic zone rounded lacunae were seen which contained the remnants of cellular material (data not shown). In contrast, chondrocytes in the proliferative zone of the growth plates of mice homozygous for p.Thr583Met were severely disorganized; they had a heterogeneous and irregular shape and did not align properly within the chondrons (Fig. 5B), which is most likely the cause of the enlarged proliferative zone seen in the mutant mice. The morphology of the growth plate in mice heterozygous for p.Thr583Met did not show any gross differences when compared with the wild-type.
The localization of key structural proteins in the growth plate cartilage is altered in mutant mice
Wild-type COMP has a predominantly pericellular localization at birth, but by 2-3-weeks of age it is also found in the interterritorial matrix of the growth plate (22). In contrast, the distribution of mutant COMP in the ECM was significantly altered in the growth plate of mice homozygous for p.Thr583Met (Fig. 6). For example, by 3 weeks of age mutant COMP was detected in greater abundance in the territorial and pericellular ECM of the growth plates of mice homozygous for p.Thr583Met, which was noticeably different from the uniform interterritorial distribution observed in the ECM of wild-type mice (Fig. 6). This change in the localization of mutant COMP became more pronounced by 6 and then 9 weeks of age.
Figure 6.

The localization of mutant COMP in the cartilage ECM is disrupted. Immunohistochemistry (IHC) using a COMP antibody revealed increasingly less interterritorial staining in the extracellular matrix (ECM) between the proliferating columns, which was visible from 3 weeks of age in the growth plates of mice carrying the p.Thr583Met COMP mutation (asterisks). Scale bar 100 μm. Key: +/+ (wild-type); m/+ (heterozygous for the mutation); m/m (homozygous for the mutation).
The localization of those proteins known to interact with COMP in vitro was also determined by IHC (Fig. 7). In the growth plates of wild-type mice, matrilin-3 was uniformly distributed in the interterritorial ECM. However, in the growth plate cartilage of littermates homozygous for p.Thr583Met, matrilin-3 appeared to have a more pronounced territorial and pericellular location. This had the effect of creating distinct spaces between the chondrocyte columns in which there was very weak or no immunostaining. A similar staining pattern was seen for type IX collagen, which was particularly evident in the late proliferative zone. Interestingly, the distribution of type II collagen, the major collagen found in the cartilage ECM, was not affected by the p.Thr583Met COMP mutation.
Figure 7.

The localization of matrilin-3 and type IX collagen, but not type II collagen, is disrupted in the growth plates of mutant mice. Representative immunohistochemistry at 6 weeks of age. Immunohistochemical analysis using a COMP antibody revealed less interterritorial staining in the ECM between the proliferating columns which was visible from 3 weeks in the growth plates of mice carrying the p.Thr583Met mutation (circled areas). Matrilin-3 follows the localization of mutant COMP in the growth plate and type IX collagen localization appears to be distinctly altered in the middle of the proliferating zone of the mutant growth plates. Type II collagen localization remains unaltered. Scale bar 100 μm. Key: +/+ (wild-type); m/+ (heterozygous for the mutation); m/m (homozygous for the mutation).
The ultrastructure of the cartilage extra cellular matrix is altered in growth plates of mutant mice
The ultrastructure of the interterritorial matrix in the proliferative zone was examined by TEM at 1 week of age (Fig. 8). The appearance of the cartilage ECM was altered in mice that were either heterozygous or homozygous for the p.Thr583Met mutation and resulted in a distinct morphological spectrum depending on the genotype. For example the interterritorial matrix from mice heterozygous for the mutation appeared to contain less proteoglycan-like amorphous material than the equivalent region of their wild-type littermates. This had the effect of making the collagen fibrils more prominent in the mutant samples when compared with wild-type. The interterritorial matrix of the mice homozygous for the mutation appeared to contain even more abundant and better defined fibrillar material.
Figure 8.

ECM ultrastructure is altered in the mutant growth plates. The ultrastructure of the interterritorial matrix at 1 week of age is altered in mutant animals and is characterized by more prominent appearing collagen fibrillar material in the growth plate ECM. Scale bar 800 nm. Highlighted area is magnified five times in the bottom panel. Key: +/+ (wild-type); m/+ (heterozygous for the mutation); m/m (homozygous for the mutation).
Chondrocytes from mutant mice exhibit a mild rough endoplasmic reticulum stress
IHC studies indicated that a significant proportion of the mutant COMP was secreted into the ECM. Nevertheless, the secretion of this mutant protein does not rule out the possibility that the mutant COMP protein elicits an unfolded protein and/or cell stress response during its passage through the cell’s ER/secretory system. We therefore used a combination of qRT-PCR and densitometric Western blot analysis to study the classical markers of UPR (such as BiP and calreticulin) and the key regulators of rER/cell stress (23).
At 3 weeks of age, qRT-PCR revealed an approximately 2.8-fold increase in BiP expression and approximately 2.2-fold increase in calreticulin expression in growth plate chondrocytes from mice that were homozygous for p.Thr583Met (Fig. 9A). We also examined the downstream mediators of the classical UPR/cell stress response, which included the phosphorylation of eukaryotic initiation factor 2α kinase (eIF2α), the cleavage of ER membrane-anchored transcription factor ATF6, the increased expression of CCAAT/enhancer-binding protein-homologous protein (CHOP), cleavage of the proapoptotic factor caspase-12 and the downregulation of the antiapoptotic factor Bcl-2 (23). Densitometric analysis of western blots detected an approximately 1.7-fold increase in cellular levels of the phosphorylated form of eIF2α (eIF2αP) in mutant chondrocytes and increased cleavage of ATF6 (approximately 2.2-fold). This was accompanied by approximately 2.5-fold increase in caspase-12 cleavage and a significant decrease in Bcl-2 expression (Fig. 9B). Furthermore, there was a modest, but statistically significant, 1.6-fold increase in CHOP expression detected by qRT-PCR (Fig. 9A). Interestingly, an upregulation of chaperone proteins was no longer detected at 6-weeks of age (data not shown), suggesting that the UPR might be a transient response to the expression of maximal levels of mutant COMP during peak levels of bone growth.
