

Abstract
Regulator of G protein signaling domain-containing Rho guanine-nucleotide exchange factors (RGS-RhoGEFs) directly link activated forms of the G12 family of heterotrimeric G protein α subunits to the small GTPase Rho. Stimulation of G12/13-coupled GPCRs or expression of constitutively activated forms of α12 and α13 has been shown to induce the translocation of the RGS-RhoGEF, p115-RhoGEF, from the cytoplasm to the plasma membrane (PM). However, little is known regarding the functional importance and mechanisms of this regulated PM recruitment, and thus PM recruitment of p115-RhoGEF is the focus of this report. A constitutively PM-localized mutant of p115-RhoGEF shows a much greater activity compared to wild type p115-RhoGEF in promoting Rho-dependent neurite retraction of NGF-differentiated PC12 cells, providing the first evidence that PM localization can activate p115-RhoGEF signaling. Next, we uncovered the unexpected finding that Rho is required for α13-induced PM translocation of p115-RhoGEF. However, inhibition of Rho did not prevent α12-induced PM translocation of p115-RhoGEF. Additional differences between α13 and α12 in promoting PM recruitment of p115-RhoGEF were revealed by analyzing RGS domain mutants of p115-RhoGEF. Activated α12 effectively recruits the isolated RGS domain of p115-RhoGEF to the PM, whereas α13 only weakly does. On the other hand, α13 strongly recruits to the PM a p115-RhoGEF mutant containing amino acid substitutions in an acidic region at the N-terminus of the RGS domain; however, α12 is unable to recruit this p115-RhoGEF mutant to the PM. These studies provide new insight into the function and mechanisms of α12/13-mediated PM recruitment of p115-RhoGEF.
Keywords: heterotrimeric G protein, Rho GTPase, guanine-nucleotide exchange factor, membrane translocation, regulator of G protein signaling (RGS)
1. INTRODUCTION
Heterotrimeric G proteins, composed of α, β and γ subunits, function at the cytoplasmic surface of the plasma membrane to couple agonist-activated G protein-coupled receptors (GPCRs) to a wide variety of effectors and downstream signaling responses [1]. G proteins are classified into four families, Gs, Gi, Gq, and G12, based on α subunit sequence identity and signaling function. α12 and α13, the α subunits of the G12 family, share 65% amino acid identity, and they share the ability to promote cellular proliferation, oncogenesis, cell migration, and cellular morphological changes. GPCRs that couple to α12 and α13 include LPA, thrombin, and thromboxane A2 receptors.
Many of the cellular responses promoted by α12 and α13 involve activation of the small GTPase Rho. α12 and α13 activate Rho by directly interacting with and activating a sub-family of Rho guanine-nucleotide exchange factors (RhoGEFs), which in turn catalyze GDP release and subsequent GTP binding to Rho. Although α12 and α13 have been shown to interact with a number of effectors [2], the RhoGEFs are the best characterized in terms of being demonstrated to clearly mediate important functions of α12 and α13.
More than 80 RhoGEFs have been identified [3], and most have the common feature of containing a tandem Dbl homology (DH) domain, which is responsible for guanine-nucleotide exchange activity, and pleckstrin homology (PH) domain, which appears to assist in catalysis and subcellular localization. Among these many RhoGEFs, only three RhoGEFs, termed p115-RhoGEF, PDZ-RhoGEF and LARG, comprise the sub-family whose members are effectors for α12 and α13. The distinguishing feature of these three RhoGEFs is the presence of a regulator of G protein signaling (RGS) domain that mediates direct binding to activated α12 and α13. These RhoGEFs are thus often termed RGS-RhoGEFs.
The mechanisms of how activated α12 or α13 stimulate the Rho guanine-nucleotide exchange activity of these RGS-RhoGEFs remain to be fully elucidated [4]. What is clear is that activated α12/13 binding to the RGS domain is essential. However, the simplest model whereby α12/13 binding to the RGS domain is sufficient for RGS-RhoGEF activation by relieving autoinhibition does not adequately account for experimental results [4]. An alternative model suggests that α12/13 also interacts with a second surface on the DH-PH domain [4-6], and this interaction is necessary for stimulation of guanine-nucleotide exchange activity. Moreover, there is evidence that in some cases covalent modification of the RGS-RhoGEF by phosphorylation is required for activation by α12 or α13 [7, 8]. To complicate matters further, in vitro reconstitution assays have revealed differences in the ability of α12 versus α13 to activate RGS-RhoGEFs. For example, α13, but not α12, can stimulate p115-RhoGEF and LARG; phosphorylation of LARG allows stimulation by α12, but how or if α12 stimulates p115-RhoGEF has not been resolved.
An additional mechanism that has been proposed to contribute to regulation of RGS-RhoGEFs by α12/13 is plasma membrane (PM) translocation. Although PDZ-RhoGEF is localized to the cortical actin cytoskeleton, both p115-RhoGEF and LARG display predominant localization in the cytoplasm. Moreover, p115-RhoGEF and LARG have been observed to re-localize from the cytoplasm to the PM upon expression of constitutively active α12 or α13 and agonist stimulation of endogenous or overexpressed G12/13-coupled GPCRs [4, 9-15]. The mechanisms of the α12/13-mediated PM translocation of RGS-RhoGEFs are not fully understood. Recruitment to the PM requires the RGS domain, but not an N-terminal acidic region of the RGS domain that is required for high affinity binding to α12/13 [4, 13]. In addition, the PH domain of p115-RhoGEF is required for its PM recruitment [12], further suggesting that multiple factors contribute to productive PM recruitment of RGS-RhoGEFs. Furthermore, although it is clear that RGS-RhoGEFs can be recruited from the cytoplasm to the PM in response to α12/13 activation, it has not been demonstrated that such PM recruitment is indeed important for RGS-RhoGEF function.
In this report, we provide new insight into the function and mechanism of PM recruitment of p115-RhoGEF. First, assaying Rho-dependent neurite retraction in PC12 cells we show that expression of a PM-targeted p115-RhoGEF induces neurite retraction, whereas cytoplasmic wild type p115-RhoGEF is mostly unable to effect neurite retraction by itself. These results are the first clear evidence that PM recruitment is sufficient to activate signaling. Next, we uncovered the unexpected finding that Rho, which is presumably downstream of p115-RhoGEF in the signaling cascade, is required for α13-induced PM translocation of p115-RhoGEF. Lastly, we demonstrated key differences between α13 and α12 in PM recruitment of p115-RhoGEF. Whereas α13 displays a novel requirement for Rho in PM recruitment of p115-RhoGEF, α12 does not. Activated α12 effectively recruits the isolated RGS domain of p115-RhoGEF to the PM, whereas α13 only weakly does. On the other hand, α13 strongly recruits to the PM a p115-RhoGEF mutant containing amino acid substitutions in an acidic region at the N-terminus of the RGS domain, but α12 is unable to recruit this p115-RhoGEF mutant to the PM.
2. MATERIALS AND METHODS
2.1 Plasmids and expression vectors
The N-terminal myc epitope (MEQKLISEED)-tagged pcDNA3-myc-p115-RhoGEF, the hemagglutinin (HA) epitope (DVPDYA)-tagged pcDNA3 Gα13QL (α13QL), untagged Gα12QL (α12QL) in pcDNAI, the C-terminal green fluorescence protein (GFP)-tagged p115-RhoGEF, and the expression vector for C3-transferase (C3), were described previously [12, 14]. The expression plasmid carrying HA epitope-tagged activated Rho cDNA (RhoV14) was obtained from P. Tsichlis (Kimmel Cancer Institute, Philadelphia). The Stratagene QuickChange Site-directed Mutagenesis Kit was used to replace Glu27, Glu29 and Asp30 of p115-RhoGEF with Ala to generate p115-RhoGEF(EED).
