

Summary
Foxg1, a winged-helix transcription factor, promotes the development of anterior neural structures; in mice lacking Foxg1, development of the cerebral hemispheres and olfactory epithelium (OE) is severely reduced. It has been suggested that Foxg1 acts by positively regulating the expression of growth factors, such as Fgf8, which support neurogenesis. However, Foxg1 also binds Smad transcriptional complexes, allowing it to negatively regulate the effects of TGFβ family ligands. Here, we provide evidence that this latter effect explains much of the ability of Foxg1 to drive neurogenesis in the OE. We show that Foxg1 is expressed in developing OE at the same time as the gene encoding growth differentiation factor 11 (Gdf11), a TGFβ family member that mediates negative-feedback control of OE neurogenesis. Mutations in Gdf11 rescue, to a considerable degree, the major defects in Foxg1-/- OE, including the early, severe loss of neural precursors and olfactory receptor neurons, and the subsequent collapse of both neurogenesis and nasal cavity formation. Rescue is gene-dosage dependent, with loss of even one allele of Gdf11 restoring substantial neurogenesis. Notably, we find no evidence for a disruption of Fgf8 expression in Foxg1-/- OE. However, we do observe both a failure of expression of follistatin (Fst), which encodes a secreted Gdf11 antagonist normally expressed in and around OE, and an increase in the expression of Gdf11 itself within the remaining OE in these mutants. Fst expression is rescued in Foxg1-/-;Gdf11-/- and Foxg1-/-;Gdf11+/- mice. These data suggest that the influence of Foxg1 on Gdf11-mediated negative feedback of neurogenesis may be both direct and indirect. In addition, defects in development of the cerebral hemispheres in Foxg1-/- mice are not rescued by mutations in Gdf11, nor is Gdf11 expressed at high levels within these structures. Thus, the pro-neurogenic effects of Foxg1 are likely to be mediated through different signaling pathways in different parts of the nervous system.
Keywords: Mouse, Olfactory epithelium, TGFβ, Follistatin, p21Cip1, Fgf8, Neuronal progenitor, Neurogenesis, Mash1, Ngn1, Sox2, Olfactory receptor neuron, Cerebral cortex, Gene dosage
INTRODUCTION
Forkhead or Fox proteins comprise a large family of `winged-helix' transcription factors that regulate diverse developmental processes in mammals (Carlsson and Mahlapuu, 2002). Foxg1 is highly expressed in anterior neural structures, and promotes their development; neural structures whose development is adversely affected in Foxg1-/- mice include the cerebral cortex, ventral telencephalon, ear, retina and olfactory epithelium (OE) (Duggan et al., 2008; Hanashima et al., 2007; Hanashima et al., 2004; Hebert and McConnell, 2000; Martynoga et al., 2005; Pauley et al., 2006; Pratt et al., 2004; Xuan et al., 1995). In mice that are null for Foxg1, the cerebral hemispheres are dramatically reduced in size, ventral telencephalic structures are lacking, and the animals die at birth (Xuan et al., 1995). Foxg1 is also expressed in the OE from an early age (Hatini et al., 1999), and Foxg1-/- mice lack an OE and most of the nasal cavity (Xuan et al., 1995). For these reasons, Foxg1 has been described as a general positive regulator of anterior nervous system development.
It has been proposed that positive effects of Foxg1 on neurogenesis are closely linked to the effects of fibroblast growth factors (FGFs) (reviewed by Hebert and Fishell, 2008). In the telencephalon, Foxg1 positively regulates expression of Fgf8 (Martynoga et al., 2005), which plays a central role in neurogenesis not only in the telencephalon, but also in the OE (Kawauchi et al., 2005). Although these data raise the possibility that Foxg1 promotes neurogenesis by inducing Fgf8, other studies indicate that FGFs such as FGF8 act upstream of Foxg1 to control Foxg1 expression and function (Regad et al., 2007; Shimamura and Rubenstein, 1997; Storm et al., 2006).
An alternative mechanism by which Foxg1 could influence neural development is through its effects on the transforming growth factor beta (TGFβ) pathway (Dou et al., 2000; Rodriguez et al., 2001; Seoane et al., 2004). TGFβ family ligands signal primarily by triggering the phosphorylation of receptor-regulated Smads, which translocate to the nucleus and interact with diverse DNA-binding proteins to influence the transcription of target genes (Massague, 2000; Moustakas et al., 2001). Experiments using cultured neuroepithelial cells and cell lines have demonstrated that, upon treatment with TGFβ1, Foxg1 binds to a Smad3-containing complex and prevents it from inducing the expression of p21Cip1 (Cdkn1a - Mouse Genome Informatics), which encodes a cyclin-dependent kinase inhibitor (CKI) that is both a Smad3 target gene and an effector of TGFβ-mediated cell cycle arrest (Dou et al., 2000; Massague and Gomis, 2006; Rodriguez et al., 2001; Seoane et al., 2004). These findings indicate that, in cells that express Foxg1, Foxg1 can interact directly with Smad-containing transcriptional complexes to block the expression of TGFβ target genes.
Recently, we discovered that growth differentiation factor 11 (Gdf11), a member of the TGFβ superfamily, is an important component of an autocrine negative-feedback loop that regulates neurogenesis in the OE (Kawauchi et al., 2004; Kawauchi et al., 2005; Wu et al., 2003). Gdf11 is made by olfactory receptor neurons (ORNs) and late-stage neuronal progenitors (immediate neuronal precursors, or INPs) within the OE proper, and is present there as early as embryonic day 10.5 (E10.5) (Nakashima et al., 1999; Wu et al., 2003) (also see Results). Tissue culture studies show that Gdf11 can both arrest the division of INPs and promote the differentiation of INP progeny, effects that are accompanied by increased expression of the CKI p27Kip1 (Lander et al., 2009; Wu et al., 2003). Moreover, Gdf11-null mice show increased OE neurogenesis in vivo, with increased numbers of proliferating INPs and an extra layer of ORNs (Wu et al., 2003).
As a member of the activin branch of the TGFβ superfamily (Nakashima et al., 1999; Newfeld et al., 1999; Schneyer et al., 2008), Gdf11 signals via the same receptor-activated Smads (Smad2 and Smad3) as Tgfβ1 (Nomura et al., 2008; Tsuchida et al., 2007). This raises the possibility that some of the effects of Foxg1 on neurogenesis in the OE are due to antagonism of Gdf11 signaling. To test this, we analyzed OE development in Foxg1;Gdf11 compound mutant mice. We observed that deficits in neurogenesis in the Foxg1-/- OE, which are apparent from the earliest times in OE development, are substantially rescued in Foxg1-/-;Gdf11-/- mice, and even in Foxg1-/-;Gdf11+/- mice. Alterations in the expression of follistatin (Fst), which encodes a secreted Gdf11 antagonist, appear to account for part of this rescue. Overall, our results imply that the pro-neurogenic effects of Foxg1 in the OE are mediated, to a large degree, by antagonism of Gdf11. Interestingly, analysis of the same animals indicates that, in the cerebral cortex, Foxg1 acts through different targets.
MATERIALS AND METHODS
Animals
Gdf11-/- mice (Gdf11tm2/tm2; Gdf11+/tm2 is the second of two reported null alleles) (Wu et al., 2003) were obtained by intercrossing Gdf11+/tm2 mice maintained on a C57bl/6J background (Jackson Labs, Bar Harbor, ME, USA). Foxg1cre/+ mice, in which the Foxg1 coding sequence is replaced by the gene encoding Cre-recombinase (Cre) (Hebert and McConnell, 2000), were maintained on an outbred Swiss Webster background (Harlan, Indianapolis, IN, USA). Although expression of Cre recombinase in the Foxg1 locus has been shown to have some effects on telencephalon development when the allele is maintained on a congenic C57bl/6 background (Eagleson et al., 2007), when maintained on an outbred background, Foxg1cre/cre mice have been shown to have nervous system phenotypes identical to those observed in another Foxg1 null strain, Foxg1lacZ/lacZ (Duggan et al., 2008; Eagleson et al., 2007; Hanashima et al., 2007; Martynoga et al., 2005; Muzio and Mallamaci, 2005; Pratt et al., 2004). Therefore, we used Foxg1cre as a null allele and Foxg1cre/cre mice are designated Foxg1-/- hereafter. Foxg1+/-;Gdf11+/- mice were obtained by crossing Gdf11+/- females with Foxg1+/- male animals. Double knockouts (Foxg1-/-;Gdf11-/-) and compound mutants (Foxg-/-;Gdf11+/-) were obtained by intercrossing the resulting Foxg1+/-;Gdf11+/- mice. Mid-day of the day of vaginal plug detection was designated embryonic day 0.5 (E0.5). All protocols for animal use were approved by the Institutional Animal Care and Use Committee of the University of California, Irvine.
Tissue culture, in situ hybridization, immunofluorescence, histology and TUNEL staining
Dissected tissues were fixed, cryoprotected, embedded and cryosectioned (12-20 μm) as described (Murray et al., 2003). Hematoxylin staining was performed using Mayer's Hematoxylin solution (Sigma MHS 16-500, St Louis, MO, USA). In situ hybridization (ISH) was performed according to published protocols (Murray et al., 2003). cRNA probes used in this study were generated from: 1.5 kb mouse Foxg1 partial cDNA (Calof et al., 2002); 1.2 kb mouse Gdf11 partial cDNA (Wu et al., 2003); 1.0 kb mouse follistatin full-length cDNA (generous gift of M. M. Matzuk, Baylor College of Medicine, Houston, TX, USA); 0.6 kb mouse Sox2 partial cDNA (Kawauchi et al., 2004); 2.0 kb mouse Mash1 full-length cDNA (Guillemot and Joyner, 1993); 2.0 kb mouse Ngn1 cDNA (Ma et al., 1999); 391 bp mouse Ncam cDNA (Barthels et al., 1987); 687 bp mouse Otx2 partial cDNA (bp 894-1581 of GenBank accession number NM144841); full-length mouse Fgf8 ORF (Kawauchi et al., 2005); full-length of P1 bacteriophage Cre recombinase cDNA (Lewandoski et al., 1997); 675 bp mouse p21Cip1 cDNA (bp 380-1055 of GenBank accession number NM007669).
