

Abstract
Interleukin (IL)–6 concentrations are positively associated with the severity of pneumonia, and this cytokine is essential to surviving experimental pneumococcal pneumonia. The role that IL-6 plays during pneumonia and its impact during gram-negative bacterial pneumonia remain to be determined. During Escherichia coli pneumonia, IL-6–deficient mice had increased bacterial burdens in their lungs, indicating compromised host defenses. Decreased neutrophil counts in alveolar air spaces, despite normal blood neutrophil counts and survival of emigrated neutrophils, suggested that defective neutrophil recruitment was responsible for exacerbating the infection. Neutrophil recruitment requires nuclear factor (NF)–κB, but IL-6 was neither sufficient nor essential to induce NF-κB–mediated gene expression in the lungs. In contrast, IL-6 induced the phosphorylation of signal transducer and activator of transcription (STAT) 1 and STAT3 in the lungs, and STAT1 and STAT3 phosphorylation during E. coli pneumonia was decreased by IL-6 deficiency. Thus, IL-6 plays essential roles in activating STAT transcription factors, enhancing neutrophil recruitment, and decreasing bacterial burdens during E. coli pneumonia.
Lower respiratory tract infections are the leading cause of disability-adjusted life years lost worldwide [1] and are the leading cause of hospitalizations and deaths due to infection in the United States [2, 3]. Gram-negative bacterial rods are common causes of pneumonia in hospitals and nursing homes [4, 5] and have especially high mortality rates [6, 7].
The cytokine interleukin (IL)–6 is induced by a wide variety of infections. During community-acquired pneumonia, IL-6 concentrations in patients’ blood and bronchoalveolar lavage (BAL) fluid are positively associated with the severity of disease [8–10]. During experimental Streptococcus pneumoniae infection in mice, IL-6 deficiency due to gene targeting increases bacterial burdens and mortality [11]. Thus, IL-6 improves defenses against S. pneumoniae in the lungs, although the responses regulated by IL-6 that are essential to the antipneumococcal host defenses remain to be determined.
Neutrophil recruitment is essential to effective host defenses against bacteria, particularly gram-negative rods, in the lungs [12, 13]. Maximal neutrophil recruitment in the lungs requires the induction of IL-6 in response to some purified bacterial products, such as peptidoglycan or pneumolysin [14, 15], but not to others, such as lipoteichoic acid or lipopolysaccharide [15, 16]. The roles that IL-6 plays in modulating neutrophil recruitment elicited by living bacteria in the lungs have yet to be identified. Furthermore, whether IL-6 impacts the host defenses against gram-negative bacteria in the lungs remains to be determined. We hypothesized that IL-6 makes essential contributions to the host defenses during E. coli pneumonia, and we tested this hypothesis using IL-6–deficient mice.
MATERIALS AND METHODS
Mice
IL-6–deficient mice [17] were on a C57BL/6 background (Jackson Laboratories); triple mutant (TM) mice [18] deficient in tumor necrosis factor (TNF) receptors 1 and 2 and the type I IL-1 receptor were on a C57BL/6x129/Sv random hybrid background; and controls were wild-type (wt) mice with a background matched to mutants. Experimental protocols were approved by the Harvard Medical Area Standing Committee on Animals.
Intratracheal instillations
Mice were anesthetized by the intramuscular injection of ketamine hydrochloride (100 mg/kg), acepromazine maleate (5 mg/kg), and atropine (0.1 mg/kg). The trachea was surgically exposed, and a total volume of 50 μL was instilled via an angiocatheter that was inserted through the trachea and into the left bronchus. Recombinant murine (rm) IL-6 (Endogen) was instilled at a dose of 0, 50, or 500 ng/mouse in sterile saline with 1% bovine serum albumin. E. coli (strain 19138; American Type Culture Collection) was instilled intratracheally at a dose of 106 cfu/mouse. Colloidal carbon (1%) was included in the instillate, to indicate deposition. At the conclusion of each experiment, mice were killed by halothane inhalation.
mRNA expression
RNA was extracted with Trizol (Life Technologies), column purified (Qiagen), and treated with DNase. For IL-6, reverse transcription (RT) was performed with 2 μg of RNA, random hexamers (Life Technologies), rRNasin ribonuclease inhibitor (Promega), and M-MLV reverse transcriptase (Promega). Real-time polymerase chain reaction (PCR) was performed using iQ SYBR Green Supermix (Biorad) and the primer set forward, 5′-ATGAAGTTCCTCTCTGCAAGAGACT-3′, and reverse, 5′-CACTAGGTTTGCCGAGTAGATCTC-3′. The primer set forward, 5′-AGAGGGAAATCGTGCGTGAC-3′, and reverse, 5′-CAATAGTGATGACCTGGCCGT-3′, was used to amplify β-actin cDNA. Copy numbers of IL-6 and β-bactin were calculated from standard curves generated from plasmid clones, and IL-6 mRNA content was normalized to that of β-actin. Intercellular adhesion molecule (ICAM)–1 RT and real-time RT-PCR was performed using the iScript One-Step RT-PCR Kit for Probes (Biorad). The primer set for amplification of ICAM-1 was forward, 5′-CACCATGCCTTAGCAGCTGAACAA-3′, and reverse, 5′-CGACCCTTCTAAGGAGTTGGAACA-3′. The primer set for amplification of 18s rRNA was forward, 5′-ATTCGAACGTCTGCCCTATCA-3′, and reverse, 5′-GTCACCCGTGGTCACCATG-3′. The probes for ICAM-1 and 18s rRNA were 5′-TCATGGTCCCAGGCGGCTCCACCTCAAAGA-3′ and 5′-TCGATGGTAGTCGCCGTGCCTACC-3′, respectively, and contained 6-FAM at the 5′ end and Black Hole Quencher–1 at the 3′ end. ICAM-1 fold induction (compared with baseline levels) [19] was normalized to 18s rRNA within each sample. Real-time RT-PCR reactions were performed using the iCycler iQ Real-Time PCR detection system (Biorad).