Figure 9.
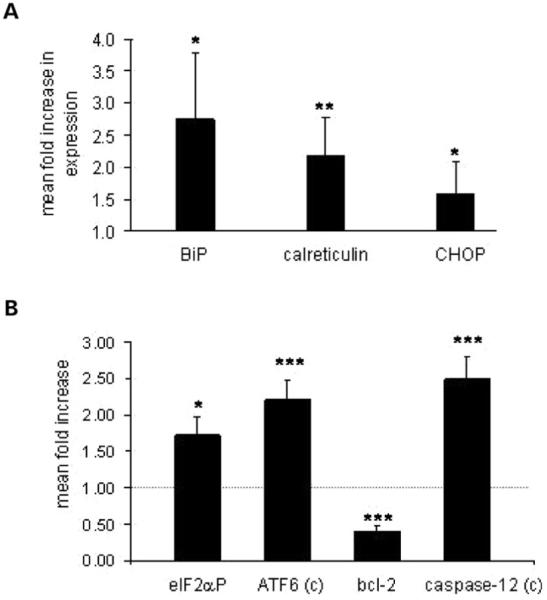
A mild ER stress is detectable in mutant chondrocytes at 3 weeks of age. (A) qRT-PCR analysis showing an approximately 2.8-fold increase in BiP expression, 2.2-fold in calreticulin and 1.6-fold increase in CHOP expression in mutant chondrocytes at 3 weeks of age (n ≥ 5, independent samples t-test). (B) Density measurement of the Western blot analysis from three independent sets of mice showing ATF6 cleavage, phosphorylation of the eIF2α, cleavage of caspase-12 and the downregulation of Bcl-2 at 3 weeks of age (independent samples t-test). All the measurements were normalized to β-actin. Key: +/+ (wild-type); m/m (homozygous for the mutation); c (cleaved); ***P < 0.001; **P < 0.01; *P < 0.05.
Chondrocyte proliferation is markedly decreased and apoptosis is increased and spatially dysregulated in mutant growth plates
In order to determine the physiological effect of the mild rER/cell stress on chondrocyte differentiation and viability. we determined the relative levels of chondrocyte proliferation (by BrdU labelling), while terminal apoptosis (i.e. fragmentation of genomic DNA) was studied with a fluorescent TUNEL assay.
Overall, there was a 24% reduction in the relative number of BrdU-labelled chondrocytes in the proliferative zone of mice homozygous for p.Thr583Met when compared with their wild-type littermates. Mice heterozygous for p.Thr583Met showed a 12% reduction in BrdU labelling (Fig. 10A; n > 20 sections from three mice per genotype; independent samples t-test).
Figure 10.

Proliferation is decreased and apoptosis is both increased and spatially dysregulated in 3 weeks old tibia of mutant mice. (A) Chondrocyte proliferation assayed by 2 h BrdU labelling appeared to be markedly reduced in growth plates of mice heterozygous (by 12%) and homozygous (by 24%) for the mutation (n ≥ 20; independent samples t-test). (B) Apoptotic cells were detected in the proliferative zone of growth plates of mice carrying the p.Thr583Met COMP mutation but not in their wild-type littermates (white circles). The white line marks the boundary between the hypertrophic zone and proliferative zone in the growth plate. DAPI staining was used as a nuclear counterstain. (C) Quantification of the TUNEL assay results showed a significant increase in apoptosis in the hypertrophic (1.8-fold and 3.3-fold, respectively) and proliferative zones (9.8-fold and 12.2-fold, respectively) of growth plates from mice heterozygous and homozygous for the mutation and a 2.5-fold increase in the apoptosis in the resting zone of the growth plates from homozygous mice (n = 18; independent samples t-test). Key: +/+ (wild-type); m/+ (heterozygous for the mutation); m/m (homozygous for the mutation); **P < 0.01; *P < 0.05.
TUNEL-positive staining was detected predominantly at the vascular invasion front and late hypertrophic zone in the growth plates of wild-type mice; approximately 0.8% of all cells in these zones were TUNEL positive, which was within normal limits (Fig. 10B and C; typically one to two TUNEL-positive cells from 150 DAPI-stained cells per section, n = 18 sections from three mice per genotype). In the growth plates from mice heterozygous and homozygous for the mutation, the apoptosis in this region was increased 1.8-fold (to 1.64%) and 3.3-fold (to 2.98%), respectively. Apoptosis in the proliferative zone was barely detectable in the wild-type growth plates; approximately 0.0081% (Fig. 10B and C; on average 0.1 TUNEL-positive cell from 1420 DAPI-stained cells per section, n = 18 sections from three mice per genotype). However, in mice either heterozygous or homozygous for p.Thr583Met, the number of TUNEL-positive cells was increased in the proliferative zone of the growth plate (Fig. 10B and C). In particular, there was a 12-fold increase in apoptosis in the proliferative zone in the homozygous mice (to 0.0997% or 1.47 cells per section) and a 9.8-fold increase in the same zone in heterozygous mice (to 0.0799% or 1.18 TUNEL-positive cells per section) (Fig. 10C; n = 18 sections from three mice per genotype, independent samples t-test). Furthermore, there was a 2.5-fold increase in apoptosis in the resting zone in the homozygous mice [from 0.6 positive cells of 190 cells in the wild-type (0.26%) to 1.125 cells (0.65%) TUNEL-positive cells] (Fig. 10C; n = 18 sections from three mice per genotype, independent samples t-test). Overall, these data confirmed that apoptosis was both increased and spatially dysregulated in the mutant growth plates.