For pulse-fix analysis of bromodeoxyuridine (BrdU) incorporation, BrdU (Sigma) was injected intraperitoneally into pregnant dams (50 μg/gm body weight) and embryos collected 30 minutes later. Tissue (12 μm cryosections) was processed for anti-BrdU immunoreactivity as described (Kawauchi et al., 2005; Kim et al., 2005). TUNEL (deoxynucleotidyl Transferase-mediated dUTP Nick End Label) staining to detect DNA fragmentation in apoptotic cells was performed as described (Kawauchi et al., 2005), except that 20 μm cryosections were used.
Explant cultures from E14.5-E15.5 CD-1 mice (Charles River, Wilmington, MA, USA) were prepared as described (DeHamer et al., 1994; Wu et al., 2003). Purified recombinant human GDF11 (20 ng/ml; obtained by agreement with Wyeth Pharmaceuticals, Cambridge, MA, USA) was added at the beginning of the culture period. After 14 hours in vitro, explants were fixed as described (Wu et al., 2003) and processed for p21Cip1 immunoreactivity using monoclonal mouse anti-p21Cip1 (1:1000; Neomarker, Fremont, CA, USA: clone # AB-6 [HJ-21]), detected with unlabeled goat anti-mouse IgG (1:300; Southern Biotech, Birmingham, AL, USA), followed by Cy2-conjugated donkey anti-goat-IgG (1:100; Jackson ImmunoResearch, West Grove, PA, USA). Cell nuclei were counterstained with Hoechst 33342 (10 μg/ml, Sigma). For quantification, total migratory cells in a minimum number of 10 randomly chosen fields were counted for each condition (n=500-1000 cells per condition). The mean fluorescence intensity for each cell was quantified as the mean pixel density over the total area of the cell, measured using Zeiss AxioVision software (Carl Zeiss, Thornwood, NY, USA). The percentage of cells with mean fluorescence intensities of >6500 (`P21+ cells') was plotted for each condition (mean fluorescence intensity for >95% of cells in both control and no-first-antibody conditions was <6500).
Quantitative RT-PCR
Total RNA (4 μg) was isolated from OE turbinates (E16.5) or frontonasal processes (E11.5; OE and underlying mesenchyme, excluding forebrain) for each of 2-3 embryos of the indicated genotypes. Aurum Total RNA Mini Kit (BioRad, Hercules, CA, USA) was used for RNA isolation, and RNA was reverse transcribed using Superscript III reverse transcriptase and random oligonucleotide hexamers (Invitrogen, Carlsbad, CA, USA). Reactions were assembled using iQ SYBR Green Supermix (BioRad). Specificity of amplification was verified for each reaction by examination of the corresponding melt curve. Normalization of Gdf11 or Sox2 transcripts was carried out using Gapdh (run in parallel reactions for all samples) to control for the amount of tissue in each sample (Nguyen et al., 2005). Possible artefacts from amplification of genomic DNA were controlled for by processing samples prepared without reverse transcriptase; in all cases these proved negative. All PCR reactions were performed on an iQ5 iCycler (BioRad). Cycling conditions were 95°C for 3 minutes followed by 35 cycles of 95°C for 30 seconds, 58°C for 30 seconds and 72°C for 45 seconds. Primers used were: mouse Gdf11 exon3 (final concentration 200 nM), 5′-CTAAGCGCTACAAGGCCAAC-3′ and 5′-AGCATGTTGATTGGGGACAT-3′; mouse Sox2 3′UTR (100 nM), 5′-AAGGGTTCTTGCTGGGTTTT-3′ and 5′-AGACCACGAAAACGGTCTTG-3′; mouse Gapdh (150 nM), 5′-TTCACCACCATGGAGAAGGC-3′ and 5′-GGCATGGACTGTGGTCATGA-3′. PCR product sizes were 151 bp, 150 bp and 237 bp, respectively.
Delta cycle time (dCT) values were obtained for each reaction by subtracting the CT value of Gapdh for that reaction from the CT value of the tested transcript (Gdf11 or Sox2) run in parallel. Means and standard errors (s.e.m.) were calculated for dCTs obtained from duplicate or triplicate reactions for each biological sample. To obtain ddCT values, the mean dCT for a given transcript in wild type was subtracted from the mean dCT for that same transcript obtained from a given experimental sample. Fold-change relative to the wild-type value was calculated as 2-(ddCT). Errors in fold-change were propagated from errors of dCT values, as the square root of the sum of the squares of the error (s.e.m.) for the dCT value of each transcript.
RESULTS
Aberrant primary neurogenesis in the OE of Foxg1-/- mice
To understand when and how deficits in nasal cavity morphogenesis and OE neurogenesis occur in Foxg1-/- mice, ISH was performed using neuronal lineage markers at E11, when the olfactory pit (OP) first begins to invaginate. Even at this early stage of OE development, sometimes referred to as primary neurogenesis (Kawauchi et al., 2005), cells at all stages of the OE neuronal lineage are evident (Fig. 1A) (Beites et al., 2005; Kawauchi et al., 2004): neural stem cells can be identified by expression of Sox2; early committed neuronal progenitors that derive from these cells express the proneural gene Mash1 (Ascl1 - Mouse Genome Informatics); cells of the next lineage stage - referred to as immediate neuronal precursors (INPs) - express neurogenin 1 (Ngn1; Neurog1 - Mouse Genome Informatics); and differentiated ORNs that derive from INPs are identified by expression of Ncam (Ncam1 - Mouse Genome Informatics).
Fig. 1.

Failure of primary neurogenesis in Foxg1-/- OE. (A) Sections of olfactory epithelium (OE) from wild-type and Foxg1-/- mouse embryos at E11, showing the decrease in the numbers of cells expressing stage-specific neuronal markers. D, dorsal; V, ventral. (B) Apoptotic cells visualized by TUNEL labeling in E11 OE from wild-type and Foxg1-/- OE. For comparison, numbers were normalized to an area of 15,000 μm2, the average area of each section of Foxg1-/- OE at this age. Mean values ±s.d. of TUNEL+ cells per 15,000 μm2 OE are: wild type, 7.51±4.24; Foxg1-/-, 6.11±0.095. Data, which showed no significant difference (Student's t-test) (Glantz, 2005), were collected from two animals of each genotype. (C) Fgf8 and Foxg1 expression at E11. Fgf8 is expressed at the rim of the olfactory pit (OP) in wild type, and the pattern is unchanged in Foxg1-/- OE (arrowheads). The Foxg1 expression domain (detected by ISH to Cre), located in the central neurogenic zone of the OE, is reduced in Foxg1-/- OE. Scale bars: 100 μm.
In Foxg1-/- embryos at E11, cells expressing these lineage markers were present, but were greatly reduced in number (Fig. 1A). Even at this early age, OPs were greatly reduced in size; the domain of Sox2-expressing neural stem cells was correspondingly reduced compared with that of wild-type littermates. In addition, the concentric arrangement of gene expression domains in the OP, reflective of cells at different states of neuronal differentiation (Cau et al., 1997; Kawauchi et al., 2005), was altered in Foxg1-/- embryos. Although some cells in the dorsal recess of Foxg1-/- OPs do express neuronal markers, the number of these cells was dramatically reduced compared with wild type, even when the relative decrease in OP size of the mutant was taken into account: only a few Mash1+ early progenitors could be detected in a restricted dorsomedial domain, and the decrease in Ngn1+ INPs and Ncam+ ORNs cells was even more dramatic. By contrast, wild-type OPs at this stage displayed a clear concentric arrangement of neuronal cells: Mash1-expressing progenitors were present near the rim, with Ngn1-expressing INPs and Ncam-expressing ORNs at progressively more central zones. To assess whether the deficits in Foxg1-/- OE reflected an increase in cell death, TUNEL staining was performed. As shown in Fig. 1B, we found no significant difference in relative numbers or density of apoptotic cells in Foxg1-/- versus wild-type OE at E11. Together, these observations indicate that, in Foxg1-/- OE, development and differentiation of neuronal cells begins at the normal time. However, because there was no marked increase in apoptosis in Foxg1 nulls, some other process must result in the markedly hypoplastic structure of Foxg1-/- OE.
Foxg1 is unlikely to act through regulation of Fgf8 expression or signaling in the OE
In the telencephalon, Foxg1 regulates the expression of Fgf8, and it has been proposed that morphological deficits due to loss of Foxg1 may be explained in part by decreases in Fgf8 expression (Martynoga et al., 2005). Fgf8 is also essential for morphogenesis of the nasal cavity, and for neurogenesis within the OE (Kawauchi et al., 2005), but we saw no obvious change in Fgf8 expression in the rim of Foxg1-/- OPs (Fig. 1C). In fact, the size of the Fgf8-expression domain appeared to be essentially normal in Foxg1 mutants, despite a radical difference in OP size (Fig. 1C). We next compared the patterns of expression of Fgf8 and Foxg1 in the OPs of wild-type and Foxg1 mutant embryos, in the latter case using a probe for Cre to detect cells with endogenous Foxg1 promoter activity (see Materials and methods). In both wild-type and mutant embryos, we observed no overlap between the expression domains of Foxg1 and Fgf8 (Fig. 1C), which provides a probable explanation for the lack of a direct transcriptional relationship.