2.2 Cell culture and transfection
PC12 cells were plated on collagen IV (Sigma, MO) coated plates in Dulbecco’s modified Eagle’s medium (DMEM) containing 10% fetal bovine serum (FBS) and 5% horse serum supplemented with penicillin and streptomycin. PC12 cells were transfected using Lipofectamine 2000 (Invitrogen) as per the manufacturer’s protocol. 24 h after transfection neurite extension was induced by exposing the cells to serum free medium containing 100 ng/ml nerve growth factor (NGF) for an additional 16 h. Transfection of HEK293 cells were performed as described before [12].
2.3 siRNA transfection
The Rho sense and anti-sense siRNA oligonucleotides 5’ (GAAGUCAAGCAUUUCUGUC)dTdT 3’ and 5’ (GACAGAAAUGCUUGACUUC)dTdT 3’, respectively, were purchased from Dharmacon Research, Inc., CO. A pair of scrambled siRNA was obtained from Jeff Benovic (Thomas Jefferson University, Philadelphia) as a control. Transfection of siRNA was achieved by using Oligofectamine (Invitrogen) as per manufacturer’s protocol. Complete silencing of Rho was achieved by transfecting the cells twice after 24 h intervals with the siRNA.
2.4 Immunofluorescence Assay
Cells were grown on collagen IV coated chamber slides (for PC12 cells) or uncoated cover slips (for HEK293 cells), and were transfected with appropriate plasmids. The cells were subjected to immunofluorescence microscopy as done earlier [12], after probing with anti-myc 9E10 (Covance or Novagen), anti-HA 12CA5 (Covance or Roche) and/or anti-α12 polyclonal antibody (Santa Cruz Biotech., CA). Goat anti-mouse antibodies conjugated with FITC (Vector Laboratories, CA), Alexa 488 or Alexa 594 (Invitrogen), and goat anti-rabbit antibodies conjugated with Texas Red (Vector Laboratories, CA) or Alexa 594 (Invitrogen) were used in our immunofluorescent labeling. For actin staining, the chamber slides were prepared for fluorescence microscopy as mentioned above, with the exception that a 1:50 dilution of Alexa594 conjugated phalliodin (Molecular Probes) was included during the secondary antibody incubation.
2.5 Subcellular fractionation
HEK 293 cells were plated at ~2 × 106 cells in 6 cm plates, grown for 24 h and transfected with 3 μg of total plasmids. Cells were fractionated into membrane bound P-fractions and cytoplasmic S-fractions as done before [12]. We used anti-myc and anti-HA antibodies to detect myc- and HA epitope-tagged proteins, respectively, on immunoblots. Expression of endogenous Rho was detected by using anti-Rho antibody (Upstate, CA) at 1:2000 dilution.
2.6 Cell Morphology
PC12 cells were transfected with either GFP vector alone or with other expression plasmids. 24 h later the cells were stimulated with 100 ng/ml NGF for an additional 16 h, before fixing. Cells were subjected to immunofluorescence assay. Two types of morphologies were detected, i) neurite-bearing cells or ii) rounded cells. The percent (%) cell rounding by overexpressed proteins was calculated and plotted as described elsewhere [16], with some modification: % cell rounding = (the number of round fluorescent labeled cells / total number of fluorescent cells counted) × 100. More than 100 cells were counted for five random fields in at least 3 independent experiments.
2.7 Co-immunoprecipitation
Co-immunoprecipitations of p115-RhoGEF with α13QL and α12QL were performed as described previously [12].
2.8 TUNEL Assay
Cell death was detected by TUNEL assay with In Situ Death Detection kit (Roche, Indianapolis, IN) as described by Wang et. al.[21]. 24h post-tranfected PC12 cells were treated with 100 ng/ml NGF for additional 16 h prior to TUNEL assay. Cell death was evaluated by detecting DNA strand break under a fluorescent microscope equipped with a set of excitation-emmission filters for fluorescein. Fluorescein positive cells were counted and compared among cells expressing GFP, p115-RhoGEF, α13QL, or their mutants. As a positive control, PC 12 cells were treated with 50 ng/ml TNFα (BD Biosciences, Palo Alto, CA) for 40 and 96 hrs. In each case cells were subjected to NGF treatment after 24 h TNFα treatment to mimic transfection experiments.
3. RESULTS
3.1 PM association of p115-RhoGEF induces Rho-dependent neurite retraction in PC12 cells
We and others have demonstrated that constitutively activated forms of α12 and α13 and stimulation of G12/13-coupled GPCRs induces the translocation of p115-RhoGEF from the cytoplasm to the plasma membrane (PM) [4, 9, 11-15]. However, no studies have yet demonstrated that PM recruitment plays a role in activation of p115-RhoGEF-mediated signaling. Here, we examined the effect of PM association of p115-RhoGEF on neurite retraction of NGF-differentiated PC12 cells, a well characterized model system in which neurite retraction induced by α12/13 and G12/13-coupled GPCRs depends on activation of Rho [17-21]. In these experiments, PC12 cells were transiently transfected with a GFP expression plasmid, in the presence or absence of additional expression vectors, and then allowed to differentiate in the presence of NGF. GFP expressing cells were scored for the presence of neurite extensions or for showing round morphology lacking neurite extensions (Figure 1). In the absence of NGF, the majority of GFP-expressing PC12 cells (57.40±3.0 %, p<0.0001) displayed a rounded morphology lacking neurites (Fig. 1B), while in the presence of NGF 80-90% of the cells contained clear neurite extensions (Fig. 1A.a and Fig. 1B). When cells were also transfected with a vector for constitutively active α13QL, a majority of GFP-expressing PC12 cells displayed a dramatic inhibition of NGF-stimulated neurite extension (Fig. 1A.c and Fig. 1B), consistent with previous reports of α13QL-stimulated neurite retraction [21]. As a control, a non-palmitoylated form of α13QL, termed α13QL-CCSS [14], had no effect on NGF-induced neurites, in agreement with our previous report showing that α13QL-CCSS is in an activated conformation but unable to activate Rho-dependent signaling [14] (Fig 1A.e). The critical dependence on Rho for neurite retraction was demonstrated by expression of C3-transferase (C3), a toxin that specifically inhibits Rho by catalyzing its ADP-ribosylation [22]. In the absence of NGF treatment, C3-mediated inhibition of Rho dramatically induced neurites (Fig. 1B), and in the presence of NGF treatment these cells showed longer and more branched neurites compared to cells expressing only GFP (Fig 1A, compare g and a). Thus, the above described experiments confirmed the suitability of the PC12 system for examining regulators of Rho.
Figure 1. Rho-dependent neurite retraction in PC12 cells by overexpression of α13QL, p115-RhoGEF and their mutants.
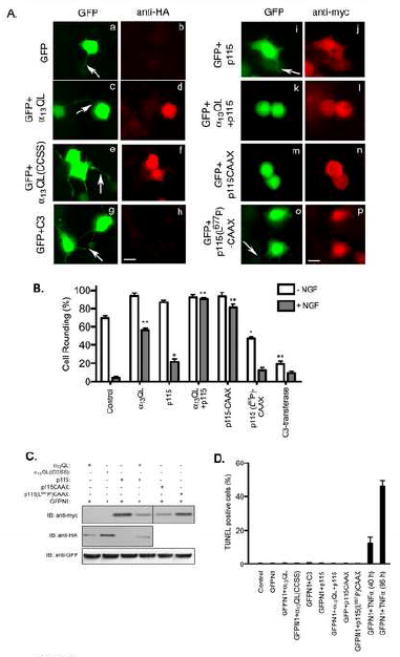
A. PC12 cells were transfected with plasmids encoding GFP along with vector (a and b), HA-α13QL (c and d), HA-α13QL(CCSS) (e and f), myc-tagged C3-transferase (g and h), myc-tagged p115-RhoGEF (i and j), myc-p115-RhoGEF and HA-α13QL (k and l), myc-p115-RhoGEF-CAAX (m and n), and myc-p115(L677P)-CAAX (o and p). 24 h post-transfection cells were transferred into serum free medium containing 100 ng/ml NGF. 16 h post-NGF-treatment cells were fixed and labeled with anti-HA antibody (anti-HA) or anti-myc antibody (anti-myc) followed by Texas Red-conjugated anti-mouse secondary antibody. The neuronal morphology was detected by observing the distribution of GFP in the cells co-expressing the indicated proteins. Representative images are presented. The arrows indicate neurite formation. Note that the GFP images are purposely overexposed so that the neurites are visible. Bar, 10 μm. B. A quantitative analysis of cell rounding is represented by plotting percent (%) of round cells calculated in each transfection experiment, as described in “Materials and Methods.” The data are average +/- S.E. of three separate experiments. Statistical significance (student’s t-test), indicated by * (p < 0.05) and ** (p < 0.001), is shown and compares untreated samples (-NGF) to control and NGF-treated (+NGF) to control. C. Total cell lysates from the indicated transfections of PC12 cells were immunoblotted with anti-myc, anti-HA and anti-GFP antibodies to determine expression levels of GFP, p115-RhoGEF, α13QL and their mutants. D. 40 h post-transfection, differentiating PC12 cells were subjected to TUNEL assay as described in the “Materials and Methods” section. The data is a representative experiment performed in triplicate.