DISCUSSION
We have generated a murine model of PSACH resulting from a p.Thr583Met mutation in the C-terminal domain of COMP for the purpose of elucidating the disease mechanism since ethical and scientific reasons prevent a similar analysis in children. The equivalent human mutation has been shown to cause either MED or mild PSACH (18,24,25). PSACH–MED patients are normal at birth but during childhood they can exhibit increasing disproportionate short stature while the head and face remain normal (9). Morphometric analysis of mice carrying the p.Thr583Met mutation revealed a similar progression of the disease. Mice that were either heterozygous or homozygous for the mutation were normal at birth with correctly proportioned skeletal elements and normal bone mineralization. However, with increasing age mutant mice were lighter than their wild-type littermates and developed mild short-limbed dwarfism as a result of disturbed endochondral ossification. Although the mutant mice showed only a 4% reduction in bone length, this is consistent with the equivalent human phenotype (i.e. that which is caused specifically by the p.Thr585Met mutation) in which the height of affected adult males ranged from 155 to 180 cm (<5th to 75th percentile) (18). Essentially, a 4% reduction in human adult height corresponds to a drop from the 50th to 15th percentile and this reduction is consistent with the final heights of affected males reported for this family (18). With increasing age mutant mice developed a hip dysplasia and also degenerative joint disease, which is reminiscent of the osteoarthritis suffered by PSACH–MED patients. In summary this is the first murine model to accurately reproduce the clinical features of mild PSACH–MED resulting from a C-terminal COMP mutation. The subsequent study of the cartilage growth plate of mutant mice allowed us to gain an insight into the morphological changes and pathomolecular mechanisms that were induced by the mutation.
Mutant mice exhibited a disordered growth plate morphology (i.e. dysplasia), which was characterized by a disruption to the spatial arrangement of proliferating chondrocytes within individual chondrons, and the subsequent misalignment of chondrons within their columns. Furthermore, columns of proliferating cells were sparser and unevenly distributed along the horizontal axis of the growth plates. Interestingly, growth plates from the xiphoid processes of mutant mice also showed a disrupted morphology (not shown). This suggests that although comp expression is induced by weight-bearing mutant (26), comp is still expressed at high enough levels in non-weight bearing tissues to induce growth plate dysplasia.
Ultrastructural analysis of growth plate cartilage revealed rounded and severely misshapen chondrocytes within the proliferative zone of mice homozygous for the mutation. These misshapen chondrocytes did not appear to be oriented correctly and were spatially misaligned within individual chondrons when compared with the tightly organized flattened cells within the growth plate chondrons of wild-type animals. Interestingly, a similar phenotype was observed in mice with a cartilage-specific deletion of integrin β1 (27) and in the α10 integrin-null mice (28) leading to the hypothesis that it is integrin binding and the subsequent signalling events that are responsible for the alignment of newly divided chondrocytes within the proliferative zone of the growth plate (27). It is interesting to speculate that in the growth plates of p.Thr583Met COMP-mutant mice the misalignment of chondrocytes could in part be due to a disruption to integrin binding, which might normally be mediated by the C-terminal domain of COMP (29-31). Alternatively, integrin signalling and/or chondrocyte alignment could be affected by dominant-negative changes in the structure of the cartilage ECM caused by the presence of abnormal COMP. It is possible that the p.Thr583Met substitution could disrupt the COMP T3-CTD protein structure and thus affect its function. The CTD has been shown to bind to type II and type IX collagen in vitro, while a potential integrin binding site (SFYVVMWK) is located only 21 amino acids from the site of the mutation (29,31). We therefore considered the possibility that the presence of mutant COMP in the ECM was exerting a dominant-negative effect on ECM composition and structure and thus contributing to the growth plate dysplasia. COMP has been reported to bind to type II collagen molecules and catalyse collagen fibrillogenesis in vitro (32). It also interacts with various matrix bridging and modulating proteins such as matrilin-3 (33), type IX collagen (19) and fibronectin (30). Interestingly, mutations in the genes encoding matrilin-3 and type IX collagen are also implicated in the aetiology of autosomal dominant MED (9). These proteins can be co-retained with COMP harbouring T3 mutations (34), thus providing some functional rationale for the non-allelic genetic heterogeneity of MED. During embryonic bone development COMP has a pericellular and territorial localization in the ECM of the cartilage growth plate, which is followed by an interterritorial localization later in development (22). IHC analysis of growth plate cartilage from newborn wild-type mice showed a typical pericellular and territorial localization of COMP. This was followed at 2 weeks of age by an interterritorial localization, which remained until adulthood. In contrast, COMP harbouring the p.Thr583Met mutation remained predominantly pericellular and territorial throughout bone growth. Furthermore, the localization of type IX collagen and matrilin-3 appeared to follow the abnormal localization of mutant COMP in the ECM and resulted in a ‘washed-out’ appearance, suggesting that the abnormal localization of mutant COMP was exerting a dominant-negative effect on the localization of its interacting partners.
The ultrastructure of the cartilage ECM was also disrupted in mutant mice when viewed by TEM. Increased fibrillar material was observed in the territorial and interterritorial ECM of the proliferative and hypertrophic zones of mutant mice when compared with their wild-type littermates. This change may be resulting from the mislocalization of COMP and the resulting changes in the ECM making the collagen fibrils more prominent and/or the effect of a C-terminal COMP mutation on collagen fibrillogenesis (32) and other macromolecular assemblies. The presence of irregular and thicker collagen fibrils in the interterritorial ECM of the proliferative zone of mutant growth plates could be explained by the mislocalization of type IX collagen and/or matrilin-3 in the ECM (35,36). It is also feasible that the altered composition and structure of the territorial ECM might affect the organization and alignment of chondrocytes within the chondrons.