Although these data do not rule out the possibility that Foxg1 acts by influencing Fgf8 signaling, rather than Fgf8 expression, this too seems unlikely given that we observe no increase in apoptosis in the Foxg1-/- OE (see above and Fig. 1B), whereas a marked increase in apoptosis of Fgf8+/Sox2+ primary neural stem cells is a hallmark phenotype when Fgf8 is inactivated in and around the OE (Kawauchi et al., 2005). We thus infer that the disruption of nasal cavity morphogenesis and OE histogenesis (neurogenesis) in the Foxg1 mutant is likely to occur through a mechanism that is distinct from a disruption in Fgf8 expression and/or signaling.
Expression of Gdf11 and Foxg1 overlap in developing OE
As the first step toward testing the hypothesis that Foxg1 promotes neurogenesis via antagonism of Gdf11 signaling, we examined whether Foxg1 and Gdf11 are expressed at appropriate times and locations to interact in this way. At E10.5, widespread expression of Foxg1 mRNA was observed in the neuroepithelium of the invaginating OP, as well as in the developing forebrain (Fig. 2A). At E11.5, Foxg1 expression was absent from the distal rim of the OP, and became restricted to the central region, where neuronal differentiation is taking place (Fig. 2B) (Kawauchi et al., 2005). As development proceeds (E12.5 to E17.5), Foxg1 expression is maintained in the OE, but becomes progressively restricted to the basal compartment of the epithelium, where stem and neuronal progenitor cells are located (Fig. 2C-E) (Beites et al., 2005).
Fig. 2.

Expression of Foxg1 and Gdf11 in developing mouse OE. (A,B) Coronal sections through heads of E10.5 and E11.5 wild-type mice. Gdf11 expression is detected both in the OE and in a subset of cells in the mesenchyme, possibly migrating pioneer neurons (arrowheads). FB, forebrain; OE, olfactory epithelium. (C-E) Horizontal sections showing the OE in one-half of the nasal region (septum is at bottom) at E12.5, E14.5 and E17.5 in wild-type mice. The expression domains of Foxg1 and Gdf11 overlap, except in regions such as the anterior OE, which has ceased planar expansion at these ages. Insets show high magnification views of the OE at posterior regions of co-expression and anterior regions where co-expression has ceased (anterior is right; posterior, left). NC, nasal cavity; BL, basal lamina. Scale bars: 200 μm.
Gdf11 expression is first evident at E10.5 in the epithelium of the OP, and is also observed outside of the OE proper, in what are probably the migrating olfactory pioneer neurons that demarcate the pathway of the developing olfactory nerve (Fig. 2A) (Astic et al., 2002). By E12.5, Gdf11 expression expands to include the entire sensory neuroepithelium of the OE, and this pattern is maintained throughout development (Fig. 2C-E) (Nakashima et al., 1999; Wu et al., 2003).
Overall, the expression domain of Gdf11 overlaps substantially with that of Foxg1 throughout pre-natal development. As Gdf11 is known to both be expressed by, and act upon, OE neuronal progenitor cells (Lander et al., 2009; Wu et al., 2003), Foxg1-expressing cells are in the correct locations and at the right times to be targets of Gdf11.
Interestingly, the expression of Foxg1 throughout the lateral extent of the OE is not uniform. By E12.5, there are clear regional differences: Foxg1 expression is highest at locations such as the recesses of the developing turbinates and the posterior recess of the nasal cavity (at the junction of the septum and the turbinates). By comparing such patterns over time, it can be seen that the locations of high Foxg1 expression represent the sites where the OE is most actively expanding into the nasal mesenchyme. By contrast, Gdf11 expression is rather uniformly expressed wherever OE is present (Fig. 2C-E, and insets).
p21Cip1 expression is regulated by Gdf11 and Foxg1 in early OE development
Fox gene products affect cell proliferation and cell cycle dynamics in several cell types, including neural cells (Dou et al., 2000; Gomis et al., 2006; Hanashima et al., 2002; Martynoga et al., 2005), and it has been proposed that the major role of Foxg1 in cortical development is to promote neural precursor proliferation (Xuan et al., 1995). To quantify stem/progenitor cell proliferation in the OE of Foxg1-/- mice, we performed pulse-fix experiments at E11 and E12, using BrdU to label cells in S-phase (Fig. 3A). At both ages, the number of BrdU-immunopositive cells in any given section through the OE was much lower in Foxg1-/- mice, as was the overall size of the OE. Even if the discrepancy in size was corrected for by normalizing the number of BrdU-immunopositive cells to the linear distance along the basal lamina of the epithelium, 44% less labeling was still observed in Foxg1-/- OE than in wild type (at E11.0: wild type, 207±4 BrdU+ cells/mm OE; Foxg1-/-, 121±6 BrdU+ cells/mm OE).
Fig. 3.

Cell proliferation and p21Cip1 expression in mutant and wild-type OE. (A) BrdU pulse-fix experiments were performed as described in the Materials and methods, with pregnant dams injected at gestational day 11 or 12. Images show representative anti-BrdU immunostaining results. Graph shows quantification (mean±s.e.m.) of total number of BrdU+ cells per OE section at each age in Foxg1-/- embryos (blue) and wild-type littermates (pink). Histogram shows average OE area per section for mutants versus wild types. The number of BrdU+ cells is significantly lower in Foxg1-/- mutants at each age, as is the average area of OE per section (P<0.01, Student's t-test). Data were collected from two animals of each genotype at each age. (B) Coronal sections of an E10.5 Foxg1-/- embryo and a wild-type littermate, processed for ISH with a p21Cip1 probe. Dorsal is up; ventral, down. OE, olfactory epithelium. Scale bar: 100 μm. (C) Horizontal sections of an E13.5 Gdf11-/- embryo and a wild-type littermate, processed as in B. nc, nasal cavity; A, anterior; P, posterior. Scale bar: 100 μm. (D, top) Immunofluorescence staining of migratory OE neuronal cells in explant cultures (after 14 hours in vitro), grown with or without Gdf11, then processed for anti-p21Cip1 immunoreactivity. (Bottom) Quantification of results from a typical culture experiment. Percentages of p21Cip1+ cells were: 2.5% of 587 counted cells in control cultures; 14.3% of 1065 counted cells in GDF11-treated cultures. P<0.05, Student's t-test.
Interestingly, this change in BrdU labeling in Foxg1-/- OE was accompanied by an expansion of expression of the CKI p21Cip1 (Fig. 3B). At E10.5, expression of p21Cip1, which at later stages of OE development correlates with neuronal differentiation (Kastner et al., 2000; Legrier et al., 2001), was normally confined to the rim of the invaginating nasal pit (Fig. 3B). In Foxg1-/- OE, however, p21Cip1 expression was expanded to include both the rim and the central region of the OE. This finding suggests that increased p21Cip1 expression in Foxg1-/- OE might contribute to the alterations in the number of cycling cells and the deficits in neuronal cell differentiation observed in these mutants.
In cultured neural cells and cell lines, it has been shown that Foxg1 can repress p21Cip1 induction effected by TGFβ signaling (Dou et al., 2000; Rodriguez et al., 2001; Seoane et al., 2004). Because Gdf11 acts through the same intracellular effectors as TGFβ, we investigated whether Gdf11 controls the expression of p21Cip1 in the OE. Two types of experiments were done. First, we performed ISH for p21Cip1 in Gdf11 nulls and their wild-type littermates. As shown in Fig. 3C, p21Cip1 levels were greatly reduced in the OE of E13.5 Gdf11-/- animals, implying that Gdf11 is a positive regulator of p21Cip1 expression in vivo. Second, we cultured OE explants from wild-type embryos at a similar age (E15.5), and examined expression of p21Cip1 by immunofluorescence after 14 hours' growth in the presence or absence of recombinant Gdf11. The results are shown in Fig. 3D. As described previously, neuronal cells (neuronal progenitors and immature ORNs) comprise virtually all of the migratory cells in explant cultures of OE purified from mouse embryos at this age (Calof and Chikaraishi, 1989; DeHamer et al., 1994; Mumm et al., 1996). Figure 3D shows that only a small percentage (2.5%) of neuronal cells in untreated control cultures showed significant p21Cip1 immunoreactivity. By contrast, the percentage of p21Cip1-immunoreactive neuronal cells was more than fivefold greater in GDF11-treated cultures (14.3%). Thus, GDF11 treatment causes an increase in p21Cip1 expression in OE neuronal progenitors and/or immature ORNs.
Inactivation of Gdf11 rescues neurogenesis in Foxg1-/- OE
The presence of Gdf11 and Foxg1 at similar times and locations in the OE, the known ability of Foxg1 to inhibit the induction of TGFβ pathway target genes that are also induced by Gdf11, and the oppositely directed effects of Gdf11 and Foxg1 mutations on OE neurogenesis, all raise the possibility that Foxg1 acts via inhibition of Gdf11 activity. To assess this directly, we compared OE development in wild type, Foxg1-/-, Gdf11-/- and Foxg1-/-;Gdf11-/- double mutants. Morphology and neuronal lineage markers were first examined at birth (the latest age to which both strains survive) in sagittal sections at equivalent mediolateral levels (Fig. 4A). Figure 4B-D shows ISH analysis of littermate wild-type, Foxg1-/- and Foxg1-/-;Gdf11-/- animals, performed using a probe to Ngn1 to highlight the neuronal progenitor layer of the OE (Beites et al., 2005; Wu et al., 2003). As shown in Fig. 4B, the nasal cavity and turbinate cartilage underlying the nasal mucosa are easily visualized in sections from wild-type mice, with Ngn1+ progenitor cells forming a distinct layer in the basal region of the OE proper. In sections from Foxg1-/- mice, however, no olfactory turbinate structures were observed: Only a small cavity filled with serous gland tubules was seen, and no expression of Ngn1 could be detected (Fig. 4C). By contrast, both the gross morphology of nasal structures and the microscopic structure of the OE are significantly rescued in Foxg1;Gdf11 double mutants (Fig. 4D). The double mutants have recognizable olfactory turbinates that are lined with OE, and this OE contains Ngn1+ cells in their normal, basal, location. This OE surrounds a substantial nasal cavity that is located dorsal and posterior to the major maxillary incisor, as is the case in wild-type animals (compare Fig. 4B and 4D). Thus, the absence of Gdf11 results in substantial rescue of the defects in nasal cavity morphogenesis and OE neuroepithelial development observed in Foxg1-/- mice.