Next, p115-RhoGEF was expressed along with GFP in PC12 cells. Only 15.81±1.7 % (p=0.0008) cells expressing p115-RhoGEF displayed the ability to induce neutrite retraction in NGF-differentiated cells (Fig. 1A.i and Fig. 1B). However, when α13QL and p115-RhoGEF were expressed together, neurite retraction (i.e., cell rounding) was strongly observed in 91±1.4 % (p= 0.0007) NGF-treated cells (Fig. 1A.k and Fig. 1B). Although α13QL recruits p115-RhoGEF to the PM in PC12 cells, these data cannot indicate that PM recruitment of p115-RhoGEF is functionally important. To test this, we generated a mutant of p115-RhoGEF that is constitutively targeted to the PM. This was accomplished by fusing the C-terminal 20 amino acids from H-Ras to the C-terminus of p115-RhoGEF, to generate p115-RhoGEF-CAAX (Fig. S1). A number of studies have demonstrated that this region of H-Ras undergoes farnesylation and palmitoylation, and is sufficient to target heterologous proteins to the PM [23, 24]. In contrast to wild type cytoplasmic p115-RhoGEF, PM-localized p115-RhoGEF-CAAX strongly induced neurite retraction (i.e. cell rounding) in 87±2.2 % (p=0.0001) of NGF-treated cells. (Fig. 1A.m and Fig. 1B). This result provides the first evidence that PM localization of p115-RhoGEF can activate Rho-dependent signaling. When p115-RhoGEF-CAAX contained a mutation in the PH domain that inhibits Rho-dependent signaling [12], NGF-stimulated neurite extensions were unaffected (Fig. 1A.o and Fig. 1B); cells showed long neurites, consistent with PM-associated p115-RhoGEF-CAAX inducing neurite retraction via a Rho-dependent pathway. Lastly, when we evaluated apoptosis by TUNEL assay, as described in a previous report [25], we observed no differences among the TUNEL positive cells expressing p115-RhoGEF or α13QL or their mutants compared to cells expressing control GFP (Fig. 1D) 40 h post-transfection, suggesting that the observed effects on neutrite retraction and cell rounding in Figure 1 are not due to cell death. As a positive control for the TUNEL assay, we treated PC12 cells with tumor necrosis factor α (TNFα) that is known to induce cell death [26]. 40 h treatment of 50 ng/ml TNFα produced ~10% TUNEL positive cells, which reached almost 50% after 96 h treatment (Fig. 1D). All immunofluorescence assays in Figure 1 were performed 40 h post-transfection. In summary, neurite retraction and cell rounding of NGF-differentiated PC12 cells provides a model system for studying Rho-dependent signaling. The key result using this system is that expression of p115-RhoGEF only weakly activates this signaling pathway, but simply PM-targeting of p115-RhoGEF (i.e., p115-RhoGEF-CAAX) is able to strongly enhance its ability to induce neurite retraction and cell rounding.
3.2 Inhibition of Rho abrogates α13-mediated PM translocation of p115-RhoGEF
During the course of these studies, we noted that p115-RhoGEF failed to be recruited to the PM by constitutively active α13QL when Rho was inhibited by C3. Rho is downstream of and directly activated by p115-RhoGEF, and it is therefore surprising and unexpected that inhibition of Rho would affect PM recruitment of p115-RhoGEF. Thus, we proceeded to examine the requirement for Rho in α13QL-mediated PM recruitment of p115-RhoGEF.
In PC12 cells, expressed p115-RhoGEF is distributed throughout the cytoplasm (Fig. 2a), and co-expression of α13QL strongly recruits p115-RhoGEF to the PM as shown by the intense staining at the cell periphery (Fig. 2d). However, when C3 is expressed together with α13QL and p115-RhoGEF, p115-RhoGEF remains in the cytoplasm and is not detected at the PM (Fig. 2g and 2j). A similar cytoplasmic localization of p115-RhoGEF is observed when C3 is co-expressed with p115-RhoGEF in the absence of α13QL (Fig 2m). C3 did not affect the localization of α13QL; α13QL displayed a subcellular distribution of both PM and cytoplasmic staining in the absence (Fig 2e) or presence of C3 (Fig 2h and 2k). The functional inhibition of Rho by C3 is evident by the observation that C3 expression induces neurite extensions (Fig. 2g-o). In this set of experiments, a GFP-tagged p115-RhoGEF was utilized; N-terminally myc-tagged and C-terminally GFP-tagged forms of p115-RhoGEF are both effectively recruited to the PM by α13QL in PC12 and HEK293 cells. Because C3 prevents the activation of Rho, these experiments suggest a novel requirement of activation of Rho in facilitating PM recruitment of p115-RhoGEF.
Figure 2. Inhibition of Rho prevents α13QL-induced PM translocation of p115-RhoGEF in PC12 cells.
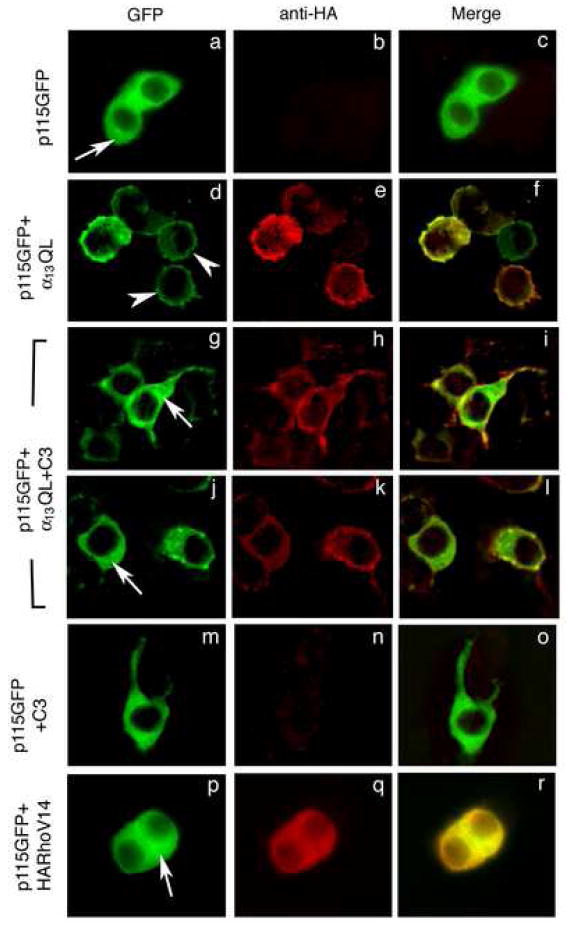
PC12 cells were transiently transfected with plasmids encoding GFP-tagged p115-RhoGEF (p115GFP), C3-transferase (C3), HA-tagged RhoV14 (HARhoV14) or α13QL (α13QL), in various combinations as indicated. 16 h after transfection, cells were fixed and stained with anti-HA monoclonal antibody (b, e, h, k, n, and q) followed by anti-mouse secondary antibody conjugated to Texas Red to detect α13QL or RhoV14. p115-RhoGEF was detected due to the intrinsic fluorescence of the fused GFP (a, d, g, j, m, and p). Arrows indicate cytoplasmic staining, and arrowheads indicate plasma membrane staining. Representative images were recorded by immunofluorescence microscopy.