We did not detect by IHC the intracellular retention of mutant COMP in chondrocytes from the growth plates of mutant mice. This is in contrast to COMP-T3 mutations in which the retention of mutant COMP has been consistently demonstrated in patient cartilage samples (11,12,14). This observation therefore suggests that the p.Thr585Met COMP–CTD mutation does not cause a significant protein trafficking defect. Indeed, these in vivo observations are consistent with studies using transfected cells in which mutant COMP harbouring CTD mutations is efficiently secreted (15,16). Interestingly, although the retention of p.Thr585Met COMP was not detected by IHC an unfolded protein response was detected in chondrocytes at 3 weeks of age. This was characterized initially by the increased expression of several chaperone proteins such as BiP and calreticulin. This in turn resulted in the subsequent activation of downstream mediators of rER/cell stress, such as eIF2α, ATF6 and caspase-12, which then normalized by 6 weeks of age suggesting a transient response to the maximal expression of mutant COMP. Interestingly, BiP, calreticulin and ERp72 have been found within abnormally dilated ER cisternae associated with COMP TSP-3 mutations and could indicate a common response to the expression of mutant COMP. However, while CTD mutations may only delay the secretion of COMP, the TSP-3 mutants perhaps prevent its secretion to a greater extent suggesting that they have a more detrimental effect on the structure of COMP.
In the growth plates of mutant mice the columns of proliferating chondrocytes appeared sparser with distinct areas of hypocellularity. BrdU-labelling experiments were performed at 3 weeks of age to determine the effect of the p.Thr583Met COMP mutation on chondrocyte proliferation within the growth plate. Cell proliferation was significantly reduced in the mutant growth plates, which may be due to the mild rER/cell stress response induced by the expression and trafficking of mutant COMP and/or the dominant-negative effect of mutant COMP present in the cartilage ECM.
A decrease in Bcl-2 expression was noted at 3 weeks of age in mutant chondrocytes, which was accompanied by increased and spatially dysregulated apoptosis. Bcl-2 is a mitochondrial membrane bound anti-apoptotic protein which is also found on ER membranes and has been implicated in general and ER stress-mediated apoptosis (37). Blc-2 has been shown to be localized mainly in the proliferative zone of the murine growth plates at 3 weeks of age, with a decrease in expression towards the hypertrophic region of the growth plate (38). Bcl-2 knock-out mice exhibit a mild short limb dwarfism and increased osteoblast numbers at a later age (39). It is therefore tempting to speculate that the downregulation of Bcl-2 is responsible in part for the increased and spatially dysregulated apoptosis observed in the growth plates of mutant mice. The downregulation of Bcl-2 may be the result of CHOP-mediated cell stress resulting from either the processing of misfolded COMP protein or compositional and structural changes in the cartilage ECM induced by the presence of mutant protein. A similar effect on the proliferation and apoptosis of chondrocytes was noted in mice carrying a cartilage-specific deletion of β1 integrin (27) and also α10 integrin null mice (28), suggesting that correct signals from the ECM microenvironment may regulate chondrocyte proliferation and survival. Furthermore, in the DTDST knock-in model, mice homozygous for the mutation also exhibited a decrease in the number of proliferating cells when compared with the wild-type littermates (40). These data suggest that reduced chondrocyte proliferation, which could be caused by a disruption to integrin signalling and/or a cell stress response to mutant protein trafficking and its dominant-negative effect in the ECM, may be a common feature of chondrodysplasias.
In summary, the mouse model presented in this paper is the first in vivo model of chondrodysplasia resulting from a mutation in COMP. The murine chondrodysplasia phenotype resembles that of mild PSACH–MED; the equivalent human disease. More importantly, our data suggests that the disease arises through a combination of both intracellular and extracellular mechanisms; however, the precise molecular pathways that lead to reduced chondrocyte proliferation and increased dysregulated apoptosis remain to be fully determined.
MATERIALS AND METHODS
Generation of p.Thr583Met COMP knock-in mice
Gene targeting was performed as described by Talts et al. (41). Briefly, a female 129S6/SvEvTac mouse spleen genomic DNA library (RPCI) was screened for a clone containing the whole of the comp gene. A loxP-flanked Neo cassette was cloned into pBluescript (pBS Neo) using an Eco RV site in the polylinker sequence. The long arm of the construct was generated by EcoRI digestion and subcloning of an 8 kb genomic fragment containing comp. The short arm of the construct (2 kb) was generated by subcloning of an EcoRI/SpeI comp positive fragment (6 kb). The p.Thr583Met change (ACA → ATG) was introduced by site-directed mutagenesis of a 1.6 kb fragment generated by restriction digestion using a SpeI site in the pBluescript polylinker and a MluI site in the construct. Primers used for the introduction of the mutation were: forward (mutation): 3′-TTC GAG GGC ATG TTC CAT GTA C-5′, reverse (mutation): 5′-TAC ATG GAA CAT GCC CTC GAA-3′, forward: 3′-GGG TGA CGC GTG TCA GGG TGA-5′, reverse: 5′-AAC TAG TGG ATC CCC CGG GC-3′. The mutation was confirmed by direct sequencing. In the short arm of the construct, an EcoRI restriction site was modified into a ClaI site by ligation of a linker oligonucleotide (5′-GAATTATCGATAATTC-3′). The pBluescript vector was modified by NotI digestion and Klenow blunting, thus generating a unique FseI site for construct linearization. R1 (129Sv) ES cells were grown on a layer of G418 resistant embryonic feeder cells in a medium supplemented with leukaemia inhibitory factor. The targeting vector (70 μg) was linearized with FseI and electroporated into 4 × 107 R1 (129Sv) ES cells using a BioRad gene pulser (0.8 kV, 3 μF, 0.1 ms). Electroporated cells were propagated in selective medium with G418 (500 μg/ml) for 5-6 days and resistant clones were picked and screened for homologous recombination by HindIII digestion and Southern blot analysis with an external 1 kb probe. A probe directed against the Neo cassette was used to confirm single incorporation of the construct sequence in the genome. The presence of the mutation was confirmed by direct sequencing and the positive clones were grown from frozen stocks and microinjected into C57BL/6 blastocysts and implanted into foster mothers. The resulting chimeric animals were assessed on fur colour and mated with a deleter Cre line of transgenic mice to delete the loxP-flanked selection cassette. Heterozygous F1 offspring were then mated to generate the animals used in all of the described experiments. Cre recombination resulted in one loxP site remaining in the intronic sequence, allowing for genotyping procedure from tail tip genomic DNA using primers spanning intron 16 (Extract-N-Amp™ Tissue PCR Kit, Sigma–Aldrich Ltd).