Cytokine concentrations
Cytokine concentrations were measured by ELISA (R&D Systems) in BAL fluid and lung homogenates collected at indicated times after E. coli infection. For BAL fluid, the trachea was cannulated, lungs were lavaged 12 times with 0.8−1.0-mL volumes of PBS, the lavageates were pooled, and cells were removed by centrifugation. For lung homogenates, excised lungs were ground using a sterile glass Duall homogenizer in a buffered salts solution containing 0.5% Triton X-100 and protease inhibitors.
Bacterial burdens
Lung homogenates were serially diluted in ice-cold sterile water, and aliquots were spread on 5% sheep blood–agar plates. After 18−24 h of incubation at 37°C, colonies were counted, and data for viable bacteria were expressed as colony-forming units per lung.
Neutrophil recruitment
Lungs and blood were collected 24 h after E. coli infection. Peripheral blood was drawn from the inferior vena cava. After erythrocyte lysis, leukocytes were counted using a hemacytometer, and differential distributions in stained blood smears were determined. Excised lungs were fixed by the intratracheal instillation of 6% glutaraldehyde at 23 cm H2O pressure. Neutrophils in alveolar air spaces were quantified by morphometric analyses of histological lung sections [18, 20, 21]. For morphometric examinations, investigators were blinded to the identities of mice.
Neutrophil survival
Cells from BAL fluid were collected [22] from wt and IL-6–deficient mice 24 h after E. coli infection. Cell counts and viability were determined using a hemacytometer and trypan blue dye (Sigma), and morphological features were assessed using cytocentrifuge slide preparations. Preparations were 83%–96% neutrophils with an initial viability of 91%–96%. Cells were suspended at a concentration of 106 cells/mL in a buffered salt solution containing divalent cations, and 200-μL samples were cultured in 96-well ultra-low-attachment plates (Corning) at 37°C in 5% CO2. Samples were withdrawn at indicated time points for assessment using a hemacytometer with trypan blue and a cytocentrifuge with Diff-Quick (Dade Behring).
Assessment of inhibitor of NF-κB (IκB) and signal transducer and activator of transcription (STAT) proteins by immunoblotting
IκB content and STAT1 and STAT3 phosphor-ylation were analyzed by immunoblotting. Excised lungs were homogenized in lysis buffer (buffered saline solution containing NP-40 (Calbiochem), sodium deoxycholate, SDS, sodium orthovanadate, and protease inhibitors) using a Kinematica Polytron, and total protein concentrations were measured using a bicinchoninic acid assay. Proteins were separated on 4%–12% gradient gels by SDS–polyacrylamide gel electrophoresis and transferred to Immobilon-P polyvinylidene fluoride membranes. Membranes were probed with the following polyclonal antibodies: sc-371 against IκB-α (Santa Cruz Biotechnology), sc-945 against IκB-β (Santa Cruz Biotechnology), 9172 against STAT1 (Cell Signaling Technology), 9171 against STAT1 phosphorylated on tyrosine 701 (Cell Signaling Technology), 9132 against STAT3 (Cell Signaling Technology), or 9131 against STAT3 phosphorylated on tyrosine 705 (Cell Signaling Technology). After washing, membrane-bound primary antibodies were detected on autoradiographic film by horseradish peroxidase–conjugated secondary antibodies and the ECL Plus chemiluminescent system (Amersham Pharmacia Biotech).
Statistics
Data are expressed as means ± SEs. Data in groups of 2 were compared using Student's t test. Sets of >12 groups were compared using factorial analysis of variance with post hoc tests as indicated. If data failed Levene's test for homogeneity of variances, they were log transformed. Differences were considered to be statistically significant if P < .05.
RESULTS
Expression of IL-6 during E. coli pneumonia
IL-6 was induced within 1 h of E. coli infection (figure 1A). Such rapid kinetics suggested that IL-6 transcription may result from signals generated by receptors for bacterial products. IL-6 mRNA concentrations peaked 6 h after infection but remained increased throughout the 24-h period that was examined (figure 1A). At later time points, IL-6 expression could be induced from a combination of receptors for bacterial products or host cytokines. IL-6 protein concentrations were measured in lung homogenates during E. coli pneumonia, and TM mice were used to determine the requirements of signaling from receptors for TNF-α and IL-1 (α and β). IL-6 concentrations were increased after infection, and concentrations in TM mice did not significantly differ from those in wt mice (figure 1B). Therefore, IL-6 expression during E. coli pneumonia does not require signaling from receptors for TNF-α and IL-1.
Figure 1.

Interleukin (IL)–6 in the lungs during Escherichia coli pneumonia. A, IL-6 mRNA in the lungs. IL-6 mRNA increased within 1 h of infection and remained increased through 24 h after infection. IL-6 and β-actin mRNA were measured in lung homogenates using quantitative reverse-transcription polymerase chain reaction. IL-6 transcripts were normalized to β-actin transcripts for each sample. The effect of time on IL-6 mRNA was statistically significant (P<.05, 1-way analysis of variance [ANOVA]). B, IL-6 protein in the lungs. IL-6 protein increased during infection of either wild-type mice (n = 5 mice/group) or triple mutant (TM) mice (n = 5 mice in the 0-h group, n = 4 mice in the 6-h group, and n = 6 mice in the 24-h group) lacking signaling receptors for tumor necrosis factor–α and IL-1. IL-6 concentrations in lung homogenates were determined using ELISA. There was a statistically significant effect of time (P<.05, 2-way ANOVA) but not of genotype.
Bacterial burden
To determine whether IL-6 deficiency impacted innate host defenses against intrapulmonary E. coli, living bacteria were quantified in lungs from wt and IL-6–deficient mice. In both genotypes, bacterial burdens were decreased over time, but IL-6–deficient mice were less effective than wt mice at decreasing bacterial burdens (figure 2A). The differences between the mice were statistically significant by 24 h, when IL-6–deficient mice already had several-fold more bacteria in their lungs than did wt mice. By 48 h, wt mice had eliminated 99.9% of the bacterial burden. IL-6–deficient mice were also controlling the infection for 48 h, but they had hundreds-fold more bacteria in their lungs than did wt mice. Bacteremia was never detected in wt mice (data not shown) but was occasionally observed in IL-6–deficient mice (20%–25% of mice at either time point after infection). Thus, IL-6 deficiency compromises antibacterial host defenses during E. coli pneumonia.
Figure 2.