To further characterize the requirement for Rho activation, we examined whether Rho activation was sufficient for PM recruitment of p115-RhoGEF and whether downstream Rho signaling to the actin cytoskeleton was necessary. When the constitutively active RhoA V14 mutant was co-expressed in PC12 cells with p115-RhoGEF, p115-RhoGEF remained in the cytoplasm (Fig. 2p) and did not translocate to the PM. This result indicates that, whereas active Rho is necessary for p115-RhoGEF PM recruitment, it is not sufficient. One of the important signaling pathways mediated by Rho leads to increases in cellular F-actin, and consequently inhibition of Rho with C3 leads to loss of F-actin structures [27]. To examine a role for an intact actin cytoskeleton in α13QL-promoted PM recruitment of p115-RhoGEF, cells were treated with latrunculin B which causes the rapid shift of a cell’s F-actin to G-actin and thereby disrupts the actin cytoskeleton. In latrunculin B treated cells, p115-RhoGEF retained the ability to be recruited to the PM in response to co-expression of α13QL (data not shown). Thus, the inhibition by C3 of α13QL-promoted PM recruitment of p115-RhoGEF together with the latrunculin B experiment suggests that Rho facilitates PM recruitment of p115-RhoGEF through a mechanism that does not involve regulated changes in the actin cytoskeleton.
A requirement for Rho in PM recruitment of p115-RhoGEF was confirmed through biochemical fractionation and the use of siRNA depletion of Rho. In these experiments, we utilized HEK293 cells because a higher transfection efficiency, compared to PC12 cells, was required to detect sufficient amounts of expressed protein in biochemical fractionation experiments. First, we confirmed by immunofluorescence that C3 inhibition of Rho showed the same effect on p115-RhoGEF PM recruitment in HEK293 cells as it did in PC12 cells. Indeed, α13QL induced PM localization of p115-RhoGEF in HEK293 cells, as reported previously [12-14], but co-expression of C3 prevented PM recruitment of p115-RhoGEF (Fig. 3A). Thus, we proceeded to biochemical fractionation experiments. Cells were lysed in a hypotonic buffer, and high-speed centrifugation separated the cell lysate into membrane-rich particulate (P) and cytoplasm-rich soluble (S) fractions. When expressed alone, p115-RhoGEF was detected almost exclusively in the soluble fraction (Fig 3B, lanes 1S and 1P), but p115-RhoGEF strongly partitioned into the P fraction (approximately 50:50, S:P) when co-expressed with α13QL (Fig 3B, lanes 2S and 2P). However, co-expression of C3-transferase with α13QL and p115-RhoGEF, abrogated the partitioning of p115-RhoGEF into the P fraction (Fig 3B, lanes 3S and 3P), consistent with fluorescence microscopy results (Fig. 2 and 3A). As a complementary technique to C3-mediated inhibition of Rho, siRNA was used to deplete RhoA protein in HEK293 cells. Using a previously reported siRNA [28], RhoA was efficiently depleted (Fig. 3B, right panel, lane 4). In these RhoA-depleted cells, p115-RhoGEF failed to be recruited to the P fraction when α13QL was co-expressed (Fig. 3B, lanes 4S and 4P). A control siRNA had no effect; p115-RhoGEF still partitioned into the P fraction when co-expressed with α13QL (Fig. 3B, lanes 5S and 5P). Taken together, the fluorescence microscopy and biochemical fractionation, using C3-mediated inhibition and siRNA-mediated depletion of Rho, indicate a novel role for Rho in regulated PM translocation of p115-RhoGEF.
Figure 3. Inhibition or depletion of Rho prevents α13QL-induced PM recruitment of p115-RhoGEF in HEK293 cells.
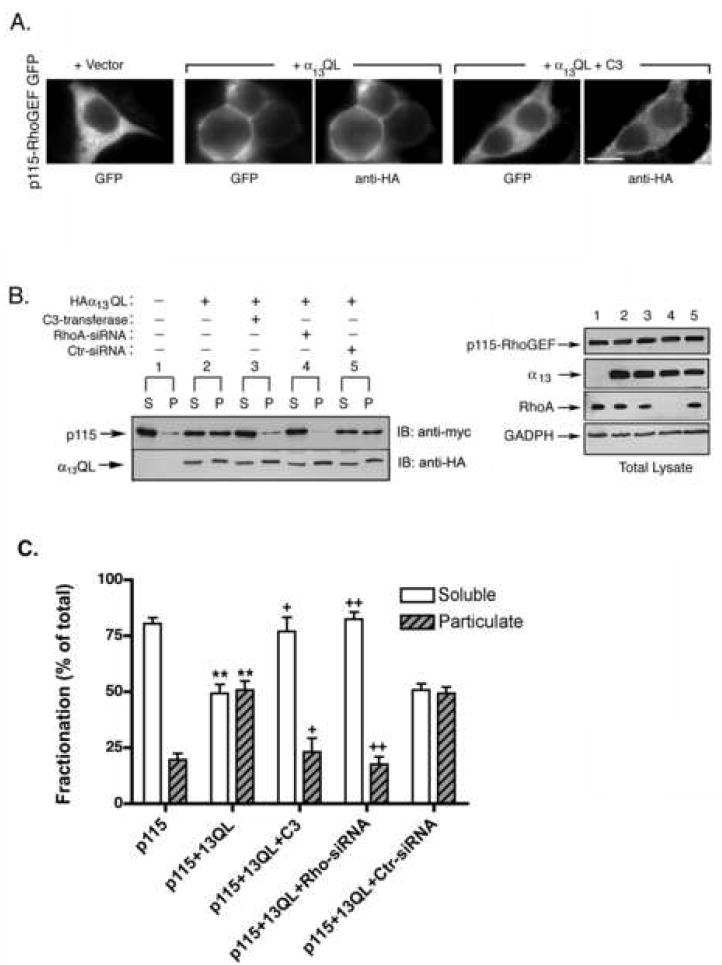
A. HEK293 cells were transfected with the expression vector for p115-RhoGEF-GFP along with empty vector, an expression plasmid for HA-tagged α13QL, or expression plasmids for both HA-tagged and α13QL myc-tagged C3-transferase (C3), as indicated. 24 h post-transfection, cells were fixed and subjected to immunofluorescence microscopy as described in the “Materials and Methods.” B. HEK293 cells were transfected with plasmids encoding myc-tagged p115-RhoGEF along with expression plasmids for HA-tagged α13QL, myc-tagged C3-transferase, siRNA specific for Rho silencing (RhoA-siRNA) or non-specific siRNA (Ctr-siRNA), as indicated (left panel). Cells were lysed and fractionated into soluble (S) and particulate (P) fractions, as described in the “Materials and Methods.” The fractions were immunoblotted (IB) with anti-myc antibody to detect p115-RhoGEF and anti-HA antibody to detect α13QL. Total cell lysates were immunoblotted (right panel) with anti-myc antibody and anti-HA antibody to show equivalent expression of p115-RhoGEF and α13QL, respectively. In addition, effective knockdown of endogenous Rho was detected by probing cell lysates with anti-Rho antibody, and immunoblotting with an anti-GADPH antibody served as a loading control. The lane numbering in the right panel indicates that the cell lysate corresponds to the transfection indicated in the left panel. C. Data from three independent fractionation experiments of p115-RhoGEF were quantitated (mean +/- S.E.). Statistical significance (student’s t-test), indicated by ** (p < 0.005), compares p115-RhoGEF alone to p115-RhoGEF in the presence of co-expressed α13QL; + (p < 0.05) and ++ (p < 0.005) indicates significance in comparison to p115-RhoGEF + α13QL. Soluble fractions (white bars) are compared to other soluble fractions, while particulate fractions (black bars) are compared to other particulate fractions.