Real-time PCR
RNA was extracted from the xiphoid process at 3 weeks of age using TriZOL solution (Ambion Inc.) following snap-freezing and tissue homogenization. The RNA was DNaseI (Ambion Inc.) treated and Superscript III™ reverse transcriptase with random hexamers (InVitrogen Ltd) was used to generate the cDNA. Real-time analysis of wild-type and mutant COMP expression was performed using SYBR® Green Kit on ABIPrism™ 7000 sequence detector system (Applied Biosystems Ltd). Each sample, including ‘no template’ controls, was run in duplicate and every loaded sample had an 18S control. Each experiment was repeated at least three times for statistical relevance and the results were analysed by independent samples t-test.
Western blotting
Xiphoid tissue at 3 weeks of age was homogenized, boiled in SDS loading buffer containing DTT and loaded on an SDS–PAGE gel. The gel was electroblotted onto a nitrocellulose membrane, which was blocked overnight with 2% skimmed milk powder in PBS-T. Primary antibodies [eIF2α, eIF2αP, caspase-12 (Cell Signalling Ltd), ATF6 (Imgenex Corp.), Bcl-2, actin (Abcam Plc.)] were diluted 1:500 in blocking solution and secondary antibodies (goat anti-rabbit 1:1000, goat anti-mouse 1:10 000, ThermoFisher Scientific Ltd) were diluted in PBS-T. An ECL detection kit (Perkin-Elmer Inc.) was used to develop the blots according to manufacturer’s protocol.
Analysis of skeleton
New born pups were sacrificed, skinned, eviscerated and fixed in 95% ethanol for 3-5 days. The preparations were then stained with Alcian Blue (cartilage, 24 h), rinsed and fixed in 95% ethanol for 2 days. The remaining muscle tissue was cleared in 1% (w:v) KOH (for 6 h) and the skeletons were stained with Alizarin Red (bone, 3 h). The specimens were then further cleared in 2% (w:v) KOH for 48 h followed by decreasing concentrations of 2% KOH in glycerol (80:20, 60:40, 40:60 and 20:80; 24 h in each) and stored in 20:80 2% KOH:glycerol (42).
Bone measurements were generated by measuring radiographic images at 3, 6 and 9 weeks of age. Angles between the tuberosity of the ischium and the pelvic region were also measured at these ages. Mice were weighed at 3, 6 and 9 weeks of age and the weights were used to construct growth curves. All the measurements were analysed by one-way ANOVA for statistical significance.
Histology and immunohistochemistry
Dissected tissue samples were fixed in ice-cold 10% neutral buffered formalin solution (Sigma–Aldrich Ltd; for histology and TUNEL assay) or in ice-cold 95% ethanol 5% acetic acid solution (for IHC), decalcified in 20% EDTA pH 7.4, paraffin embedded and sectioned (6 μm sections). For H&E staining, the slides were dewaxed in xylene, rehydrated and H&E stained using a ThermoShandon Ltd automated stainer, dehydrated in increasing concentrations of ethanol and in xylene and mounted using a xylene-based mounting solution. Safranin O staining was performed by rehydrating the samples and staining in 0.02% (w/v) Fast Green (Sigma–Aldrich Ltd) followed by staining in 0.1% (w/v) Safranin O (Sigma–Aldrich Ltd). For IHC analysis, slides were dewaxed and rehydrated, endogenous peroxidase activity was quenched in H2O2/MetOH followed by antigen unmasking in 0.2% bovine testes hyaluronidase (Sigma–Aldrich Ltd) in PBS. Samples were blocked in goat serum and BSA in PBS for 1 h and immediately incubated with primary antibody in PBS/BSA for 1 h. The antibodies used were COMP, matrilin-3, type IX collagen, type II collagen (Calbiolchem Ltd) and Grp78 (BiP, Santa Cruz Biotechnology Inc.), calreticulin (Stressgen Bioreagents) and Erp72 (Santa Cruz Biotechnology Ltd). Slides were washed in PBS/BSA and incubated with biotinylated goat anti-rabbit IgG (Dako Cytomation Ltd) in PBS with goat serum for 1 h, followed by incubation with ABC/HRP reagent (Dako Cytomation Ltd) for 30 min and developed using DAB chromogen (Dako Cytomation Ltd), with methyl green as counter stain (Vector Labs Ltd). Vectamount™ (Vector Labs Ltd) xylene-free mounting medium was used. Apoptosis was analysed using Dead-End™ Fluorimetric TUNEL assay (Promega). In order to lower the background and the number of false positives the unmasking was performed using citrate buffer heat treatment instead of standard proteinase K unmasking (43,44). The slides were incubated in citrate buffer (pH 6.0) and treated for 15 min in a microwave oven (700, 300 and 150 W for 5 min each). Positive controls were generated by DNaseI (Ambion Inc.) treatment of the samples. The slides were mounted in Vectashield™ with DAPI (Vector Labs Ltd). The number of TUNEL-positive cells in the hypertrophic zone was expressed as a proportion of the total population of cells in the hypertrophic zone. Independent samples t-test was used for statistical analysis of TUNEL assay results.