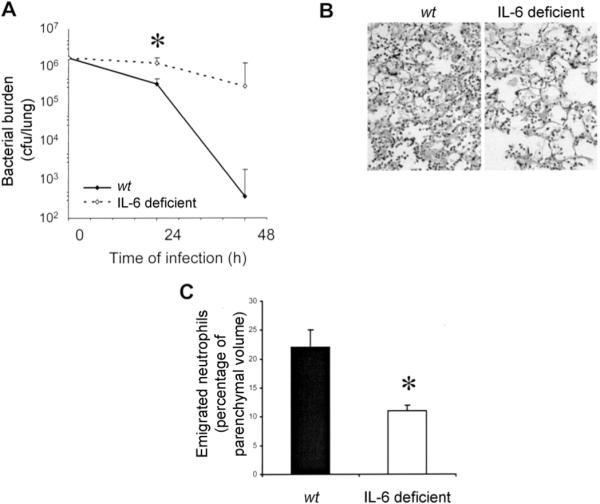
Effects of interleukin (IL)–6 deficiency on neutrophil accumulation and bacterial clearance during Escherichia coli pneumonia. A, Bacterial clearance and IL-6 deficiency. Living bacteria were quantified in lung homogenates collected from wild-type (wt) and IL-6–deficient mice 24 h after infection (n = 5−9 mice/group). The effect of time on colony-forming units was statistically significant (P<.05 , 2-way analysis of variance [ANOVA]), and so was the effect of genotype (denoted by asterisk; P<.05, 2-way ANOVA). B, Effect of IL-6 deficiency on accumulation of neutrophils in alveolar air spaces. Representative images are shown of pulmonary parenchyma from lungs of wt and IL-6–deficient mice. Left lung lobes were collected 24 h after infection, fixed by the instillation of 6% glutaraldehyde at 23 cm H2O pressure, and embedded in paraffin, and 5-mm sections were stained with hematoxylin-eosin. Histological analyses revealed acute inflammation with a diffuse and patchy distribution throughout the left lung lobe parenchyma. Polymerized proteinaceous material was prominent in alveolar air spaces. Emigrated neutrophils were present in alveolar air spaces, with more apparent in lungs from wt mice than in those from IL-6–deficient mice. C, Emigrated neutrophils in alveolar air spaces of IL-6–deficient mice. Emigrated neutrophils in lung sections (n = 7 mice/group) 24 h after infection, as in panel B, were quantified using standard point-counting morphometric techniques. The relative volumes of the parenchymal regions occupied by emigrated neutrophils were calculated by investigators blinded to the identities of the mice and were expressed as a percentage of the total parenchymal region volume (including both tissue and air spaces). *P<.05, Student's t test.
Neutrophil recruitment
Neutrophils are essential to clearing gram-negative bacterial rods from the lungs [12, 13]. E. coli infection induced neutrophil recruitment in both wt and IL-6– deficient mice (figure 2B), demonstrating that some neutrophils emigrate in the absence of IL-6. However, only one-half as many neutrophils were present in alveolar air spaces of IL-6–deficient mice, compared with those of wt mice (figure 2C). These data indicate that IL-6 is essential to maximal neutrophil accumulation in alveolar air spaces during E. coli pneumonia.
Because IL-6 can promote granulopoiesis and neutrophil mobilization from the bone marrow [23–25], we determined whether IL-6 deficiency decreased blood neutrophil counts during E. coli pneumonia. Circulating neutrophil counts did not significantly differ between genotypes, in mice with or without E. coli pneumonia (figure 3A). These data suggest that roles of IL-6 in promoting granulopoiesis and mobilizing neutrophils from the bone marrow were unlikely to be responsible for the decrease in emigrated neutrophils during E. coli pneumonia.
Figure 3.

Effect of interleukin (IL)–6 deficiency on blood neutrophils and survival of emigrated neutrophils. A, Neutrophils in venous blood from wild-type (wt) and IL-6–deficient mice with Escherichia coli pneumonia. Neutrophils were quantified in venous blood samples from mice 0, 6, or 24 h after infection. There was a significant effect of time (P<.05, 2-way analysis of variance [ANOVA]) but not of genotype on blood neutrophil counts. B, IL-6 deficiency and the life span of emigrated neutrophils. Emigrated neutrophils were collected from alveolar air spaces of wt and IL-6–deficient mice by bronchoalveolar lavage 24 h after infection and were cultured in vitro for the designated time. Viability was assessed using trypan blue exclusion, with neutrophils collected from 4 mice/group. The viability of emigrated neutrophils decreased over time but did not differ between genotypes (2-way ANOVA).
Because IL-6 can prolong the life span of neutrophils in vitro [26, 27], we determined whether the life span of emigrated neutrophils was altered by IL-6 deficiency. After E. coli infection, neutrophils cultured from wt and IL-6–deficient mice developed pyknosis and karyorhexis during a similar time frame (data not shown). Loss of plasma membrane integrity, as measured by trypan blue exclusion, did not differ between genotypes (figure 3B). These data suggest that the life span of neutrophils that emigrated during E. coli pneumonia was not shortened by IL-6 deficiency. Decreased neutrophil counts in alveolar air spaces, coupled with normal blood neutrophil counts and normal survival of emigrated neutrophils, suggest that IL-6 was necessary for maximal recruitment of neutrophils from the pulmonary capillaries to alveolar air spaces during E. coli pneumonia.
NF-κB–regulated genes essential to neutrophil recruitment
ICAM-1, KC, and macrophage inflammatory protein (MIP)–2 are necessary for neutrophil recruitment [13, 28–32] and are regulated by NF-κB [20, 21, 33–36] in response to gram-negative stimuli in the lungs. Because IL-6 can activate NF-κB in vitro [37], we determined whether IL-6 contributed to expression of these genes or NF-κB activity during E. coli pneumonia.