3.3 α12-mediated PM translocation of p115-RhoGEF does not require Rho
Since both α12 and α13 promote PM translocation of p115-RhoGEF [12], we tested whether inhibition of Rho also prevented α12QL-mediated PM recruitment of p115-RhoGEF. Surprisingly, expression of C3-transferase did not prevent α12QL-mediated PM recruitment of p115-RhoGEF, in contrast to what was observed for α13QL-mediated PM recruitment of p115-RhoGEF. In both PC12 cells (not shown) and HEK293 cells, co-expression of α12QL with p115-RhoGEF promoted strong PM localization of p115-RhoGEF regardless of whether C3 transferase was also expressed (Fig. 4A). Biochemical fractionation confirmed the lack of effect of C3. Expression of α12QL promoted a strong shift of p115-RhoGEF to the P fraction (Fig. 4B, lanes 2S and 2P) and this recruitment to the P fraction was not perturbed by expression of C3 (Fig. 4B, lanes 3S and 3P). Thus, these results provide the first indication that there are mechanistic differences in the PM recruitment of p115-RhoGEF that is mediated by α12 versus α13.
Figure 4. α12QL-induced PM recruitment of p115-RhoGEF is refractory to inhibition of Rho.
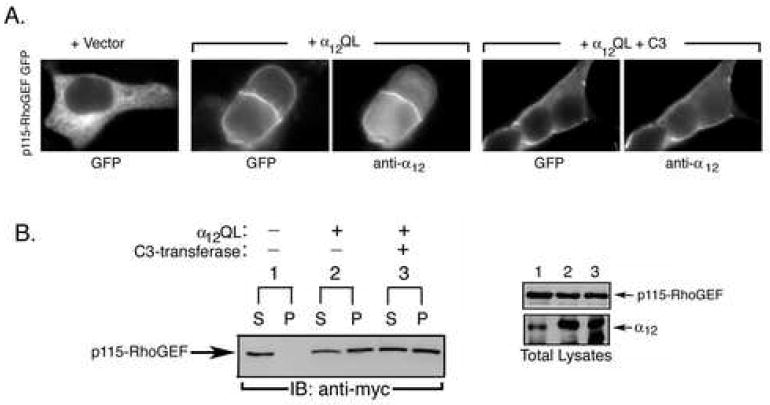
A. HEK293 cells were transfected with the expression vector for p115-RhoGEF-GFP along with empty vector, an expression plasmid for α12QL, or expression plasmids for both α12QL and myc-tagged C3-transferase (C3), as indicated. 24 h post-transfection, cells were fixed and subjected to immunofluorescence microscopy as described in the “Materials and Methods.” B. HEK293 cells were transfected with plasmids encoding myc-tagged p115-RhoGEF along with expression plasmids for α12QL and/or myc-tagged C3-transferase, as indicated (left panel). Cells were lysed and fractionated into soluble (S) and particulate (P) fractions, as described in the “Materials and Methods.” The fractions were immunoblotted (IB) with anti-myc antibody to detect p115-RhoGEF. Total cell lysates were immunoblotted (right panel) with anti-myc antibody and anti-α12 antibody to show expression of p115-RhoGEF and α12QL, respectively. The lane numbering in the right panel indicates that the cell lysate corresponds to the transfection indicated in the left panel.
3.4 RGS domain mutants of p115-RhoGEF reveal differences in α12 and α13-induced PM recruitment
To further explore differences in α12-mediated versus α13-mediated PM recruitment of p115-RhoGEF, we analyzed previously described [12] p115-RhoGEF mutants. A p115-RhoGEF mutant lacking the N-terminal RGS domain, (246-912)p115-RhoGEF remained cytoplasmic when co-expressed with either α13QL [12] or α12QL (not shown), confirming that the RGS domain is essential, presumably due to its ability to interact with α13QL and α12QL. However, a clear difference between α13QL and α12QL was observed when PM recruitment of the isolated N-terminal RGS domain of p115-RhoGEF was assayed. (1-246)p115-RhoGEF co-immunoprecipitates with both α12QL and α13QL (Fig. 5A, lane 4; and 5B, lane 4), consistent with previous reports showing interaction of the RGS domain of p115-RhoGEF with both α13 and α12 in an activation-dependent manner [6, 29, 30]. As described previously [12], (1-246)p115-RhoGEF is only weakly recruited from the cytoplasm to the PM when co-expressed with α13QL (Fig. 6A) and shows little or no shift to the P fraction in biochemical fractionation assays (Fig. 6C). Thus, the RGS domain is not sufficient for α13QL-mediated PM recruitment. On the other hand, we now find that α12QL, compared to α13QL, induces a more robust recruitment of (1-246)p115-RhoGEF to the PM (Fig. 6A) and promotes a substantial shift of (1-246)p115-RhoGEF to the P fraction (Fig. 6B). In other words, the N-terminal RGS domain of p115-RhoGEF is both necessary and sufficient for α12QL-promoted PM recruitment.
Figure 5. Co-immunoprecipitation of α12QL and α13QL with p115-RhoGEF and p115-RhoGEF RGS domain mutants.
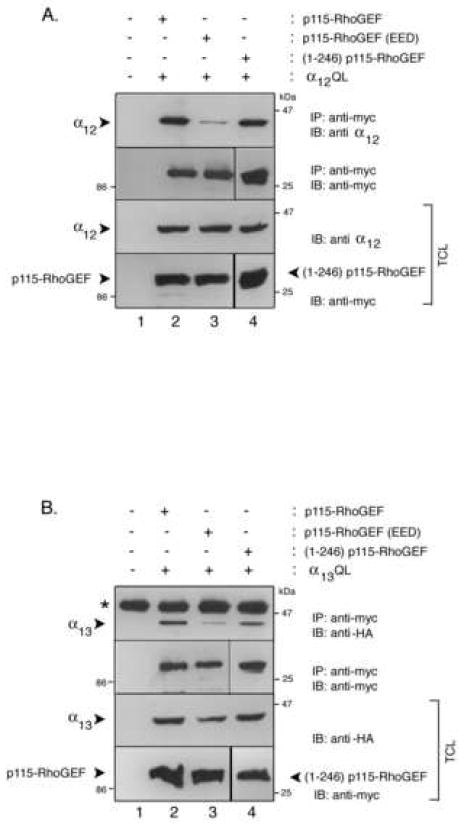
A. HEK293 cells were transfected with empty vector (lane 1), expression plasmids for α12QL and p115-RhoGEF (lane 2), expression plasmids for α12QL and p115-RhoGEF(EED) (lane 3), or expression plasmids for α12QL and (1-246)p115-RhoGEF (lane 4). Cells were lysed and the lysates were subjected to immunoprecipitation (IP) by monoclonal anti-myc antibody to precipitate p115-RhoGEF and its mutants. The immunoprecipitates were immunoblotted (IB) with polyclonal anti-α12 antibody (upper panel) and anti-myc antibody (second panel). The corresponding total cell lysates were immunoblotted and probed with anti-myc or anti-α12 antibodies to show expression of p115-RhoGEF and its mutants (lower panel), and α12QL (third panel). B. HEK293 cells were transfected with empty vector (lane 1), expression plasmids for α13QL and p115-RhoGEF (lane 2), expression plasmids for α13QL and p115-RhoGEF(EED) (lane 3), or expression plasmids for α13QL and (1-246)p115-RhoGEF (lane 4). Cells were lysed and the lysates were subjected to immunoprecipitation (IP) by monoclonal anti-myc antibody to precipitate p115-RhoGEF and its mutants. The immunoprecipitates were immunoblotted (IB) with polyclonal anti-HA antibody (upper panel) to detect α13QL and anti-myc antibody (second panel). The corresponding total cell lysates were immunoblotted and probed with anti-myc or anti-HA antibodies to show expression of p115-RhoGEF and its mutants (lower panel), and α13QL (third panel).