Fig. 4.

Rescue of the Foxg1-/- OE phenotype by loss of Gdf11. (A) Cartoon of normal frontonasal structures in mice at P0, shown as a mid-sagittal section. Red square indicates the region photographed for ISH images shown in B-D. G, serous gland; I, right upper incisor; OB, olfactory bulb; OE, olfactory epithelium; T, turbinate bone; NC, nasal cavity. (B) ISH for Ngn1 to show OE neuronal cells in wild-type animals at P0. (C) Olfactory turbinate structures and Ngn1-expressing OE are not observed in Foxg1-/- animals. (D) Foxg1-/-;Gdf11-/- mice show recovery of turbinate structures and OE expressing Ngn1. Note that there is no OB present in either Foxg1-/- or Foxg1-/-;Gdf11-/-; note also that the telencephalon is drastically reduced in Foxg1-/- mice and is not rescued by loss of Gdf11 (see Results). Scale bar: 500 μm.
To determine when such rescue occurs, we examined OE neuronal lineage markers in horizontal sections of heads from E13.5 embryos that were null for Gdf11, Foxg1, or both genes, and compared these with sections from wild-type littermates. The results are shown in Fig. 5. In wild-type mice at E13.5, cells expressing all four major stage-specific markers (Sox2, Mash1, Ngn1 and Ncam) were present in the OE (Fig. 5A). In Gdf11-/- mice at this age, the OE was essentially identical to wild type; increases in neuronal cells above wild-type numbers was not evident until a later stage of development in these mutants (Fig. 5B) (cf. Wu et al., 2003). In contrast to wild-type and Gdf11-/- embryos, Foxg1-/- embryos at E13.5 have essentially no OE (Fig. 5C). When viewed at a dorsoventral level equivalent to the wild-type example (note presence of retina in all sections), the Foxg1-/- embryo shown in Fig. 5C shows reduction of the entire frontonasal region, an absence of nasal cavities, and an aberrant (apparently enlarged) optic recess of the third ventricle. When Foxg1-/- embryos were also made null for Gdf11, however, both the OE and the frontonasal region were significantly rescued at E13.5 (Fig. 5D). The OE of Foxg1-/-;Gdf11-/- embryos was of normal thickness, and contained cells expressing all major neuronal lineage markers (Fig. 5D). Moreover, a developing nasal cavity could be seen, as well as lateral folds of mesenchyme covered by OE, evidence that basic turbinate structures are also rescued in the double mutant (Fig. 5D). Thus, the failure of both OE neurogenesis and nasal cavity morphogenesis observed in Foxg1-/- animals is significantly rescued by E13.5 when Gdf11 is absent.
Fig. 5.

Significant rescue of the Foxg1-/- OE phenotype occurs by E13.5. Horizontal sections through heads of E13.5 wild-type and Gdf11-/- embryos, hybridized with probes for indicated OE neuronal lineage markers. (A,B) Identical patterns of expression are observed in wild-type and Gdf11-/- OE at this age. OE, olfactory epithelium; nc, nasal cavity; R, retina; Sep, septum; T, turbinate. (C) Foxg1-/- embryos lack OE, a nasal cavity, and all OE neuronal lineage markers by E13.5. III, ventral portion of third ventricle; OC, optic chiasm. (D) Substantial rescue of the OE and nasal cavity structures, as well as cells expressing all OE neuronal lineage markers, is observed in Foxg1-/-;Gdf11-/- embryos. Scale bar: 200 μm.
Gdf11 dosage regulates the ability of Foxg1 to maintain OE neurogenesis
Genes of the TGFβ superfamily often show dose dependence in their effects on development (Dunn et al., 1997; Eldar et al., 2002; Lawson et al., 1999; Sutherland et al., 2003). We tested whether Gdf11 might show such activity in its effects on OE development. Mice null for Gdf11, Foxg1, as well as Foxg1-/-;Gdf11+/- and Foxg1-/-;Gdf11-/- mice, were examined at E16.5 (Fig. 6). At this stage in normal development, olfactory turbinates are well developed and all cell types in the OE can be recognized easily by their laminar position and molecular marker expression; at this stage, the OE also possesses an apical layer of sustentacular cells, the intrinsic glial cells of the OE (Beites et al., 2005; Cuschieri and Bannister, 1975a; Cuschieri and Bannister, 1975b; Murray et al., 2003; Smart, 1971). As demonstrated previously (Wu et al., 2003), Gdf11 nulls at this age have greater numbers of Ngn1-expressing INPs and Ncam-expressing ORNs than wild types, but show no obvious changes in the number of Mash1-expressing early neuronal progenitors (Fig. 6A,B). We have found that Otx2, an orthodenticle homolog that is expressed in the developing olfactory placode (Mallamaci et al., 1996), is a marker for sustentacular cells at E16.5 and beyond in the mouse OE. Figure 6B shows that the layer of Otx2-expressing sustentacular cells appears to be complete in Gdf11 mutants, as expected.
Fig. 6.

Rescue of OE neurogenesis in Foxg1-/- mutants is dependent on Gdf11 gene dosage. ISH for OE neuronal lineage markers (Mash1, Ngn1 and Ncam) and sustentacular cells (Otx2), performed on horizontal sections through the OE of E16.5 wild-type and mutant littermates. Insets show high magnification views of septal OE. (A,B) OE and cell types within it are similar in wild-type and Gdf11-/- mice at this age, except that Ngn1- and Ncam-expressing cell layers (and hence OE overall) are thicker, as reported previously (Wu et al., 2003). (C) No OE structure (apart from a truncated septum), nor any cell type-specific markers, are evident in sections from Foxg1-/- mice at the same dorsoventral level. (D) Loss of one Gdf11 allele (Foxg1-/-;Gdf11+/-) rescues all cell types in the OE, and the OE appears to be of normal thickness, although planar expanse of the OE and morphogenesis of nasal cavity are clearly deficient in the compound mutant. (E) Rescue is more pronounced in Foxg1-/-;Gdf11-/- double mutants, particularly in terms of OE planar expanse and nasal cavity morphogenesis. For all panels, posterior is left, anterior is right. Sus, sustentacular cells; BL, basal lamina. Scale bar: 400 μm.
In contrast to what was seen in wild-type and Gdf11-/- embryos at E16.5, the OE neuroepithelium itself, the nasal cavity, and molecular markers of OE neuronal and sustentacular cells were largely absent from Foxg1-/- embryos at this age, despite the fact that a septal structure could often be observed (Fig. 6C) (the presence of neural retina in all sections indicates that the horizontal sections shown have been taken at approximately the same dorsoventral level). Notably, when just one allele of Gdf11 was inactivated in Foxg1 nulls (Foxg1-/-;Gdf11+/- embryos), bilateral nasal cavities formed and were easily recognizable in the compound mutants at E16.5 (Fig. 6D). Although the surfaces of the nasal cavities were not as elaborately folded as in wild-type OE, they were lined by an OE of normal thickness. Moreover, the OE in these compound mutants contained cells expressing all neuronal and sustentacular markers tested, and these cells were present in their appropriate apical-basal positions within the OE (compare Fig. 6D to 6A). Indeed, even the layer of Ncam-expressing ORNs appeared to be as thick as that seen in wild types (Fig. 6D, inset).
When two alleles of Gdf11 were inactivated, the rescue of nasal cavity morphogenesis and OE neurogenesis was even more pronounced. As shown in Fig. 6E, cells expressing neuronal and sustentacular markers were found in their appropriate layers, and turbinates had developed as folds of differentiating mesenchyme that protrude into the nasal cavities; these were substantially larger and more elaborate in shape than those seen in compound mutants in which only one allele of Gdf11 was inactivated. Interestingly, the pattern of Otx2 expression in the layer of sustentacular cells of Foxg1-/-;Gdf11-/- OE was noticeably diffuse when compared with that observed in wild-type and Gdf11-/- OE (compare Fig. 6E with 6A,B). This observation suggests that Foxg1 may be involved in sustentacular cell differentiation.
Altogether, the observations described above support the conclusion that Foxg1 exerts its effects on OE neurogenesis through quantitative antagonism of Gdf11 activity. In view of the known interaction of Foxg1 with Smad transcriptional complexes, it is reasonable to propose that the effects of Foxg1 are mediated through a direct, cell-autonomous influence on signaling downstream of Gdf11. However, as described below, additional effects may come into play as well.
Loss of follistatin expression in Foxg1-/- nasal mucosa and its rescue by loss of Gdf11
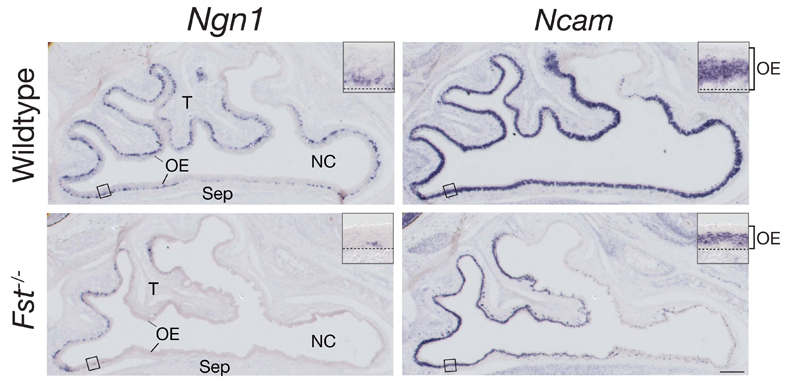
The secreted protein follistatin (Fst) is an antagonist of activins and Gdf11 that competes for binding of these factors to their receptors (Gamer et al., 2001; Nakamura et al., 1990; Schneyer et al., 1994; Schneyer et al., 2008; Wu et al., 2003). Fst is expressed in the nasal mucosa, both in the OE proper and in its underlying mesenchymal stroma (Lander et al., 2009; Wu et al., 2003); during embryonic development, stromal expression of Fst is particularly strong (Fig. 7A). The importance of Fst as an endogenous inhibitor of GDF11 is evidenced by the phenotype of Fst-/- mice, which display an OE at birth that is very thin and that is markedly depleted of neurons (Wu et al., 2003).