ICAM-1 mRNA concentrations were increased 6 h after E. coli infection, with no significant differences between genotypes (figure 4A). By 24 h, ICAM-1 mRNA concentrations were greater in IL-6–deficient mice than in wt mice (figure 4A). Thus, IL-6 was not essential for induction of ICAM-1. Concentrations of KC and MIP-2, which are particularly important in BAL fluid [38], peaked 6 h after E. coli infection [20]. KC and MIP-2 concentrations did not differ between genotypes (figure 4B). Circulating KC also enhances neutrophil recruitment [39]. However, circulating KC concentrations did not significantly differ between wt and IL-6–deficient mice 6 h after infection (870 ± 84 vs. 1899 ± 469 pg/mL). Thus, none of these mediators required IL-6 during E. coli pneumonia.
Figure 4.

Interleukin (IL)–6 and NF-κB–mediated gene expression during Escherichia coli pneumonia. A, Intercellular adhesion molecule (ICAM)–1 RNA concentrations. Steady-state concentrations of ICAM-1 RNA did not differ between wild-type (wt) and IL-6–deficient mice. Lungs were collected from 10 mice/genotype at 6 h or from 3 mice/genotype at 0 and 24 h after infection. ICAM-1 transcripts were measured by quantitative reverse-transcription polymerase chain reaction, normalized to 18s rRNA, and then expressed as the fold induction from 0-h levels, which did not differ between genotypes. Both time and genotype had significant effects (denoted by asterisk; P<.05 , 2-way analysis of variance and Bonferroni post hoc test). B, Concentrations of the NF-κB–dependent chemokines KC and macrophage inflammatory protein (MIP)–2. Chemokine concentrations did not differ between wt and IL-6–deficient mice (n = 5−6 mice/group). Lungs were collected 6 h after infection, and chemokine concentrations in bronchoalveolar lavage fluid (BAL) were determined using ELISA. There was no significant effect of genotype (Student's t test). C, Inhibitor of NF-κB (IκB) content in the lungs. IκB content decreased during E. coli pneumonia but did not differ between genotypes. The IκB content was assessed by immunoblotting at the indicated time after infection. Each lane contains protein from an individual mouse that was either homozygous (+/+) or mutant (−/−) for the Il6 gene. D, IκB content in the lungs and IL-6. IκB content was not affected by the intratracheal instillation of recombinant murine (rm) IL-6. IκB content in wt mice that received the indicated dose of rmIL-6 was assessed by immunoblotting. Each lane contains protein from an individual mouse.
NF-κB activation by lipopolysaccharide in the lungs is mediated by the degradation of IκB-α and IκB-β [40]. Similarly, E. coli infection decreased concentrations of both IκB-α and IκB-β (figure 4C) in mice. IL-6 deficiency did not alter the IκB protein content in the lungs during E. coli pneumonia (figure 4C), suggesting that IL-6 was not necessary for IκB degradation. Furthermore, IκB-α and IκB-β were unaffected by the instillation of up to 500 ng of rmIL-6 (figure 4D), suggesting that IL-6 is not sufficient to induce the degradation of lung IκB proteins. Combined with the NF-κB–dependent gene expression data given above, these results argue against regulation of NF-κB by IL-6 during E. coli pneumonia.
Other gene products regulating neutrophil recruitment in the lungs
We examined whether other cytokines relevant to acute neutrophil recruitment during bacterial pneumonia were dependent on IL-6. The cytokine IL-17 is necessary for neutrophil recruitment during gram-negative bacterial pneumonia [41, 42]. Because IL-17 expression is delayed during pneumonia and requires multiple upstream cytokines [43, 44], we measured IL-17 concentrations in lung homogenates collected 24 h after E. coli infection. Compared with lungs from wt mice, lungs from IL-6–deficient mice had increased IL-17 concentrations (table 1). Maximal expression of lipopolysaccharide-induced CXC chemokine (LIX), which contributes to neutrophil recruitment in the lungs [45], requires IL-6 under some conditions [46]. Because LIX expression during E. coli pneumonia has not been characterized, we measured LIX concentrations 6 and 24 h after E. coli infection. LIX concentrations were unchanged (at 6 h) or increased (at 24 h) by IL-6 deficiency (table 1). Altogether, these data indicate that decreased neutrophil recruitment in IL-6–deficient mice with E. coli pneumonia does not result from decreased concentrations of LIX or IL-17.
Table 1.
Cytokine concentrations in the lungs.
| Cytokine, time point, sample | wt mice | IL-6–deficient mice | P |
|---|---|---|---|
| IL-17, 24 h, lung | 15 ± 2 | 67 ± 11 | <.01 |
| LIX, 6 h, BAL fluid | 87 ± 12 | 122 ± 22 | .2 |
| LIX, 24 h, lung | 550 ± 31 | 860 ± 56 | <.01 |
NOTE. Cytokine concentrations in bronchoalveolar lavage (BAL) fluid or lung homogenates collected 6 or 24 h after the intratracheal instillation of Escherichia coli were measured by ELISA. Data are expressed in picograms per milliliter and are the means ± SE from 5−6 mice/group. IL, interleukin; LIX, lipopolysaccharide-induced CXC chemokine; wt, wild-type.
STAT1 and STAT3 phosphorylation
IL-6 activates the transcription factors STAT1 and STAT3 in many cells [47]. To determine whether IL-6 activates STAT1 and STAT3 in the lungs, rmIL-6 was intratracheally instilled, and tyrosine phosphorylation, which is essential for the transcriptional activity of these factors [48], was assessed by immunoblotting. Both STAT1 and STAT3 were phosphorylated in the lungs in response to IL-6 in a dose-dependent fashion (figure 5A), indicating that IL-6 in the lungs was sufficient for phosphorylation of these transcription factors.
Figure 5.