Figure 6. Differential α12QL- and α13QL-mediated PM recruitment of p115-RhoGEF RGS domain mutants.
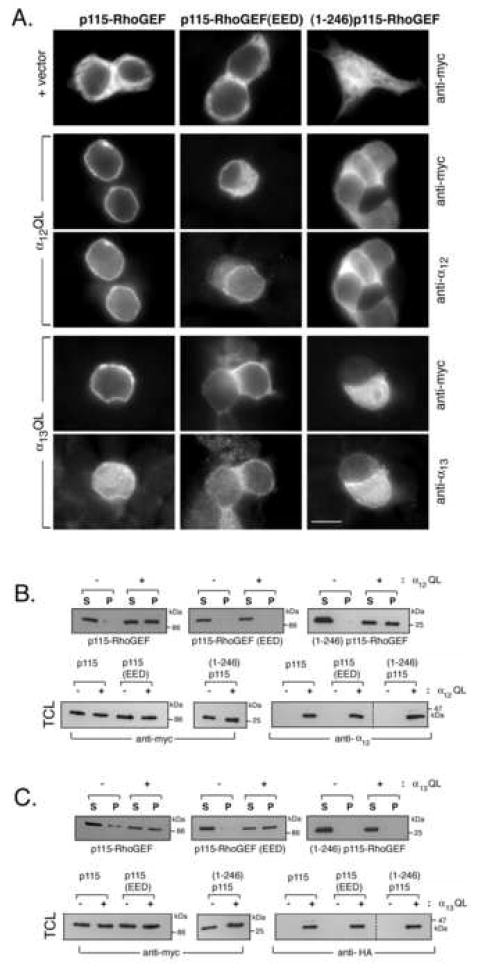
A. HEK293 cells were transfected with either p115-RhoGEF (left column), p115-RhoGEF(EED) (middle column), or (1-246)p115-RhoGEF (right column). Each p115-RhoGEF wt or mutant was co-transfected with empty vector (upper row), α12QL (rows 2 and 3), or α13QL (rows 4 and 5). 24 h post-transfection, cells were fixed and subjected to immunofluorescence microscopy as described in the “Materials and Methods,” using the indicated antibodies. B. HEK293 cells were transfected with either p115-RhoGEF, p115-RhoGEF(EED), or (1-246)p115-RhoGEF. Each p115-RhoGEF wt or mutant was co-transfected with empty vector or α12QL, as indicated. Cells were lysed and fractionated into soluble (S) and particulate (P) fractions, as described in the “Materials and Methods.” The fractions were immunoblotted (IB) with anti-myc antibody to detect p115-RhoGEF (upper panels). Total cell lysates (TCL) were immunoblotted with anti-myc antibody and anti-α12 antibody to show expression of p115-RhoGEF and α12QL, respectively (lower panels). C. HEK293 cells were transfected with either p115-RhoGEF, p115-RhoGEF(EED), or (1-246)p115-RhoGEF. Each p115-RhoGEF wt or mutant was co-transfected with empty vector or α13QL, as indicated. Cells were lysed and fractionated into soluble (S) and particulate (P) fractions, as described in the “Materials and Methods.” The fractions were immunoblotted (IB) with anti-myc antibody to detect p115-RhoGEF (upper panels). Total cell lysates were immunoblotted with anti-myc antibody and anti-HA antibody to show expression of p115-RhoGEF and α13QL, respectively (lower panels). The dashed lines (lower right panels in B and C) indicate that the samples are from the same gel; intervening lanes that were irrelevant to the figure were simply removed.
An analysis of regulated PM recruitment of a RGS domain mutant of p115-RhoGEF reveals another difference between α13 and α12. The RGS domain of p115-RhoGEF consists not only of the conserved RGS core (amino acids 44-233) but also a short N-terminal extension marked by a stretch of acidic residues (amino acids 27-34); a recent crystal structure indicates that α13 interacts with a surface in the RGS core and with the N-terminal acid region [31]. Moreover, mutagenesis studies have indicated that the N-terminal acidic residues are required for high affinity interaction between α13 and p115-RhoGEF [4, 13, 31, 32]. We previously reported the surprising result that whereas activated α13 failed to co-immunoprecipitate with an acidic region mutant of full-length p115-RhoGEF, the acidic region mutant of p115-RhoGEF retained the ability to be strongly recruited to membranes by α13QL [13]. This was interpreted as a loss of high affinity interaction between α13 and p115-RhoGEF due to loss of the critical acidic residues and thus consequent loss of stable interaction in a co-immunoprecipitation; however, weaker interactions between α13 and the core RGS domain of p115-RhoGEF would be sufficient to recruit the mutant p115-RhoGEF to the PM where additional factors, such as α13 binding to additional domains (e.g., the DH domain) of p115-RhoGEF, interactions of the PH domain with membrane lipids, and likely a Rho-dependent factor, as described herein, would allow for stable membrane binding of p115-RhoGEF [4, 13]. Consistent with our previous report, we show that an acidic mutant, termed p115-RhoGEF(EED) in which glutamic acids 27 and 29 and aspartic acid 30 are all changed to alanines, does not efficiently co-immunoprecipitate with α13QL (Fig. 5B, lane 3) but is strongly recruited to the PM (Fig. 6A) and to the P fraction in biochemical fractionation assays (Fig. 6C). Like α13QL, α12QL is severely impaired in interacting with p115-RhoGEF(EED) as evidenced by poor co-immunoprecipitation (Fig. 5A, lane 3), suggesting that α12 also requires the N-terminal acidic region of the RGS domain for strong interaction with p115-RhoGEF. However, in contrast to α13QL, α12QL expression failed to recruit p115-RhoGEF(EED) to the PM (Fig. 6A) and failed to cause a shift of p115-RhoGEF(EED) to the P fraction (Fig. 6B). Taken together results in Figure 6 demonstrate fundamental differences in how α13 and α12 promote the translocation of p115-RhoGEF to the PM.
4. DISCUSSION
Regulated and reversible translocation of proteins from one subcellular location to another is a common feature of signal transduction pathways. One example of protein translocation is the activation-dependent recruitment of specific proteins from the cytoplasm to the PM in order to form localized signaling complexes. The results described herein provide several novel insights into the function and mechanism of α12/13-mediated PM recruitment of p115-RhoGEF.
This report provides the first evidence that PM localization is actually important for Rho-dependent signaling functions of p115-RhoGEF. Although it has been thought that PM recruitment might be important for p115-RhoGEF function, results to support this have been lacking previously. We employed a well-characterized cell-based assay for Rho function in which neurite retraction of NGF-differentiated PC12 cells has been shown to be induced by α12/13 and G12/13-coupled GPCRs in a Rho-dependent manner [17-21]. To test for a role for PM localization in p115-RhoGEF function, expression of wild type p115-RhoGEF was compared with a constitutively PM-localized p115-RhoGEF-CAAX, in which the C-terminal 20 amino acids of H-Ras was appended to the C-terminus of p115-RhoGEF to provide a strong PM targeting signaling. Whereas expression of wild type p115-RhoGEF showed very weak ability to reverse the NGF-differentiated phenotype of neurite extensions in PC12 cells, p115-RhoGEF-CAAX completely reversed the NGF-promoted neurite extensions. This key result indicates that, at least for some Rho-dependent signaling functions, PM recruitment of an RGS-RhoGEF is sufficient to activate a signaling pathway. Importantly, PM recruitment of expressed or endogenous p115-RhoGEF has been observed in response to extracellular ligand activation of several G12/13-coupled GPCRs, such as LPA, thromboxane A2, sphingosine-1-phosphate and thrombin receptors [9, 12, 15], indicating that PM recruitment of p115-RhoGEF occurs under acute GPCR and G protein activation. Although it is likely that PM recruitment is not the only way in which α12/13 activates p115-RhoGEF, the results presented here, combined with previous studies [14], confirm the importance of regulated PM localization for p115-RhoGEF function.