Fig. 7.

Downregulation of Fst expression in Foxg1-/- nasal mucosa is rescued by loss of Gdf11. (A) ISH for Fst performed on coronal sections through heads of wild-type and Foxg1-/- mice at E10.5 and E12.5, and on horizontal sections at E16.5. At E16.5, when Fst is expressed in both the OE and stroma of nasal mucosa in wild types (A, top right panel), Fst expression is undetectable in Foxg1-/- embryos in rare instances when remnants of nasal mucosa are observed (A, bottom right panel). NE, nasal epithelium; OE, olfactory epithelium; BL, basal lamina; nc, nasal cavity; Str, stroma; fb, forebrain. Scale bars: 100 μm in E10.5 and E12.5; 50 μm in E16.5. (B) Fst expression is restored in the OE and underlying stroma of Foxg1-/-;Gdf11+/- and Foxg1-/-;Gdf11-/-. Scale bar: 50 μm.
Intriguingly, we found that Foxg1-/- embryos lack Fst expression in and around the OE from the earliest developmental stages (Fig. 7A). Even when OE remnants could be detected at late stages in Foxg1-/- embryos (e.g. example in Fig. 7A at E16.5), no Fst expression was detected. These results suggest an additional mechanism by which Foxg1 could antagonize Gdf11 activity: by promoting expression of Fst, a Gdf11 antagonist, Foxg1 could lower the effective concentration of Gdf11 in the extracellular milieu. Although this latter activity might contribute to the Foxg1-/- phenotype in the OE, however, it cannot explain it entirely, as that phenotype is both qualitatively and quantitatively different from the OE phenotype observed in Fst-/- mice (see Fig. S1 in the supplementary material; see also Discussion). It seems more likely that the Foxg1-/- phenotype arises as the result of a combination of intracellular (cell-autonomous) and extracellular (non-cell-autonomous) effects. This may help to explain why the phenotype is so severe.
Interestingly, both stromal and intraepithelial expression of Fst were completely rescued not only in Foxg1-/-;Gdf11-/- embryos, but also in compound Foxg1-/-;Gdf11+/- mutants in which only one allele of Gdf11 was inactivated (Fig. 7B). This demonstrates that Foxg1 is not itself required for Fst expression, i.e. Foxg1 does not itself induce Fst. The most parsimonious explanation is that it is the OE itself that induces Fst in surrounding tissue, with Foxg1 being required to generate an OE that is competent to do so. Exactly what signal from the OE induces Fst is unknown, but from the data in Fig. 7B, we can rule out Gdf11.
Does Foxg1 regulate Gdf11 expression?
In view of the fact that Foxg1 is a transcriptional regulator, we also considered the possibility that a third mechanism - a repressive effect of Foxg1 on Gdf11 expression - might also be at play in the OE. To investigate this, we measured the expression of Gdf11 in the different mutants discussed above. The results of these experiments, shown in Fig. 8, revealed several interesting findings. First, we found no evidence that Gdf11 autoregulates its own expression, as is the case for some TGFβ superfamily members (Fig. 8A) (Chen and Schier, 2002; Forbes et al., 2006). Thus, in a normal (wild-type) genetic background, inactivation of one allele of Gdf11 led to a reduction in Gdf11 transcript levels to 35-49% of wild type; loss of both alleles led to essentially undetectable expression of Gdf11 (Fig. 8A). Second, in the frontonasal process of E12 Foxg1-/- embryos, where loss of OE tissue was already significant, Gdf11 expression was still detectable in the remaining OE by ISH (Fig. 8B).
Fig. 8.

Analysis of Gdf11 mRNA expression in the OE of embryos of different genotypes. (A) Gdf11 does not regulate its own transcription. Gdf11 and Gapdh transcript levels at E16.5 and E11.5 were quantified by Q-RT-PCR, and dCT and ddCT values with errors were calculated as described in the Materials and methods. For presentation purposes, data are normalized to wild-type values. Statistics [Dunnett's test for multiple comparisons against a single control (DT) (Glantz, 2005)] were performed using mean dCT values and corresponding errors (s.e.m.), which were: E16.5, wild type=5.32±0.0233; Gdf11+/-=6.89±0.0523, Gdf11-/-=13.53±0.4933; E11.5, wild type=5.22±0.200, Gdf11+/-=6.24±0.009, Gdf11-/-=12.22±0.142. (B) Gdf11 is expressed in Foxg1-/- OE at E12.0. ISH for Gdf11 was performed on coronal cryosections of wild type and Foxg1-/-. Scale bar: 50 μm. (C-E) Relative Gdf11 and Sox2 expression values in OE from E11.5 embryos of different genotypes. Gdf11, Sox2, and Gapdh transcript levels were quantified by Q-RT-PCR as described in the Materials and methods. In C and D, Gdf11:Gapdh and Sox2:Gapdh expression levels are normalized to wild-type values for presentation purposes. Statistics (DT) were performed using mean dCT values and corresponding errors (s.e.m.), which were as follows: Gdf11:Gapdh, wild type=5.22±0.200, n=3; Gdf11+/-=6.24±0.009, n=2; Gdf11-/-=12.22±0.142, n=3; Foxg1-/-=6.07±0.088, n=3; Foxg1-/-;Gdf11+/-=6.39±0.422, n=3. Sox2:Gapdh: wild type=6.81±0.217, n=3; Gdf11+/-=6.30±0.116, n=3; Gdf11-/-=6.42±0.230, n=3; Foxg1-/-=9.09±0.083, n=3; Foxg1-/-;Gdf11+/-=8.85±0.176, n=3. (E) Gdf11 expression plotted as the ratio of the mean Gdf11 transcript level to the mean Sox2 level. Values that differ significantly from wild type (P<0.05, DT) are denoted by asterisks.
Third, Q-RT-PCR experiments, performed to determine transcript levels of Gdf11 in E11.5 frontonasal tissue (this age was chosen because there is still a reasonable amount of OE remaining in Foxg1-null animals), showed that Gdf11 transcript levels in Foxg1-/- and Foxg1-/-;Gdf11+/- mutants were significantly lower than in wild type (Fig. 8C). This was not surprising, given that Gdf11 is mainly expressed in the OE, and there is substantially less OE tissue in such mutants compared with wild type. Indeed, Q-RT-PCR showed that levels of Sox2, a marker for neuroepithelium (Fig. 1A), were also markedly decreased in Foxg1-/- and Foxg1-/-;Gdf11+/- mutants (Fig. 8D). By normalizing Gdf11 transcript levels to Sox2 transcript levels in the same samples, we could attempt to correct for the differing amounts of OE in different samples. The results (Fig. 8E) indicated that Gdf11 levels were 2- to 3-fold higher, per unit of OE, in Foxg1 nulls than in wild-type animals.
Defects in cerebral cortex development in Foxg1-/- embryos are not rescued by loss of Gdf11
Because Gdf11 is expressed in some regions of the telencephalon (Nakashima et al., 1999), we considered the possibility that absence of Gdf11 might also rescue some of the defects in telencephalic neurogenesis that are observed in Foxg1 mutants (Hanashima et al., 2007; Hanashima et al., 2004; Hanashima et al., 2002; Hebert and McConnell, 2000; Xuan et al., 1995). When we examined Foxg1-/-;Gdf11-/- embryos, however, we found no such rescue. This is illustrated in Fig. 9A, which shows Hematoxylin/Eosin-stained horizontal sections through the brains of E13.5 mice of the three different genotypes. As noted previously (Xuan et al., 1995), the developing cerebral hemispheres were much smaller in Foxg1-/- embryos than in wild types and, instead of a smooth cortical neuroepithelium, the cortical surface was buckled and uneven, and cortical thickness differed in different areas. In Foxg1-/-;Gdf11-/- double mutants, there was no rescue of this phenotype: the cerebral hemispheres were still much smaller than in wild-type embryos, and the cortex showed the same structural aberrations that are apparent in Foxg1-/- embryos. Thus, absence of Gdf11 fails to rescue the defects in cortical development seen in the Foxg1-null mouse.
Fig. 9.

Absence of Gdf11 does not rescue defects in Foxg1-/- cerebral cortex. (A) Hematoxylin and Eosin staining of horizontal sections through the brains of wild type, Foxg1-/-, and Foxg1-/-;Gdf11-/- double mutants at E13.5. The cortex is severely reduced in Foxg1-/- embryos; absence of Gdf11 does not rescue this phenotype. Scale bar: 200 μm. (B) Expression of Foxg1 and Gdf11 in coronal sections through developing mouse brain. Foxg1 is abundantly expressed in the telencephalon (except for the cortical hem) at E11.5 (expression boundary indicated by arrowheads). Gdf11 expression is apparent in the ventral telencephalon and OE at E11.5, but by E12.5 is restricted to ventral midline of the telencephalon and the nascent hippocampus (arrows). At E15.5, no Gdf11 expression is apparent in the rostral telencephalon, whereas Foxg1 levels are high, especially in dorsal areas. Scale bars: 400 μm. C, cortex; CB, cerebral cortex; CH, cortical hem; CP, cortical plate; Di, diencephalon; H, hippocampus; LV, lateral ventricle; OE, olfactory epithelium; S, striatum; POA, preoptic area; SVZ, subventricular zone.