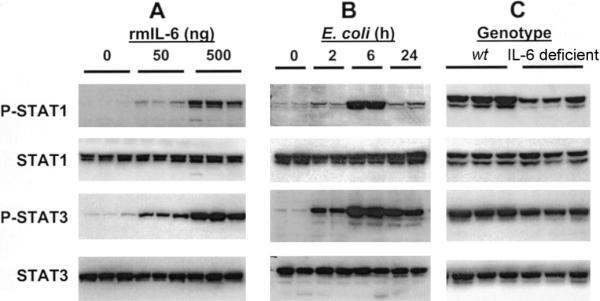
Interleukin (IL)–6 and activation of signal transducer and activator of transcription (STAT) proteins in the lungs during Escherichia coli pneumonia. Concentrations of tyrosine-phosphorylated (P) and total STAT proteins in the lungs were assessed by immunoblotting. Each lane contains protein from an individual mouse. A, IL-6 and phosphorylation of STAT1 and STAT3 in the lungs. Recombinant murine (rm) IL-6 was instilled intratracheally to wild-type (wt) mice at the indicated dose per mouse, and lungs were collected 1 h later. The instillation of rmIL-6 was sufficient to induce phosphorylation. B, Phosphorylation of STAT1 and STAT3 during E. coli pneumonia. Lungs were collected from wt mice at the indicated time after infection, and phosphorylation was assessed. C, IL-6 deficiency and STAT1 and STAT3 phosphorylation. Lungs were collected from wt and IL-6–deficient mice 6 h after infection, and phosphorylation was assessed. IL-6 deficiency decreased phosphorylation.
STAT1 and STAT3 were also activated in the lungs during E. coli pneumonia (figure 5B). Phosphorylation of both factors peaked 6 h after infection (figure 5B), which was concomitant with peak concentrations of IL-6 mRNA and protein (figure 1). At the 6-h time point, concentrations of phosphorylated STAT1 and phosphorylated STAT3 were decreased in the lungs from IL-6–deficient mice, compared with those from wt mice (figure 5C). Concentrations of phosphorylated STAT3 and STAT1 were similarly decreased 15 h after infection (data not shown). At the 2- and 24-h time points, there were lower phosphorylated STAT protein concentrations and no discernible differences between genotypes (data not shown). These data demonstrate that maximal STAT activation required IL-6 during E. coli pneumonia. In contrast to the phospho-specific signals, the total concentrations of STAT1 and STAT3 were unaffected by rmIL-6, E. coli infection, and/or IL-6 deficiency (figure 5A–5C), indicating that phosphorylation, rather than expression, is the level of regulation. These data demonstrate that activation of STAT1 and STAT3 was partially dependent on IL-6 during E. coli pneumonia, and peak STAT concentrations that occurred 6−15 h after infection required IL-6.
Effects of exogenous IL-6 during E. coli pneumonia
To determine whether exogenous IL-6 was sufficient to restore responses of IL-6–deficient mice during pneumonia, rmIL-6 or vehicle was instilled with the bacteria. A single 100-ng dose of IL-6 per mouse, which was sufficient to cause the phosphorylation of STAT1 and STAT3 in the lungs (figure 5A), was able to restore neutrophil recruitment in IL-6–deficient mice with E. coli pneumonia (table 2). These results indicate an acute role for this cytokine early during the course of E. coli pneumonia. In contrast, including 100 ng of rmIL-6 with E. coli was not sufficient to restore bacterial clearance in IL-6–deficient mice (table 2). Optimal host defenses against E. coli in the lungs may require a more prolonged or coordinated signaling from IL-6 than was achieved by instilling a single bolus of this cytokine at the time of infection.
Table 2.
Effect of interleukin (IL)–6 on neutrophils and Escherichia coli.
| wt mice and vehicle | IL-6–deficient mice and vehicle | IL-6–deficient mice and rmIL-6 | Pa | |
|---|---|---|---|---|
| Neutrophils | 17 ± 2 | 10 ± 2 | 17 ± 4 | .9 |
| E. coli | 36 ± 16 | 202 ± 51 | 257 ± 107 | <.02 |
NOTE. Either vehicle or recombinant murine (rm) IL-6 at a dose of 100 ng/mouse was included with E. coli in the intratracheal instillates to wild-type (wt) or IL-6–deficient mice, and lungs were collected 24 h after infection. Emigrated neutrophils in alveolar air spaces are expressed as a percentage of the volume of alveolar parenchyma, and living bacteria are expressed as thousands of colony-forming units per lung. Data are the means ± SE from 3−4 mice/group (neutrophils) or 5−8 mice/group (bacteria). Analyses suggest that impaired neutrophil recruitment, but not bacterial clearance, in IL-6–deficient mice was restored by the administration of exogenous IL-6.
wt mice and vehicle vs. IL-6–deficient mice and rmIL-6 (analysis of variance and Duncan's test).
DISCUSSION
IL-6 concentrations are positively associated with the severity of disease in patients with pneumonia [8–10]. IL-6 is essential to overcoming S. pneumoniae infection in the lungs [11] or E. coli infection or polymicrobial sepsis in other tissues [49–51]. The results of the present study indicate that bacterial clearance is compromised by IL-6 deficiency during E. coli pneumonia, and this cytokine directs host responses to this infection by activating STAT transcription factors and enhancing neutrophil recruitment.
IL-6 plays a complex role in inflammation, because it can both promote and limit neutrophil emigration [14–16]. To our knowledge, the roles of IL-6 in neutrophil recruitment elicited by E. coli or other living bacteria in the lungs have yet to be determined. In the present study, neutrophil recruitment during E. coli pneumonia in C57BL/6 mice became detectable by 6 h, reached peak concentrations by 24 h, and was maintained to at least 48 h after infection. During this period, bacterial burdens decreased in both wt and IL-6–deficient mice but did so less effectively in the absence of IL-6. We observed the neutrophil recruitment elicited during E. coli pneumonia to be decreased by IL-6 deficiency, indicating that neutrophil recruitment during this infection depends, in part, on IL-6. Neutrophils are needed to kill gram-negative bacteria in the lungs [12, 13], so decreased neutrophil recruitment may contribute to the increased bacterial burdens of IL-6–deficient mice with E. coli pneumonia. The restoration of neutrophil recruitment, but not bacterial clearance, by a bolus of exogenous IL-6 delivered at the time of infection suggests that optimal host defenses require not only the presence of neutrophils but also prolonged IL-6 signaling. The enhancement of neutrophil bactericidal functions after transepithelial migration requires IL-6 signaling [52], suggesting that there may have been defects in bacterial killing by neutrophils in alveolar air spaces of IL-6– deficient mice. Therefore, E. coli clearance may be compromised in IL-6–deficient mice because of a combination of decreased neutrophil recruitment and suboptimal activation of recruited neutrophils.