BrdU labelling
Mice were injected peritoneally with 0.1 ml of Cell Proliferation Labelling Reagent (Amersham) per 10 g of weight at 3 weeks of age and the proliferating cells were labelled for 2 h. Dissected tissues were fixed and analysed by IHC using anti-BrdU antibody (1:100; Abcam Plc.). Antigen unmasking was performed in 4 m HCl for 15 min, neutralized with 0.1 m borate buffer. The number of BrdU-labelled cells was expressed as a proportion of the total population of cells in the proliferative zone. Independent samples t-test was applied for statistical analysis of results.
Ultrastructural analysis
Tibia were dissected at 1 week and immediately fixed in 2.5% glutaraldehyde in 1 M sodium cacodylate buffer for 2 h at 4°C. The tissues were washed three times in 0.1 M sodium cacodylate buffer and fixed in 2% OsO4 in 0.1 M cacodylate buffer for 2 h. They were washed in distilled water and incubated for 2 h in 2% aqueous uranyl acetate at 4°C. The tibia were washed in distilled water, dehydrated in increasing concentrations of acetone: 50, 70, 90 and 100% for 30 min, incubated in propylene oxide to improve resin penetration and 1:1 solution of resin:propylene oxide, and embedded in TAAB medium slow resin (TAAB Laboratories Equipment Ltd). Thin 70-80 nm sections were cut with a diamond knife on a Leica ultra-microtome and placed on electron microscope grids. Sections on the grids were stained with silver citrate solution and viewed in a FEI Tecnai 12 Twin transmission electron microscope operated at an accelerating voltage of 80 kV.
ACKNOWLEDGEMENTS
This work was supported by grants from the Wellcome Trust (Michael Briggs is the recipient of a Wellcome Trust Senior Research Fellowship in Basic Biomedical Science; Grant 071161/Z/03/Z), the Arthritis Research Campaign (Grant 16025 to M.D.B., and R.B.H.) and National Institute of Health (RO1 AR49547-01 to M.D.B., R.B.H., D.J.T. and K.E.K.). The research was undertaken in the Wellcome Trust Centre for Cell-Matrix Research and the Histology and Transgenic Core Facilities of the Faculty of Life Sciences at the University of Manchester. We would like to thank Raimund Wagener (University of Cologne, Germany) for the matrilin-3 antibody.
Footnotes
Conflict of Interest statement. There are no conflicts of interest to declare.
REFERENCES
- 1.Rimoin DL, Rasmussen IM, Briggs MD, Roughley PJ, Gruber HE, Warman ML, Olsen BR, Hsia YE, Yuen J, Reinker K, et al. A large family with features of pseudoachondroplasia and multiple epiphyseal dysplasia: exclusion of seven candidate gene loci that encode proteins of the cartilage extracellular matrix. Hum. Genet. 1994;93:236–242. doi: 10.1007/BF00212015. [DOI] [PubMed] [Google Scholar]
- 2.McKeand J, Rotta J, Hecht JT. Natural history study of pseudoachondroplasia. Am. J. Med. Genet. 1996;63:406–410. doi: 10.1002/(SICI)1096-8628(19960517)63:2<406::AID-AJMG16>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]
- 3.Briggs MD, Hoffman SM, King LM, Olsen AS, Mohrenweiser H, Leroy JG, Mortier GR, Rimoin DL, Lachman RS, Gaines ES, et al. Pseudoachondroplasia and multiple epiphyseal dysplasia due to mutations in cartilage oligomeric matrix protein gene. Nat. Genet. 1995;10:330–336. doi: 10.1038/ng0795-330. [DOI] [PubMed] [Google Scholar]
- 4.Hecht JT, Nelson LD, Crowder E, Wang Y, Elder FFB, Harrison WR, Francomano CA, Prange CK, Lennon GG, Deere M, Lawler J. Mutations in exon 17B of cartilage oligomeric matrix protein (COMP) cause pseudoachondroplasia. Nat. Genet. 1995;10:325–329. doi: 10.1038/ng0795-325. [DOI] [PubMed] [Google Scholar]
- 5.Maddox BK, Mokashi A, Keene DR, Bachinger HP. A cartilage oligomeric matrix protein mutation associated with pseudoachondroplasia changes the structural and functional properties of the type 3 domain. J. Biol. Chem. 2000;275:11412–11417. doi: 10.1074/jbc.275.15.11412. [DOI] [PubMed] [Google Scholar]
- 6.DiCesare PE, Carlson CS, Stollerman ES, Chen FS, Leslie M, Perris R. Expression of cartilage oligomeric matrix protein by human synovium. FEBS Lett. 1997;412:249–252. doi: 10.1016/s0014-5793(97)00789-8. [DOI] [PubMed] [Google Scholar]
- 7.Oldberg A, Antonsson P, Lindblom K, Heinegard D. COMP (cartilage oligomeric matrix protein) is structurally related to the thrombospondins. J. Biol. Chem. 1992;267:22346–22350. [PubMed] [Google Scholar]
- 8.Bornstein P, Sage EH. Thrombospondins. Methods Enzymol. 1994;245:62–85. doi: 10.1016/0076-6879(94)45006-4. [DOI] [PubMed] [Google Scholar]
- 9.Briggs MD, Chapman KL. Pseudoachondroplasia and multiple epiphyseal dysplasia: mutation review, molecular interactions and genotype to phenotype correlations. Hum. Mutat. 2002;19:465–478. doi: 10.1002/humu.10066. [DOI] [PubMed] [Google Scholar]
- 10.Unger S, Hecht JT. Pseudoachondroplasia and multiple epiphyseal dysplasia: new etiologic developments. Am. J. Hum. Genet. 2001;106:244–250. [PubMed] [Google Scholar]