To understand why an absence of Gdf11 should fail to rescue cortical development, while OE development is rescued so dramatically, we performed ISH to examine the expression of Gdf11 and Foxg1 in the developing forebrain from E11.5 through E15.5. As shown in Fig. 9B, Foxg1 was initially expressed throughout the telencephalon, but gradually became restricted to more dorsal structures, including the developing cerebral cortex. By contrast, Gdf11 expression at E11.5 was primarily found in a subset of the most ventral cells, but gradually disappeared by E15.5. Thus, cells that express Gdf11 appear not to be located in the vicinity of the developing cortical neuroepithelium.
DISCUSSION
Foxg1 activity supports a self-sustaining neurogenic network
The experiments described above demonstrate that Foxg1 promotes OE neurogenesis in large part through the antagonism of Gdf11. Of the possible mechanisms by which this effect may be achieved, the simplest, and most direct, involves the known interaction of Foxg1 with Smad signaling complexes to inhibit Smad-dependent transcription (Dou et al., 2000; Massague and Gomis, 2006; Rodriguez et al., 2001; Seoane et al., 2004). Although that interaction was discovered through the study of p21Cip1 induction by TGFβ1 in cultured cells and cell lines, Gdf11 acts through the same Smads as TGFβ1 (Nomura et al., 2008; Tsuchida et al., 2007), and, we have shown that Gdf11 induces p21Cip1 in cells of the OE (Fig. 3). Recent studies in the zebrafish, in which morpholino oligonucleotides were used to reduce Foxg1 levels in a mosaic fashion (Duggan et al., 2008), also showed that Foxg1 influences OE neurogenesis in a cell-autonomous fashion, a result opposite to what would have been expected if Foxg1 acts by controlling the expression of a secreted growth factor, such as an FGF.
The fact that we observe a higher level of Gdf11 transcripts, relative to Sox2 transcripts, in Foxg1 mutant OE (Fig. 8E) raises the additional possibility that Foxg1 regulates the expression of Gdf11 at the transcriptional level, although we cannot rule out alternative interpretations of the data. For example, loss of Foxg1 in Foxg1-/- OE might increase the number of Gdf11-expressing cells (relative to Sox2-expressing cells), rather than the level of Gdf11 transcripts per cell. Conversely, loss of Foxg1 might decrease the expression of Sox2. Even if Foxg1 does regulate Gdf11 transcription, the mechanism need not be direct (i.e. it could be mediated through other Foxg1 target genes). Likewise, loss of expression of the extracellular Gdf11 antagonist Fst in and around the OE of Foxg1-/- mice almost certainly reflects an indirect effect, as Foxg1 is not expressed in the stroma (where most Fst expression occurs), and Fst expression can be achieved in the complete absence of Foxg1 OE, by removing copies of Gdf11.
Regardless of the underlying mechanism(s), however, the finding that loss of Foxg1 is capable of leading to increased Gdf11 signaling, increased Gdf11 expression, and decreased expression of a Gdf11 antagonist collectively suggest that the relationship between Gdf11 activity and Foxg1 activity is a highly sensitive one.
At the same time, a highly sensitive relationship also appears to exist between Gdf11 activity and the capacity of OE cells for neurogenesis. The fact that removal of a single Gdf11 allele transforms a Foxg1-/- embryo from one in which no OE or nasal cavity develops, into one with an OE of normal thickness and composition, suggests that there is a threshold level of Gdf11 activity below which neurogenesis proceeds fairly normally, and above which neurogenesis fails completely.
Threshold responses to signaling molecules usually imply cooperativity (or some other form of ultrasensitivity), such that a doubling in gene dosage produces more than a doubling in signaling. Because Gdf11 does not appear to regulate its own expression (Fig. 8A), we believe the likely source of ultrasensitivity lies elsewhere. If, as we suggest, the OE induces expression of Fst in its underlying stroma, then a positive-feedback loop emerges: an increase in Gdf11 activity would lead to a decrease in OE size, which would cause a decrease in Fst expression, which would in turn cause an increase in Gdf11 activity. A decrease in Gdf11 activity would be similarly self-enhancing. According to this view, Gdf11 in the embryonic OE is less of a graded regulator of neuronal production than a switch-like controller of a self-sustaining program of neurogenesis, with Foxg1 regulating when and where the switch is thrown.
Foxg1 and olfactory epithelium morphogenesis
During embryonic development of the OE, the process of neurogenesis can be viewed as serving two distinct ends: histogenesis and morphogenesis. By histogenesis we mean the generation of an appropriate complement and number of OE cells at each location along the epithelium. By morphogenesis we mean the planar growth and invagination of the epithelium to produce a deep, characteristically folded nasal cavity. In Foxg1-/- embryos, both processes fail from early stages. Yet when Foxg1 mutants are rescued through loss of Gdf11, the two processes are restored to very different degrees. Histogenesis is nearly normal in both Foxg1-/-;Gdf11-/- and Foxg1-/-;Gdf11+/- mutants, but morphogenesis is still impaired in Foxg1-/-;Gdf11-/- mutants, and even more so in Foxg1-/-;Gdf11+/- animals (Figs 5 and 6). These results stand in marked contrast to the phenotype of Fst-/- mice, which exhibit severely defective histogenesis (an extremely thin OE), but relatively normal morphogenesis (see Fig. S1 in the supplementary material). How could excessive Gdf11 activity disrupt morphogenesis in one situation but not the other?
We believe that the answer lies in the expression pattern of Foxg1 in the developing OE. As shown in Fig. 2, Foxg1 is initially found throughout the OE, but soon becomes localized primarily to those areas in which planar expansion of the epithelium is occurring. This implies that Gdf11 levels in most of the OE are normally low enough to permit a constant, steady accumulation of ORNs, driving normal histogenesis. At locations where Foxg1 is strongly expressed, however, potent inhibition of Gdf11 signaling might allow the tissue to switch into a mode of more dramatic expansion. Recently, we used mathematical modeling to show that the only change needed to convert a tissue that adds cells at a constant rate to one that adds cells at an exponentially increasing rate is adjustment of the `replication probability' of a stem or transit-amplifying cell to a level above 50% (Lander et al., 2009). As Gdf11 demonstrably lowers INP replication probabilities (Lander et al., 2009; Wu et al., 2003), the idea that a sufficient reduction in Gdf11 activity could switch the OE into an exponential growth mode is very plausible. In the Fst-/- mutant, excessive levels of free extracellular Gdf11 everywhere could account for a reduction in steady-state neurogenesis throughout the OE (and thereby a very thin epithelium), but in regions of Foxg1 expression, even this higher level of Gdf11 signaling might still be effectively blocked (through the cell autonomous action of Foxg1 on Gdf11 signaling). The result would be that planar expansion, and thus morphogenesis, could proceed normally in the Fst-/- mutant. By contrast, in the Foxg1-/- OE, unopposed Gdf11 activity would occur everywhere, leading to a failure of both histogenesis and morphogenesis.
In summary, we propose that a major role of Gdf11 is to set a balance between the proliferation and differentiation of progenitor cells, whereas the primary role of Foxg1 is to tip that balance in favor of tissue expansion. A schematic depiction of the balancing act between Gdf11 and Foxg1 activities is presented in Fig. 10. It will be interesting to see whether the model of Foxg1 action through regulation of TGFβ family signaling applies to the OE only, or to other developing neural structures as well. Although Foxg1 clearly does not act through Gdf11 in the cerebral cortex (Fig. 9), a potential role for other TGFβ family ligands cannot be ruled out. Alternatively, it may be that, in some neural structures, the positive regulation of pro-neurogenic signals, such as FGFs, is of greater importance than the negative regulation of anti-neurogenic ones.
Fig. 10.