Gene expression and neutrophil recruitment elicited by gram-negative bacterial stimuli in the lungs are mediated by NF-κB [20, 21, 33, 34, 36]. IL-6 transcription is regulated by NF-κB [53, 54]. NF-κB can be activated by receptors for bacterial products or by endogenous factors such as cytokines. To determine the importance of receptors for the early response cytokines IL-1 (α and β) and TNF-α in IL-6 expression during E. coli pneumonia, we measured IL-6 concentrations in lungs from TM mice and found that IL-6 did not require signaling from these cytokines. IL-6 activated NF-κB in an intestinal epithelial cell line in vitro [37], suggesting the possibility that IL-6 is upstream of NF-κB activation during pneumonia. However, data in the present study demonstrate that IL-6 in alveolar air spaces is not sufficient to activate NF-κB in the lungs. NF-κB could be activated by IL-6 in the interstitial or vascular compartments of the lungs or by IL-6:soluble IL-6 receptor complexes formed during pneumonia. However, IκB degradation and the expression of NF-κB–dependent chemokines (ICAM-1, KC, and MIP-2) were unaffected by IL-6 deficiency during E. coli pneumonia, suggesting that IL-6 is not necessary for NF-κB activation during pneumonia. These data cannot preclude a role for IL-6 in activating NF-κB during E. coli pneumonia, but, altogether, they suggest that IL-6 is more likely to be downstream, rather than upstream, of NF-κB during this infection.
Because NF-κB was not affected by instillation of rmIL-6 or IL-6 deficiency, we considered other transcription factors that might be regulated by IL-6. The STAT family of transcription factors is regulated by cytokines [55]. STAT1 and STAT3 can be activated by IL-6 [47], but, to our knowledge, there is little or no evidence indicating that IL-6 activates other STAT proteins. Therefore, we examined STAT1 and STAT3 in the lungs. The present study demonstrates that STAT1 and STAT3 transcription factors are activated during E. coli pneumonia. In contrast to NF-κB, rmIL-6 activated STAT1 and STAT3 in the lungs. Furthermore, STAT activation after E. coli infection was partially dependent on endogenous IL-6. Therefore, IL-6 functions to activate these 2 STAT transcription factors during E. coli pneumonia.
The roles that STAT proteins play during pneumonia are an important focus for future research. STAT1 and STAT3 can regulate cell migration [56–59], suggesting that IL-6 activation of STAT1 and STAT3 in neutrophils may contribute to neutrophil emigration during pneumonia. Furthermore, STAT1 and STAT3 regulate the expression of extracellular mediators that may contribute to neutrophil recruitment, including CC and CXC chemokines [60–62], adhesion molecules [61, 62], S100 proteins [63], acute-phase response proteins [64, 65], and complement proteins [63]. Therefore, IL-6 activation of STATs may contribute to neutrophil recruitment during pneumonia by inducing genes in neutrophils (mediating migration) or by inducing genes in lung parenchymal cells (guiding migration). STAT3 also has antiapoptotic functions [66]. Loss of STAT3 function in the lungs [67] or other tissues [66] predisposes mice to premature death, perhaps because of excessive cell death, when challenged. Therefore, IL-6 activation of STAT3 may be protective during pneumonia by both facilitating the host defenses and preventing inflammatory injury.
Data in the present study demonstrate that IL-6 plays several roles during E. coli pneumonia: activating STAT1 and STAT3, facilitating neutrophil recruitment, and improving bacterial clearance. These findings suggest a causal relationship between IL-6–induced STAT activation and neutrophil-dependent killing of bacteria in the lungs, although the findings must be tested with further studies. It is also likely that IL-6 regulates transcription factors other than STAT1 and STAT3 and that they contribute to the phenotype of IL-6–deficient mice with E. coli pneumonia. Because data in the present study show that IL-6 facilitates both STAT activation and pulmonary host defenses, manipulating IL-6 and STAT signaling pathways deserves further consideration in the modification of innate immunity and protection of the lungs during bacterial pneumonia.
Acknowledgments
Financial support: National Institutes of Health (grants HL-68153, HL-79392, HL-07118, and ES-00002); American Physiological Society (M.R.J. is an American Physiological Society Postdoctoral Fellow in Physiological Genomics).
Footnotes
Presented in part: 100th International Conference of the American Thoracic Society, Orlando, Florida, 23 May 2004 (abstract G11).
Potential conflicts of interest: none reported.
References
- 1.Michaud CM, Murray CJL, Bloom BR. Burden of disease—implications for future research. JAMA. 2001;285:535–9. doi: 10.1001/jama.285.5.535. [DOI] [PubMed] [Google Scholar]
- 2.Simonsen L, Conn LA, Pinner RW, Teutsch S. Trends in infectious disease hospitalizations in the United States, 1980−1994. Arch Intern Med. 1998;158:1923–8. doi: 10.1001/archinte.158.17.1923. [DOI] [PubMed] [Google Scholar]
- 3.Armstrong GL, Conn LA, Pinner RW. Trends in infectious disease mortality in the United States during the 20th century. JAMA. 1999;281:61–6. doi: 10.1001/jama.281.1.61. [DOI] [PubMed] [Google Scholar]
- 4.Marrie TJ, Fine MJ, Obrosky DS, Coley C, Singer DE, Kapoor WN. Community-acquired pneumonia due to Escherichia coli. Clin Microbiol Infect. 1998;4:717–23. doi: 10.1111/j.1469-0691.1998.tb00657.x. [DOI] [PubMed] [Google Scholar]