- 11.Hecht JT, Hayes E, Snuggs M, Decker G, Montufar-Solis D, Doege K, Mwalle F, Poole R, Stevens J, Duke PJ. Calreticulin, PDI, Grp94 and BiP chaperone proteins are associated with retained COMP in pseudoachondroplasia chondrocytes. Matrix Biol. 2001;20:251–262. doi: 10.1016/s0945-053x(01)00136-6. [DOI] [PubMed] [Google Scholar]
- 12.Vranka J, Mokashi A, Keene DR, Tufa S, Corson G, Sussman M, Horton WA, Maddox K, Sakai L, Bachinger HP. Selective intracellular retention of extracellular matrix proteins and chaperones associated with pseudoachondroplasia. Matrix Biol. 2001;20:439–450. doi: 10.1016/s0945-053x(01)00148-2. [DOI] [PubMed] [Google Scholar]
- 13.Delot E, King LM, Briggs MD, Wilcox WR, Cohn DH. Trinucleotide expansion mutations in the cartilage oligomeric matrix protein (COMP) gene. Hum. Mol. Genet. 1999;8:123–128. doi: 10.1093/hmg/8.1.123. [DOI] [PubMed] [Google Scholar]
- 14.Hecht JT, Montufar-Solis D, Decker G, Lawler J, Daniels K, Duke PJ. Retention of cartilage oligomeric matrix protein (COMP) and cell death in redifferentiated pseudoachondroplasia chondrocytes. Matrix Biol. 1998;17:625–633. doi: 10.1016/s0945-053x(98)90113-5. [DOI] [PubMed] [Google Scholar]
- 15.Spitznagel L, Nitsche DP, Paulsson M, Maurer P, Zaucke F. Characterization of a pseudoachondroplasia-associated mutation (His 587 → Arg) in the C-terminal, collagen- binding domain of cartilage oligomeric matrix protein (COMP) Biochem. J. 2004;377:479–487. doi: 10.1042/BJ20031179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schmitz M, Becker A, Schmitz A, Weirich C, Paulsson M, Zaucke F, Dinser R. Disruption of extracellular matrix structure may cause pseudoachondroplasia phenotypes in the absence of impaired cartilage oligomeric matrix protein secretion. J. Biol. Chem. 2006;281:32587–32595. doi: 10.1074/jbc.M601976200. [DOI] [PubMed] [Google Scholar]
- 17.Kennedy J, Jackson GC, Barker FS, Nundlall S, Bella J, Wright MJ, Mortier GR, Neas K, Thompson E, Elles R, Briggs MD. Novel and recurrent mutations in the C-terminal domain of COMP cluster in two distinct regions and result in a spectrum of phenotypes within the pseudoachondroplasia—multiple epiphyseal dysplasia disease group. Hum. Mutat. 2005;25:593–594. doi: 10.1002/humu.9342. [DOI] [PubMed] [Google Scholar]
- 18.Briggs MD, Mortier GR, Cole WG, King LM, Golik SS, Bonaventure J, Nuytinck L, De Paepe A, Leroy JG, Biesecker L, et al. Diverse mutations in the gene for cartilage oligomeric matrix protein in the pseudoachondroplasia-multiple epiphyseal dysplasia disease spectrum. Am. J. Hum. Genet. 1998;62:311–319. doi: 10.1086/301713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Holden P, Meadows RS, Chapman KL, Grant ME, Kadler KE, Briggs MD. Cartilage oligomeric matrix protein interacts with type IX collagen, and disruptions to these interactions identify a pathogenetic mechanism in a bone dysplasia family. J. Biol. Chem. 2001;276:6046–6055. doi: 10.1074/jbc.M009507200. [DOI] [PubMed] [Google Scholar]
- 20.Carlson CB, Bernstein DA, Annis DS, Misenheimer TM, Hannah BL, Mosher DF, Keck JL. Structure of the calcium-rich signature domain of human thrombospondin-2. Nat. Struct. Mol. Biol. 2005;12:910–914. doi: 10.1038/nsmb997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kvansakul M, Adams JC, Hohenester E. Structure of a thrombospondin C-terminal fragment reveals a novel calcium core in the type 3 repeats. EMBO J. 2004;23:1223–1233. doi: 10.1038/sj.emboj.7600166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shen Z, Heinegard D, Sommarin Y. Distribution and expression of cartilage oligomeric matrix protein and bone sialoprotein show marked changes during rat femoral head development. Matrix Biol. 1995;14:773–781. doi: 10.1016/s0945-053x(05)80020-4. [DOI] [PubMed] [Google Scholar]
- 23.Rutkowski DT, Kaufman RJ. A trip to the ER: coping with stress. Trends Cell Biol. 2004;14:20–28. doi: 10.1016/j.tcb.2003.11.001. [DOI] [PubMed] [Google Scholar]
- 24.Czarny-Ratajczak M, Lohiniva J, Rogala P, Kozlowski K, Perala M, Carter L, Spector TD, Kolodziej L, Seppanen U, Glazar R, et al. A mutation in COL9A1 causes multiple epiphyseal dysplasia: further evidence for locus heterogeneity. Am. J. Hum. Genet. 2001;69:969–980. doi: 10.1086/324023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Song HR, Lee KS, Li QW, Koo SK, Jung SC. Identification of cartilage oligomeric matrix protein (COMP) gene mutations in patients with pseudoachondroplasia and multiple epiphyseal dysplasia. J. Hum. Genet. 2003;48:222–225. doi: 10.1007/s10038-003-0013-7. [DOI] [PubMed] [Google Scholar]
- 26.Kersting UG, Stubendorff JJ, Schmidt MC, Bruggemannet GP. Changes in knee cartilage volume and serum COMP concentration after running exercise. Osteoarth. Cartilage. 2005;13:925–934. doi: 10.1016/j.joca.2005.06.005. [DOI] [PubMed] [Google Scholar]