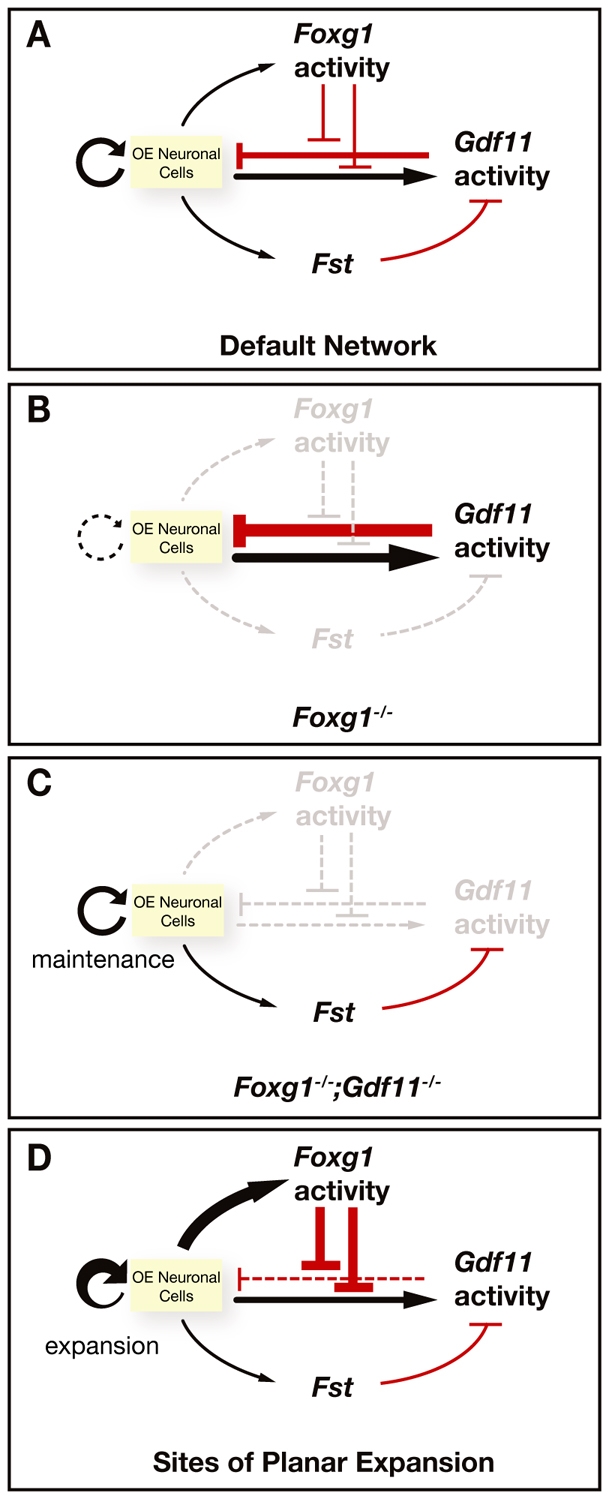
Schematic model of Foxg1-Gdf11 interactions controlling OE neurogenesis. (A) In wild-type OE, Foxg1 and Gdf11 are both produced by OE neuronal cells, but Foxg1 pro-neurogenic activity antagonizes both the anti-neurogenic activity of Gdf11 and the production of Gdf11 by OE neuronal cells. Fst is also expressed by OE neuronal cells, and Fst action antagonizes Gdf11 activity. This default network of gene activities controls the normal steady-state level of neurogenesis in the OE. (B) In Foxg1-/- OE, Foxg1 activity is absent, Fst expression is downregulated, and Gdf11 expression is upregulated, resulting in hypersensitivity of the frontonasal region and OE to the action of Gdf11. Both OE neurogenesis and planar expansion of the OE fail. (C) In the Foxg1-/-;Gdf11-/- double mutant, Fst expression is restored and histogenesis (neurogenesis) within the OE is rescued, as the anti-neurogenic activity of Gdf11 is now removed and any similar anti-neurogenic factors are antagonized by Fst. (D) Foxg1 activity strongly inhibits both Gdf11 activity and expression, which would allow the OE to undergo planar expansion in sites where Foxg1 is highly expressed in wild-type OE (e.g. posterior recess of the nasal cavity). Once expansive growth is finished, Foxg1 expression is downregulated (e.g. in the anterior septum), and OE neurogenesis returns to its default state.
Supplementary material
Supplementary material for this article is available at http://dev.biologists.org/cgi/content/full/136/9/1453/DC1
Supplementary Material
Acknowledgments
We thank members of the Calof laboratory for critical comments on the manuscript and Mr Dan L. Phan and Ms Michelle Hoang for technical support. We thank Drs Jean Hebert and Susan McConnell for providing Foxg1cre mice, and Dr Martin Matzuk for providing the Fst probe and Fst+/- mice.
This work was funded by NIH grant DC03583 (to A.L.C.) and P50 GM0-76516 (to A.D.L. and A.L.C.). R.S. is supported by a pre-doctoral supplement to NIH grant 1P01-HD052860 (sub2, to A.D.L. and A.L.C.). Deposited in PMC for release after 12 months.
References
- Astic, L., Pellier-Monnin, V., Saucier, D., Charrier, C. and Mehlen, P. (2002). Expression of netrin-1 and netrin-1 receptor, DCC, in the rat olfactory nerve pathway during development and axonal regeneration. Neuroscience 109, 643-656. [DOI] [PubMed] [Google Scholar]
- Barthels, D., Santoni, M. J., Wille, W., Ruppert, C., Chaix, J. C., Hirsch, M. R., Fontecilla-Camps, J. C. and Goridis, C. (1987). Isolation and nucleotide sequence of mouse NCAM cDNA that codes for a Mr 79,000 polypeptide without a membrane-spanning region. EMBO J. 6, 907-914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beites, C. L., Kawauchi, S., Crocker, C. E. and Calof, A. L. (2005). Identification and molecular regulation of neural stem cells in the olfactory epithelium. Exp. Cell Res. 306, 309-316. [DOI] [PubMed] [Google Scholar]
- Calof, A. L. and Chikaraishi, D. M. (1989). Analysis of neurogenesis in a mammalian neuroepithelium: proliferation and differentiation of an olfactory neuron precursor in vitro. Neuron 3, 115-127. [DOI] [PubMed] [Google Scholar]
- Calof, A. L., Bonnin, A., Crocker, C., Kawauchi, S., Murray, R. C., Shou, J. and Wu, H. H. (2002). Progenitor cells of the olfactory receptor neuron lineage. Microsc. Res. Tech. 58, 176-188. [DOI] [PubMed] [Google Scholar]
- Carlsson, P. and Mahlapuu, M. (2002). Forkhead transcription factors: key players in development and metabolism. Dev. Biol. 250, 1-23. [DOI] [PubMed] [Google Scholar]
- Cau, E., Gradwohl, G., Fode, C. and Guillemot, F. (1997). Mash1 activates a cascade of bHLH regulators in olfactory neuron progenitors. Development 124, 1611-1621. [DOI] [PubMed] [Google Scholar]
- Chen, Y. and Schier, A. F. (2002). Lefty proteins are long-range inhibitors of squint-mediated nodal signaling. Curr. Biol. 12, 2124-2128. [DOI] [PubMed] [Google Scholar]
- Cuschieri, A. and Bannister, L. H. (1975a). The development of the olfactory mucosa in the mouse: electron microscopy. J. Anat. 119, 471-498. [PMC free article] [PubMed] [Google Scholar]
- Cuschieri, A. and Bannister, L. H. (1975b). The development of the olfactory mucosa in the mouse: light microscopy. J. Anat. 119, 277-286. [PMC free article] [PubMed] [Google Scholar]
- DeHamer, M. K., Guevara, J. L., Hannon, K., Olwin, B. B. and Calof, A. L. (1994). Genesis of olfactory receptor neurons in vitro: regulation of progenitor cell divisions by fibroblast growth factors. Neuron 13, 1083-1097. [DOI] [PubMed] [Google Scholar]
- Dou, C., Lee, J., Liu, B., Liu, F., Massague, J., Xuan, S. and Lai, E. (2000). BF-1 interferes with transforming growth factor beta signaling by associating with Smad partners. Mol. Cell. Biol. 20, 6201-6211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duggan, C. D., Demaria, S., Baudhuin, A., Stafford, D. and Ngai, J. (2008). Foxg1 is required for development of the vertebrate olfactory system. J. Neurosci. 28, 5229-5239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn, N. R., Winnier, G. E., Hargett, L. K., Schrick, J. J., Fogo, A. B. and Hogan, B. L. (1997). Haploinsufficient phenotypes in Bmp4 heterozygous null mice and modification by mutations in Gli3 and Alx4. Dev. Biol. 188, 235-247. [DOI] [PubMed] [Google Scholar]
- Eagleson, K. L., Schlueter McFadyen-Ketchum, L. J., Ahrens, E. T., Mills, P. H., Does, M. D., Nickols, J. and Levitt, P. (2007). Disruption of Foxg1 expression by knock-in of cre recombinase: effects on the development of the mouse telencephalon. Neuroscience 148, 385-399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eldar, A., Dorfman, R., Weiss, D., Ashe, H., Shilo, B. Z. and Barkai, N. (2002). Robustness of the BMP morphogen gradient in Drosophila embryonic patterning. Nature 419, 304-308. [DOI] [PubMed] [Google Scholar]
- Forbes, D., Jackman, M., Bishop, A., Thomas, M., Kambadur, R. and Sharma, M. (2006). Myostatin auto-regulates its expression by feedback loop through Smad7 dependent mechanism. J. Cell Physiol. 206, 264-272. [DOI] [PubMed] [Google Scholar]
- Gamer, L. W., Cox, K. A., Small, C. and Rosen, V. (2001). Gdf11 is a negative regulator of chondrogenesis and myogenesis in the developing chick limb. Dev. Biol. 229, 407-420. [DOI] [PubMed] [Google Scholar]
- Glantz, S. A. (2005). Primer of Biostatistics. New York: McGraw-Hill.