The studies in this report also describe an unexpected role for Rho in the α13-promoted PM recruitment of p115-RhoGEF. Immunofluorescence microscopy and biochemical fractionation (Fig. 2 and 3) strikingly show that inhibition of Rho by the toxin C3 transferase or that siRNA depletion of Rho completely abrogates the α13QL-mediated PM recruitment of p115-RhoGEF. This is surprising because Rho is activated by p115-RhoGEF and thus is downstream of the α13/p115-RhoGEF interaction. The mechanism of the requirement for Rho is unclear and is the subject of future studies. Loss of α13-mediated PM recruitment of p115-RhoGEF upon Rho inhibition or depletion does not appear to be an indirect effect of disruption of the cell’s actin cytoskeleton since depolymerization of F-actin by treatment with latrunculin B did not prevent α13-dependent PM translocation of p115-RhoGEF (not shown). Moreover, the presence of overexpressed, constitutively active Rho is not sufficient to induce PM localization of p115-RhoGEF (Figure 2), suggesting that the requirement for Rho is one of several factors that can influence the important PM recruitment of p115-RhoGEF. We can speculate on several potential mechanisms of how Rho could promote the regulated PM localization of p115-RhoGEF. Rho may be required to help tether p115-RhoGEF to the PM through direct binding or indirectly via additional scaffolding proteins, and this function could be promoted in a feedback manner. Alternatively, activation of Rho, either directly by p115-RhoGEF or in a parallel pathway initiated by active α13, could activate a positive feedback signaling response to enhance p115-RhoGEF PM recruitment. Interestingly, a recent report uncovered a positive feedback response in the activation of LARG [33]. The Rho effector Dia1 was shown to bind LARG and enhance LARG’s in vitro guanine-nucleotide exchange activity, thus providing a positive feedback loop. It will be important to test whether Dia1 plays a role in α13-promoted PM recruitment of LARG and p115-RhoGEF.
The results in this report also describe novel mechanistic differences in α13-mediated versus α12-mediated PM recruitment of p115-RhoGEF. First, α12-mediated PM recruitment of p115-RhoGEF was not prevented by C3 transferase inhibition of Rho (Fig. 4). This result is in striking contrast to the requirement for Rho in α13-mediated PM recruitement of p115-RhoGEF. Second, our studies revealed p115-RhoGEF RGS domain-based differences in α13-mediated versus α12-mediated PM recruitment. Whereas α13QL shows little or no ability to recruit to the PM the isolated N-terminal RGS domain (aa 1-246) of p115-RhoGEF (Fig. 6A and 6B) [12], α12QL strongly recruited the isolated RGS domain to the PM and to the membrane fraction, as assayed by immunofluorescence microscopy and biochemical fractionation (Fig. 6A and 6C). This result suggests that α12QL compared to α13QL interacts more strongly with p115-RhoGEF’s RGS domain. However, in co-immunoprecipitation or pull-down experiments, the RGS domain of p115-RhoGEF appears to interact equally well with activated α12 and α13 (Fig. 5) [6]. Another set of experiments examined the ability of α12QL versus α13QL to recruit to the PM a mutant p115-RhoGEF in which an N-terminal acidic region of the RGS domain (aa 27-32) was disrupted by substitution with alanines. This N-terminal acidic stretch provides a second distinct site of RGS domain interaction with α13 [31, 32]; the other contact surface utilizes multiple loops within amino acids 44-233 of the RGS domain. The N-terminal acidic region is necessary for the GAP activity of the RGS domain towards α13 [31, 32] and for high-affinity binding of p115-RhoGEF to α13 [4, 13, 32]. However, mutational disruption of the acidic region does not inhibit the ability of α13 to stimulate the Rho exchange activity of p115-RhoGEF [32], and, importantly for the studies herein, does not prevent the α13-mediated PM recruitment of p115-RhoGEF [13]. In agreement with other reports, we show that α13QL poorly interacts, via co-immunoprecipitation (Fig. 5B), with an N-terminal acidic region EED mutant of p115-RhoGEF; additionally, we now show a similar weak interaction of p115-RhoGEF(EED) with α12QL (Fig. 5A). Surprisingly, however, whereas α13QL efficiently recruits p115-RhoGEF(EED) to the PM and the membrane fraction, α12QL fails to do so (Fig. 6). Taken together, the results show clear differences in the mechanisms of α13 versus α12 to mediate the PM recruitment of p115-RhoGEF and support the idea that PM recruitment mediated by α12 relies predominantly on interactions with the RGS domain, but PM recruitment mediated by α13 utilizes multiple mechanisms.
Although these studies are the first to describe differences between α13 and α12 in terms of PM recruitment of an RGS-RhoGEF, some important differences in interaction have been noted. For example, α13 is a better substrate than is α12 for the RGS domain-mediated GAP activity of p115-RhoGEF or LARG [6, 29]. Moreover, activated α13 stimulates the RhoGEF activity of p115-RhoGEF or LARG in assays in vitro, but activated α12 is unable to do so [6, 34]. In fact, α12 can inhibit the ability of α13 to activate p115-RhoGEF in vitro [34]. It has thus been suggested that α13 activates an RGS-RhoGEF by interacting with two distinct surfaces, i.e., the RGS domain and the DH/PH domain, while α12 would prevent α13 interaction by binding only to the RGS domain [4, 6]. Our results, examining the requirements for PM recruitment of p115-RhoGEF by α13 or α12, are consistent with this model. However, the exact role in cells for α13 versus α12 in regulating p115-RhoGEF remains to be established, and whether α12 activates or inhibits p115-RhoGEF in vivo is still an open question. So far, there exists no clear evidence to prove or refute the proposal that α12 indeed activates Rho via p115-RhoGEF. Interesting, α12 is unable to activate LARG in vitro unless LARG has undergone tyrosine phosphorylation; it is possible that p115-RhoGEF also undergoes a covalent modification in cells that confers it with the ability to then be positively regulated by α12.
5. CONCLUSION
In summary, the results presented here not only indicate the importance of regulated PM localization for p115-RhoGEF function, but also describe an unexpected role for Rho in the PM recruitment of p115-RhoGEF and unexpected differences in the mechanisms used by α13 and α12 to promoted the PM recruitment of p115-RhoGEF.
Supplementary Material
PC12 cells were transfected with plasmids encoding myc-p115-RhoGEF (a), myc-p115-RhoGEF and HA-α13QL (b), myc-p115-RhoGEF-CAAX (c), and myc-p115(L677P)-CAAX (d). 24 h post-transfection cells were transferred into serum free medium containing 100 ng/ml NGF. 16 h post-NGF-treatment cells were fixed and anti-myc antibody (anti-myc) followed by Texas Red-conjugated anti-mouse secondary antibody. Representative images are presented. The arrows indicate neurite formation.
Acknowledgments
This work was supported by NIH grant (to K. K.), NIH grant GM62884 (to P. W.) and a fellowship from the American Heart Association Pennsylvania-Delaware Affiliate (to R. B.). We thank Katarzyna Urbanska for her assistance in performing the TUNEL assay.