- 27.Aszódi A, Hunziker E, Brakebusch C, Fassler R. Beta1 integrins regulate chondrocyte rotation, G1 progression, and cytokinesis. Genes Dev. 2003;17:2465–2479. doi: 10.1101/gad.277003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bengtsson T, Aszódi A, Nicolae C, Hunziker E, Lundgren-Akerlund E, Fassler R. Loss of alpha10beta1 integrin expression leads to moderate dysfunction of growth plate chondrocytes. J. Cell Sci. 2005;118:929–936. doi: 10.1242/jcs.01678. [DOI] [PubMed] [Google Scholar]
- 29.DiCesare PE, Morgelin M, Mann K, Paulsson M. Cartilage oligomeric matrix protein and thrombospondin 1. Purification from articular cartilage, electron microscopic structure, and chondrocyte binding. Eur. J. Biochem. 1994;223:927–937. doi: 10.1111/j.1432-1033.1994.tb19070.x. [DOI] [PubMed] [Google Scholar]
- 30.DiCesare PE, Chen FS, Moergelin M, Carlson CS, Leslie MP, Perris R, Fang C. Matrix-matrix interaction of cartilage oligomeric matrix protein and fibronectin. Matrix Biol. 2002;21:461–470. doi: 10.1016/s0945-053x(02)00015-x. [DOI] [PubMed] [Google Scholar]
- 31.Chen FH, Thomas AO, Hecht JT, Goldring MB, Lawler J. Cartilage oligomeric matrix protein/thrombonspondin-5 supports chondrocyte attachment through interaction with integrins. J. Biol. Chem. 2005;280:32655–32661. doi: 10.1074/jbc.M504778200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sodersten F, Ekman S, Eloranta ML, Heinegard D, Dudhia J, Hultenby K. Ultrastructural immunolocalization of cartilage oligomeric matrix protein (COMP) in relation to collagen fibrils in the equine tendon. Matrix Biol. 2005;24:376–385. doi: 10.1016/j.matbio.2005.06.003. [DOI] [PubMed] [Google Scholar]
- 33.Mann HH, Ozbek S, Engel J, Paulsson M, Wagener R. Interactions between the cartilage oligomeric matrix protein and matrilins. Implications for matrix assembly and the pathogenesis of chondrodysplasias. J. Biol. Chem. 2004;279:25294–25298. doi: 10.1074/jbc.M403778200. [DOI] [PubMed] [Google Scholar]
- 34.Hecht JT, Makitie O, Hayes E, Haynes R, Susic M, Montufar-Solis D, Duke PJ, Cole WG. Chondrocyte cell death and intracellular distribution of COMP and type IX collagen in the pseudoachondroplasia growth plate. J. Orthop. Res. 2004;22:759–767. doi: 10.1016/j.orthres.2003.11.010. [DOI] [PubMed] [Google Scholar]
- 35.Hagg R, Bruckner P, Hedbom E. Cartilage fibrils of mammals are biochemically heterogeneous: differential distribution of decorin and collagen IX. J. Cell Biol. 1998;142:285–294. doi: 10.1083/jcb.142.1.285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Budde B, Blumbach K, Ylostalo J, Zaucke F, Ehlen HW, Wagener R, Ala-Kokko L, Paulsson M, Bruckner P, Grassel S. Altered integration of matrilin-3 into cartilage extracellular matrix in the absence of collagen IX. Mol. Cell. Biol. 2005;25:10465–10478. doi: 10.1128/MCB.25.23.10465-10478.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Breckenridge DG, Germain M, Mathai JP, Nguyen M, Shore GC. Regulation of apoptosis by endoplasmic reticulum pathways. Oncogene. 2003;22:8608–8618. doi: 10.1038/sj.onc.1207108. [DOI] [PubMed] [Google Scholar]
- 38.Amling M, Neff L, Tanaka S, Inoue D, Kuida K, Weir E, Philbrick WM, Broadus AE, Baron R. Bcl-2 lies downstream of parathyroid hormone-related peptide in a signaling pathway that regulates chondrocyte maturation during skeletal development. J. Cell Biol. 1997;136:205–213. doi: 10.1083/jcb.136.1.205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Boot-Handford RP, Michaelidis TM, Hillarby MC, Zambelli A, Denton J, Hoyland JA, Freemont AJ, Grant ME, Wallis GA. The bcl-2 knockout mouse exhibits marked changes in osteoblast phenotype and collagen deposition in bone as well as a mild growth plate phenotype. Int. J. Exp. Pathol. 1998;79:329–335. doi: 10.1046/j.1365-2613.1998.790411.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Forlino A, Piazza R, Tiveron C, Della Torre S, Tatangelo L, Bonafe L, Gualeni B, Romano A, Pecora F, Superti-Furga A, Cetta G, Rossi A. A diastrophic dysplasia sulfate transporter (SLC26A2) mutant mouse: morphological and biochemical characterization of the resulting chondrodysplasia phenotype. Hum. Mol. Genet. 2005;14:859–871. doi: 10.1093/hmg/ddi079. [DOI] [PubMed] [Google Scholar]
- 41.Kimmel CA, Trammell C. A rapid procedure for routine double staining of cartilage and bone in fetal and adult animals. Stain Technol. 1981;56:271–273. doi: 10.3109/10520298109067325. [DOI] [PubMed] [Google Scholar]
- 42.Talts JF, Brakenbush C, Fässler R. Integrin gene targeting. Methods Mol. Biol. 129:153–187. doi: 10.1385/1-59259-249-X:153. [DOI] [PubMed] [Google Scholar]
- 43.Shi S-R, Cote RJ, Taylor CR. Antigen retrieval immunohistochemistry: past, present and future. J. Histochem. Cytochem. 1997;45:327–343. doi: 10.1177/002215549704500301. [DOI] [PubMed] [Google Scholar]
- 44.Gal I, Varga T, Szilagyi I, Balazs M, Schlammadinger J, Szabo G., Jr Protease-elicited TUNEL positivity of non-apoptotic fixed cells. J. Histochem. Cytochem. 2000;48:963–970. doi: 10.1177/002215540004800709. [DOI] [PubMed] [Google Scholar]