- Gomis, R. R., Alarcon, C., Nadal, C., Van Poznak, C. and Massague, J. (2006). C/EBPbeta at the core of the TGFbeta cytostatic response and its evasion in metastatic breast cancer cells. Cancer Cell 10, 203-214. [DOI] [PubMed] [Google Scholar]
- Guillemot, F. and Joyner, A. L. (1993). Dynamic expression of the murine Achaete-Scute homologue Mash-1 in the developing nervous system. Mech. Dev. 42, 171-185. [DOI] [PubMed] [Google Scholar]
- Hanashima, C., Shen, L., Li, S. C. and Lai, E. (2002). Brain factor-1 controls the proliferation and differentiation of neocortical progenitor cells through independent mechanisms. J. Neurosci. 22, 6526-6536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanashima, C., Li, S. C., Shen, L., Lai, E. and Fishell, G. (2004). Foxg1 suppresses early cortical cell fate. Science 303, 56-59. [DOI] [PubMed] [Google Scholar]
- Hanashima, C., Fernandes, M., Hebert, J. M. and Fishell, G. (2007). The role of Foxg1 and dorsal midline signaling in the generation of Cajal-Retzius subtypes. J. Neurosci. 27, 11103-11111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatini, V., Ye, X., Balas, G. and Lai, E. (1999). Dynamics of placodal lineage development revealed by targeted transgene expression. Dev. Dyn. 215, 332-343. [DOI] [PubMed] [Google Scholar]
- Hebert, J. M. and McConnell, S. K. (2000). Targeting of cre to the Foxg1 (BF-1) locus mediates loxP recombination in the telencephalon and other developing head structures. Dev. Biol. 222, 296-306. [DOI] [PubMed] [Google Scholar]
- Hebert, J. M. and Fishell, G. (2008). The genetics of early telencephalon patterning: some assembly required. Nat. Rev. Neurosci. 9, 678-685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kastner, A., Moyse, E., Bauer, S., Jourdan, F. and Brun, G. (2000). Unusual regulation of cyclin D1 and cyclin-dependent kinases cdk2 and cdk4 during in vivo mitotic stimulation of olfactory neuron progenitors in adult mouse. J. Neurochem. 74, 2343-2349. [DOI] [PubMed] [Google Scholar]
- Kawauchi, S., Beites, C. L., Crocker, C. E., Wu, H. H., Bonnin, A., Murray, R. and Calof, A. L. (2004). Molecular signals regulating proliferation of stem and progenitor cells in mouse olfactory epithelium. Dev. Neurosci. 26, 166-180. [DOI] [PubMed] [Google Scholar]
- Kawauchi, S., Shou, J., Santos, R., Hebert, J. M., McConnell, S. K., Mason, I. and Calof, A. L. (2005). Fgf8 expression defines a morphogenetic center required for olfactory neurogenesis and nasal cavity development in the mouse. Development 132, 5211-5223. [DOI] [PubMed] [Google Scholar]
- Kim, J., Wu, H.-H., Lander, A. D., Lyons, K. M., Matzuk, M. M. and Calof, A. L. (2005). GDF11 controls the timing of progenitor cell competence in developing retina. Science 308, 1927-1930. [DOI] [PubMed] [Google Scholar]
- Lander, A. D., Gokoffski, K. K., Wan, F. Y. M., Nie, Q. and Calof, A. L. (2009). Cell Lineages and the Logic of Proliferative Control. PLoS Biol. 7, e1000015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawson, K. A., Dunn, N. R., Roelen, B. A., Zeinstra, L. M., Davis, A. M., Wright, C. V., Korving, J. P. and Hogan, B. L. (1999). Bmp4 is required for the generation of primordial germ cells in the mouse embryo. Genes Dev. 13, 424-436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Legrier, M. E., Ducray, A., Propper, A., Chao, M. and Kastner, A. (2001). Cell cycle regulation during mouse olfactory neurogenesis. Cell Growth Differ. 12, 591-601. [PubMed] [Google Scholar]
- Lewandoski, M., Meyers, E. N. and Martin, G. R. (1997). Analysis of Fgf8 gene function in vertebrate development. Cold Spring Harb. Symp. Quant. Biol. 62, 159-168. [PubMed] [Google Scholar]
- Ma, Q., Fode, C., Guillemot, F. and Anderson, D. J. (1999). Neurogenin1 and neurogenin2 control two distinct waves of neurogenesis in developing dorsal root ganglia. Genes Dev. 13, 1717-1728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallamaci, A., Di Blas, E., Briata, P., Boncinelli, E. and Corte, G. (1996). OTX2 homeoprotein in the developing central nervous system and migratory cells of the olfactory area. Mech. Dev. 58, 165-178. [DOI] [PubMed] [Google Scholar]
- Martynoga, B., Morrison, H., Price, D. J. and Mason, J. O. (2005). Foxg1 is required for specification of ventral telencephalon and region-specific regulation of dorsal telencephalic precursor proliferation and apoptosis. Dev. Biol. 283, 113-127. [DOI] [PubMed] [Google Scholar]
- Massague, J. (2000). How cells read TGF-beta signals. Nat. Rev. Mol. Cell Biol. 1, 169-178. [DOI] [PubMed] [Google Scholar]
- Massague, J. and Gomis, R. R. (2006). The logic of TGFbeta signaling. FEBS Lett. 580, 2811-2820. [DOI] [PubMed] [Google Scholar]
- Moustakas, A., Souchelnytskyi, S. and Heldin, C. H. (2001). Smad regulation in TGF-beta signal transduction. J. Cell Sci. 114, 4359-4369. [DOI] [PubMed] [Google Scholar]
- Mumm, J. S., Shou, J. and Calof, A. L. (1996). Colony-forming progenitors from mouse olfactory epithelium: evidence for feedback regulation of neuron production. Proc. Natl. Acad. Sci. USA 93, 11167-11172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray, R. C., Navi, D., Fesenko, J., Lander, A. D. and Calof, A. L. (2003). Widespread defects in the primary olfactory pathway caused by loss of Mash1 function. J. Neurosci. 23, 1769-1780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muzio, L. and Mallamaci, A. (2005). Foxg1 confines Cajal-Retzius neuronogenesis and hippocampal morphogenesis to the dorsomedial pallium. J. Neurosci. 25, 4435-4441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura, T., Takio, K., Eto, Y., Shibai, H., Titani, K. and Sugino, H. (1990). Activin-binding protein from rat ovary is follistatin. Science 247, 836-838. [DOI] [PubMed] [Google Scholar]
- Nakashima, M., Toyono, T., Akamine, A. and Joyner, A. (1999). Expression of growth/differentiation factor 11, a new member of the BMP/TGFbeta superfamily during mouse embryogenesis. Mech. Dev. 80, 185-189. [DOI] [PubMed] [Google Scholar]
- Newfeld, S. J., Wisotzkey, R. G. and Kumar, S. (1999). Molecular evolution of a developmental pathway: phylogenetic analyses of transforming growth factor-beta family ligands, receptors and Smad signal transducers. Genetics 152, 783-795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen, T. T., Cho, K., Stratton, S. A. and Barton, M. C. (2005). Transcription factor interactions and chromatin modifications associated with p53-mediated, developmental repression of the alpha-fetoprotein gene. Mol. Cell. Biol. 25, 2147-2157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nomura, T., Ueyama, T., Ashihara, E., Tateishi, K., Asada, S., Nakajima, N., Isodono, K., Takahashi, T., Matsubara, H. and Oh, H. (2008). Skeletal muscle-derived progenitors capable of differentiating into cardiomyocytes proliferate through myostatin-independent TGF-beta family signaling. Biochem. Biophys. Res. Commun. 365, 863-869. [DOI] [PubMed] [Google Scholar]
- Pauley, S., Lai, E. and Fritzsch, B. (2006). Foxg1 is required for morphogenesis and histogenesis of the mammalian inner ear. Dev. Dyn. 235, 2470-2482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pratt, T., Tian, N. M., Simpson, T. I., Mason, J. O. and Price, D. J. (2004). The winged helix transcription factor Foxg1 facilitates retinal ganglion cell axon crossing of the ventral midline in the mouse. Development 131, 3773-3784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Regad, T., Roth, M., Bredenkamp, N., Illing, N. and Papalopulu, N. (2007). The neural progenitor-specifying activity of FoxG1 is antagonistically regulated by CKI and FGF. Nat. Cell Biol. 9, 531-540. [DOI] [PubMed] [Google Scholar]
- Rodriguez, C., Huang, L. J., Son, J. K., McKee, A., Xiao, Z. and Lodish, H. F. (2001). Functional cloning of the proto-oncogene brain factor-1 (BF-1) as a Smad-binding antagonist of transforming growth factor-beta signaling. J. Biol. Chem. 276, 30224-30230. [DOI] [PubMed] [Google Scholar]
- Schneyer, A. L., Rzucidlo, D. A., Sluss, P. M. and Crowley, W. F., Jr (1994). Characterization of unique binding kinetics of follistatin and activin or inhibin in serum. Endocrinology 135, 667-674. [DOI] [PubMed] [Google Scholar]
- Schneyer, A. L., Sidis, Y., Gulati, A., Sun, J. L., Keutmann, H. and Krasney, P. A. (2008). Differential antagonism of activin, myostatin and GDF11 by wild type and mutant follistatin. Endocrinology 149, 4589-4595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seoane, J., Le H. V., Shen, L., Anderson, S. A. and Massague, J. (2004). Integration of Smad and forkhead pathways in the control of neuroepithelial and glioblastoma cell proliferation. Cell 117, 211-223. [DOI] [PubMed] [Google Scholar]
- Shimamura, K. and Rubenstein, J. L. (1997). Inductive interactions direct early regionalization of the mouse forebrain. Development 124, 2709-2718. [DOI] [PubMed] [Google Scholar]
- Smart, I. H. (1971). Location and orientation of mitotic figures in the developing mouse olfactory epithelium. J. Anat. 109, 243-251. [PMC free article] [PubMed] [Google Scholar]
- Storm, E. E., Garel, S., Borello, U., Hebert, J. M., Martinez, S., McConnell, S. K., Martin, G. R. and Rubenstein, J. L. (2006). Dose-dependent functions of Fgf8 in regulating telencephalic patterning centers. Development 133, 1831-1844. [DOI] [PubMed] [Google Scholar]
- Sutherland, D. J., Li, M., Liu, X. Q., Stefancsik, R. and Raftery, L. A. (2003). Stepwise formation of a SMAD activity gradient during dorsal-ventral patterning of the Drosophila embryo. Development 130, 5705-5716. [DOI] [PubMed] [Google Scholar]
- Tsuchida, K., Nakatani, M., Uezumi, A., Murakami, T. and Cui, X. (2007). Signal transduction pathway through activin receptors as a therapeutic target of musculoskeletal diseases and cancer. Endocr. J. 55, 11-21. [DOI] [PubMed] [Google Scholar]
- Wu, H. H., Ivkovic, S., Murray, R. C., Jaramillo, S., Lyons, K. M., Johnson, J. E. and Calof, A. L. (2003). Autoregulation of neurogenesis by GDF11. Neuron 37, 197-207. [DOI] [PubMed] [Google Scholar]
- Xuan, S., Baptista, C. A., Balas, G., Tao, W., Soares, V. C. and Lai, E. (1995). Winged helix transcription factor BF-1 is essential for the development of the cerebral hemispheres. Neuron 14, 1141-1152. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}