- 5.Ruiz M, Ewig S, Marcos MA, et al. Etiology of community-acquired pneumonia: impact of age, comorbidity, and severity. Am J Respir Crit Care Med. 1999;160:397–405. doi: 10.1164/ajrccm.160.2.9808045. [DOI] [PubMed] [Google Scholar]
- 6.Loeb M, Mandell LA. Microbiology of hospital-acquired pneumonia. Semin Respir Crit Care Med. 1997;18:111–20. [Google Scholar]
- 7.Ahmed QA, Niederman MS. Respiratory infection in the chronically critically ill patient: ventilator-associated pneumonia and tracheobronchitis. Clin Chest Med. 2001;22:71–85. doi: 10.1016/s0272-5231(05)70026-5. [DOI] [PubMed] [Google Scholar]
- 8.Marik PE. The clinical features of severe community-acquired pneumonia presenting as septic shock. Norasept II Study Investigators. J Crit Care. 2000;15:85–90. doi: 10.1053/jcrc.2000.16460. [DOI] [PubMed] [Google Scholar]
- 9.Antunes G, Evans SA, Lordan JL, Frew AJ. Systemic cytokine levels in community-acquired pneumonia and their association with disease severity. Eur Respir J. 2002;20:990–5. doi: 10.1183/09031936.02.00295102. [DOI] [PubMed] [Google Scholar]
- 10.Kolsuz M, Erginel S, Alatas O, et al. Acute phase reactants and cytokine levels in unilateral community-acquired pneumonia. Respiration. 2003;70:615–22. doi: 10.1159/000075208. [DOI] [PubMed] [Google Scholar]
- 11.van der Poll T, Keogh CV, Guirao X, Buurman WA, Kopf M, Lowry SF. Interleukin-6 gene–deficient mice show impaired defense against pneumococcal pneumonia. J Infect Dis. 1997;176:439–44. doi: 10.1086/514062. [DOI] [PubMed] [Google Scholar]
- 12.Rehm SR, Gross GN, Pierce AK. Early bacterial clearance from murine lungs: species-dependent phagocyte response. J Clin Invest. 1980;66:194–9. doi: 10.1172/JCI109844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tsai WC, Strieter RM, Mehrad B, Newstead MW, Zeng X, Standiford TJ. CXC chemokine receptor CXCR2 is essential for protective innate host response in murine Pseudomonas aeruginosa pneumonia. Infect Immun. 2000;68:4289–96. doi: 10.1128/iai.68.7.4289-4296.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rijneveld AW, van den Dobbelsteen GP, Florquin S, et al. Roles of interleukin-6 and macrophage inflammatory protein–2 in pneumolysin-induced lung inflammation in mice. J Infect Dis. 2002;185:123–6. doi: 10.1086/338008. [DOI] [PubMed] [Google Scholar]
- 15.Leemans JC, Vervoordeldonk MJ, Florquin S, Van Kessel KP, Van Der Poll T. Differential role of interleukin-6 in lung inflammation induced by lipoteichoic acid and peptidoglycan from Staphylococcus aureus. Am J Respir Crit Care Med. 2002;165:1445–50. doi: 10.1164/rccm.2106045. [DOI] [PubMed] [Google Scholar]
- 16.Xing Z, Gauldie J, Cox G, et al. IL-6 is an antiinflammatory cytokine required for controlling local or systemic acute inflammatory responses. J Clin Invest. 1998;101:311–20. doi: 10.1172/JCI1368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kopf M, Baumann H, Freer G, et al. Impaired immune and acute-phase responses in interleukin-6-deficient mice. Nature. 1994;368:339–42. doi: 10.1038/368339a0. [DOI] [PubMed] [Google Scholar]
- 18.Mizgerd JP, Lupa MM, Hjoberg J, et al. Roles for early response cytokines during Escherichia coli pneumonia revealed by mice with combined deficiencies of all signaling receptors for TNF and IL-1. Am J Physiol Lung Cell Mol Physiol. 2004;286:L1302–10. doi: 10.1152/ajplung.00353.2003. [DOI] [PubMed] [Google Scholar]
- 19.Schmittgen TD, Zakrajsek BA, Mills AG, Gorn V, Singer MJ, Reed MW. Quantitative reverse transcription-polymerase chain reaction to study mRNA decay: comparison of endpoint and real-time methods. Anal Biochem. 2000;285:194–204. doi: 10.1006/abio.2000.4753. [DOI] [PubMed] [Google Scholar]
- 20.Mizgerd JP, Lupa MM, Kogan MS, Warren HB, Kobzik L, Topulos GP. Nuclear factor-κB p50 limits inflammation and prevents lung injury during Escherichia coli pneumonia. Am J Respir Crit Care Med. 2003;168:810–7. doi: 10.1164/rccm.200303-412OC. [DOI] [PubMed] [Google Scholar]
- 21.Mizgerd JP, Lupa MM, Spieker MS. NF-kappaB p50 facilitates neutrophil accumulation during LPS-induced pulmonary inflammation. BMC Immunol. 2004;5:10. doi: 10.1186/1471-2172-5-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mizgerd JP, Horwitz BH, Quillen HC, Scott ML, Doerschuk CM. Effects of CD18-deficiency on the emigration of murine neutrophils during pneumonia. J Immunol. 1999;163:995–9. [PubMed] [Google Scholar]
- 23.Liu F, Poursine-Laurent J, Wu HY, Link DC. Interleukin-6 and the granulocyte colony-stimulating factor receptor are major independent regulators of granulopoiesis in vivo but are not required for lineage commitment or terminal differentiation. Blood. 1997;90:2583–90. [PubMed] [Google Scholar]
- 24.Zhang P, Iwama A, Datta MW, Darlington GJ, Link DC, Tenen DG. Upregulation of interleukin 6 and granulocyte colony-stimulating factor receptors by transcription factor CCAAT enhancer binding protein alpha (C/EBP alpha) is critical for granulopoiesis. J Exp Med. 1998;188:1173–84. doi: 10.1084/jem.188.6.1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Suwa T, Hogg JC, English D, Van Eeden SF. Interleukin-6 induces demargination of intravascular neutrophils and shortens their transit in marrow. Am J Physiol Heart Circ Physiol. 2000;279:H2954–60. doi: 10.1152/ajpheart.2000.279.6.H2954. [DOI] [PubMed] [Google Scholar]