Abbreviations used
- G protein
guanine nucleotide-binding protein
- PM
plasma membrane
- HEK293 cells
human embryonic kidney cells
- HA
hemagglutinin
- PAGE
polyacrylamide gel electrophoresis
- PVDF
polyvinylidene difluoride
- RGS
regulator of G protein signaling
- GEF
guanine-nucleotide exchange factor
- NGF
nerve growth factor
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Cabrera-Vera TM, Vanhauwe J, Thomas TO, Medkova M, Preininger A, Mazzoni MR, Hamm HE. Endocr Rev. 2003;24(6):765–781. doi: 10.1210/er.2000-0026. [DOI] [PubMed] [Google Scholar]
- 2.Kelly P, Casey PJ, Meigs TE. Biochemistry. 2007;46(23):6677–6687. doi: 10.1021/bi700235f. [DOI] [PubMed] [Google Scholar]
- 3.Rossman KL, Der CJ, Sondek J. Nat Rev Mol Cell Biol. 2005;6(2):167–180. doi: 10.1038/nrm1587. [DOI] [PubMed] [Google Scholar]
- 4.Sternweis PC, Carter AM, Chen Z, Danesh SM, Hsiung YF, Singer WD. Adv Protein Chem. 2007;74:189–228. doi: 10.1016/S0065-3233(07)74006-8. [DOI] [PubMed] [Google Scholar]
- 5.Wells CD, Liu MY, Jackson M, Gutowski S, Sternweis PM, Rothstein JD, Kozasa T, Sternweis PC. J Biol Chem. 2002;277(2):1174–1181. doi: 10.1074/jbc.M105274200. [DOI] [PubMed] [Google Scholar]
- 6.Kreutz B, Hajicek N, Yau DM, Nakamura S, Kozasa T. Cell Signal. 2007;19(8):1681–1689. doi: 10.1016/j.cellsig.2007.03.004. [DOI] [PubMed] [Google Scholar]
- 7.Suzuki N, Nakamura S, Mano H, Kozasa T. Proc Natl Acad Sci U S A. 2003;100(2):733–738. doi: 10.1073/pnas.0234057100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Holinstat M, Mehta D, Kozasa T, Minshall RD, Malik AB. J Biol Chem. 2003;278(31):28793–28798. doi: 10.1074/jbc.M303900200. [DOI] [PubMed] [Google Scholar]
- 9.Wells CD, Gutowski S, Bollag G, Sternweis PC. J Biol Chem. 2001;276(31):28897–28905. doi: 10.1074/jbc.M102913200. [DOI] [PubMed] [Google Scholar]
- 10.Grabocka E, Wedegaertner PB. Mol Pharmacol. 2007;72(4):993–1002. doi: 10.1124/mol.107.035162. [DOI] [PubMed] [Google Scholar]
- 11.Grabocka E, Wedegaertner PB. Oncogene. 2005;24(13):2155–2165. doi: 10.1038/sj.onc.1208414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bhattacharyya R, Wedegaertner PB. Biochem J. 2003;371(Pt 3):709–720. doi: 10.1042/BJ20021897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bhattacharyya R, Wedegaertner PB. FEBS Lett. 2003;540(13):211–216. doi: 10.1016/s0014-5793(03)00267-9. [DOI] [PubMed] [Google Scholar]
- 14.Bhattacharyya R, Wedegaertner PB. Journal of Biological Chemistry. 2000;275(20):14992–14999. doi: 10.1074/jbc.M000415200. [DOI] [PubMed] [Google Scholar]
- 15.Meyer BH, Freuler F, Guerini D, Siehler S. J Cell Biochem. 2008;104(5):1660–1670. doi: 10.1002/jcb.21732. [DOI] [PubMed] [Google Scholar]
- 16.Park SY, Li H, Avraham S. Cell Signal. 2007;19(2):289–300. doi: 10.1016/j.cellsig.2006.07.005. [DOI] [PubMed] [Google Scholar]
- 17.Aoki J, Katoh H, Yasui H, Yamaguchi Y, Nakamura K, Hasegawa H, Ichikawa A, Negishi M. Biochem J. 1999;340(Pt 2):365–369. [PMC free article] [PubMed] [Google Scholar]
- 18.Nusser N, Gosmanova E, Makarova N, Fujiwara Y, Yang L, Guo F, Luo Y, Zheng Y, Tigyi G. Cell Signal. 2006;18(5):704–714. doi: 10.1016/j.cellsig.2005.06.010. [DOI] [PubMed] [Google Scholar]
- 19.Jalink K, van Corven EJ, Hengeveld T, Morii N, Narumiya S, Moolenaar WH. J Cell Biol. 1994;126(3):801–810. doi: 10.1083/jcb.126.3.801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tigyi G, Fischer DJ, Sebok A, Yang C, Dyer DL, Miledi R. J Neurochem. 1996;66(2):537–548. doi: 10.1046/j.1471-4159.1996.66020537.x. [DOI] [PubMed] [Google Scholar]
- 21.Katoh H, Aoki J, Yamaguchi Y, Kitano Y, Ichikawa A, Negishi M. Journal of Biological Chemistry. 1998;273(44):28700–28707. doi: 10.1074/jbc.273.44.28700. [DOI] [PubMed] [Google Scholar]
- 22.Aktories K, Wilde C, Vogelsgesang M. Rev Physiol Biochem Pharmacol. 2004;152:1–22. doi: 10.1007/s10254-004-0034-4. [DOI] [PubMed] [Google Scholar]
- 23.Choy E, Chiu VK, Silletti J, Feoktistov M, Morimoto T, Michaelson D, Ivanov IE, Philips MR. Cell. 1999;98(1):69–80. doi: 10.1016/S0092-8674(00)80607-8. [DOI] [PubMed] [Google Scholar]
- 24.Apolloni A, Prior IA, Lindsay M, Parton RG, Hancock JF. Mol Cell Biol. 2000;20(7):2475–2487. doi: 10.1128/mcb.20.7.2475-2487.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang JY, Grabacka M, Marcinkiewicz C, Staniszewska I, Peruzzi F, Khalili K, Amini S, Reiss K. J Neurosci Res. 2006;83(1):7–18. doi: 10.1002/jnr.20712. [DOI] [PubMed] [Google Scholar]
- 26.Royuela M, Rodriguez-Berriguete G, Fraile B, Paniagua R. Histol Histopathol. 2008;23(10):1279–1290. doi: 10.14670/HH-23.1279. [DOI] [PubMed] [Google Scholar]
- 27.Copeland JW, Treisman R. Mol Biol Cell. 2002;13(11):4088–4099. doi: 10.1091/mbc.02-06-0092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Deroanne C, Vouret-Craviari V, Wang B, Pouyssegur J. J Cell Sci. 2003;116(Pt 7):1367–1376. doi: 10.1242/jcs.00308. [DOI] [PubMed] [Google Scholar]
- 29.Kozasa T, Jiang X, Hart MJ, Sternweis PM, Singer WD, Gilman AG, Bollag G, Sternweis PC. Science. 1998;280(5372):2109–2111. doi: 10.1126/science.280.5372.2109. [DOI] [PubMed] [Google Scholar]
- 30.Kreutz B, Yau DM, Nance MR, Tanabe S, Tesmer JJ, Kozasa T. Biochemistry. 2006;45(1):167–174. doi: 10.1021/bi051729t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chen Z, Singer WD, Sternweis PC, Sprang SR. Nat Struct Mol Biol. 2005;12(2):191–197. doi: 10.1038/nsmb888. [DOI] [PubMed] [Google Scholar]
- 32.Chen Z, Singer WD, Wells CD, Sprang SR, Sternweis PC. J Biol Chem. 2003;278(11):9912–9919. doi: 10.1074/jbc.M212695200. [DOI] [PubMed] [Google Scholar]
- 33.Kitzing TM, Sahadevan AS, Brandt DT, Knieling H, Hannemann S, Fackler OT, Grosshans J, Grosse R. Genes Dev. 2007;21(12):1478–1483. doi: 10.1101/gad.424807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hart MJ, Jiang X, Kozasa T, Roscoe W, Singer WD, Gilman AG, Sternweis PC, Bollag G. Science. 1998;280(5372):2112–2114. doi: 10.1126/science.280.5372.2112. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PC12 cells were transfected with plasmids encoding myc-p115-RhoGEF (a), myc-p115-RhoGEF and HA-α13QL (b), myc-p115-RhoGEF-CAAX (c), and myc-p115(L677P)-CAAX (d). 24 h post-transfection cells were transferred into serum free medium containing 100 ng/ml NGF. 16 h post-NGF-treatment cells were fixed and anti-myc antibody (anti-myc) followed by Texas Red-conjugated anti-mouse secondary antibody. Representative images are presented. The arrows indicate neurite formation.