- 26.Daffern PJ, Jagels MA, Hugli TE. Multiple epithelial cell-derived factors enhance neutrophil survival: regulation by glucocorticoids and tumor necrosis factor-alpha. Am J Respir Cell Mol Biol. 1999;21:259–67. doi: 10.1165/ajrcmb.21.2.3605. [DOI] [PubMed] [Google Scholar]
- 27.Ottonello L, Frumento G, Arduino N, et al. Differential regulation of spontaneous and immune complex-induced neutrophil apoptosis by proinflammatory cytokines: role of oxidants, Bax and caspase-3. J Leukoc Biol. 2002;72:125–32. [PubMed] [Google Scholar]
- 28.Frevert CW, Huang S, Danaee H, Paulauskis JD, Kobzik L. Functional characterization of the rat chemokine KC and its importance in neutrophil recruitment in a rat model of pulmonary inflammation. J Immunol. 1995;154:335–44. [PubMed] [Google Scholar]
- 29.Kumasaka T, Quinlan WM, Doyle NA, et al. The role of ICAM-1 in endotoxin-induced pneumonia evaluated using ICAM-1 antisense oligonucleotides, anti-ICAM-1 monoclonal antibodies, and ICAM-1 mutant mice. J Clin Invest. 1996;97:2362–9. doi: 10.1172/JCI118679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Qin L, Quinlan WM, Doyle NA, et al. The roles of CD11/CD18 and ICAM-1 in acute Pseudomonas aeruginosa-induced pneumonia in mice. J Immunol. 1996;157:5016–21. [PubMed] [Google Scholar]
- 31.Schmal H, Shanley TP, Jones ML, Friedl HP, Ward PA. Role for macrophage inflammatory protein-2 in lipopolysaccharide-induced lung injury in rats. J Immunol. 1996;156:1963–72. [PubMed] [Google Scholar]
- 32.Greenberger MJ, Strieter RM, Kunkel SL, et al. Neutralization of macrophage inflammatory protein–2 attenuates neutrophil recruitment and bacterial clearance in murine Klebsiella pneumonia. J Infect Dis. 1996;173:159–65. doi: 10.1093/infdis/173.1.159. [DOI] [PubMed] [Google Scholar]
- 33.Alcamo EA, Mizgerd JP, Horwitz BH, et al. Targeted mutation of tumor necrosis factor 1 rescues the RelA-deficient mouse and reveals a critical role for NF-κB in leukocyte recruitment. J Immunol. 2001;167:1592–600. doi: 10.4049/jimmunol.167.3.1592. [DOI] [PubMed] [Google Scholar]
- 34.Poynter ME, Irvin CG, Janssen-Heininger YM. A prominent role for airway epithelial NF-kappa B activation in lipopolysaccharide-induced airway inflammation. J Immunol. 2003;170:6257–65. doi: 10.4049/jimmunol.170.12.6257. [DOI] [PubMed] [Google Scholar]
- 35.Sadikot RT, Han W, Everhart MB, et al. Selective I kappa B kinase expression in airway epithelium generates neutrophilic lung inflammation. J Immunol. 2003;170:1091–8. doi: 10.4049/jimmunol.170.2.1091. [DOI] [PubMed] [Google Scholar]
- 36.Skerrett SJ, Liggitt HD, Hajjar AM, Ernst RK, Miller SI, Wilson CB. Respiratory epithelial cells regulate lung inflammation in response to inhaled endotoxin. Am J Physiol Lung Cell Mol Physiol. 2004;287:L143–52. doi: 10.1152/ajplung.00030.2004. [DOI] [PubMed] [Google Scholar]
- 37.Wang L, Walia B, Evans J, Gewirtz AT, Merlin D, Sitaraman SV. IL-6 induces NF-kappa B activation in the intestinal epithelia. J Immunol. 2003;171:3194–201. doi: 10.4049/jimmunol.171.6.3194. [DOI] [PubMed] [Google Scholar]
- 38.Li Q, Park PW, Wilson CL, Parks WC. Matrilysin shedding of syndecan-1 regulates chemokine mobilization and transepithelial efflux of neutrophils in acute lung injury. Cell. 2002;111:635–46. doi: 10.1016/s0092-8674(02)01079-6. [DOI] [PubMed] [Google Scholar]
- 39.Quinton LJ, Nelson S, Zhang P, et al. Selective transport of cytokine-induced neutrophil chemoattractant from the lung to the blood facilitates pulmonary neutrophil recruitment. Am J Physiol Lung Cell Mol Physiol. 2004;286:L465–72. doi: 10.1152/ajplung.00153.2003. [DOI] [PubMed] [Google Scholar]
- 40.Mizgerd JP, Scott ML, Spieker MR, Doerschuk CM. Functions of IκB proteins in inflammatory responses to E. coli LPS in mouse lungs. Am J Respir Cell Mol Biol. 2002;27:575–82. doi: 10.1165/rcmb.2002-0015OC. [DOI] [PubMed] [Google Scholar]
- 41.Ye P, Garvey PB, Zhang P, et al. Interleukin-17 and lung host defense against Klebsiella pneumoniae infection. Am J Respir Cell Mol Biol. 2001;25:335–40. doi: 10.1165/ajrcmb.25.3.4424. [DOI] [PubMed] [Google Scholar]
- 42.Ye P, Rodriguez FH, Kanaly S, et al. Requirement of interleukin 17 receptor signaling for lung CXC chemokine and granulocyte colony-stimulating factor expression, neutrophil recruitment, and host defense. J Exp Med. 2001;194:519–27. doi: 10.1084/jem.194.4.519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ferretti S, Bonneau O, Dubois GR, Jones CE, Trifilieff A. IL-17, produced by lymphocytes and neutrophils, is necessary for lipopolysaccharide-induced airway neutrophilia: IL-15 as a possible trigger. J Immunol. 2003;170:2106–12. doi: 10.4049/jimmunol.170.4.2106. [DOI] [PubMed] [Google Scholar]
- 44.Happel KI, Zheng M, Young E, et al. Cutting edge: roles of Toll-like receptor 4 and IL-23 in IL-17 expression in response to Klebsiella pneumoniae infection. J Immunol. 2003;170:4432–6. doi: 10.4049/jimmunol.170.9.4432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jeyaseelan S, Chu HW, Young SK, Worthen GS. Transcriptional profiling of lipopolysaccharide-induced acute lung injury. Infect Immun. 2004;72:7247–56. doi: 10.1128/IAI.72.12.7247-7256.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.McLoughlin RM, Hurst SM, Nowell MA, et al. Differential regulation of neutrophil-activating chemokines by IL-6 and its soluble receptor isoforms. J Immunol. 2004;172:5676–83. doi: 10.4049/jimmunol.172.9.5676. [DOI] [PubMed] [Google Scholar]
- 47.Heinrich PC, Behrmann I, Muller-Newen G, Schaper F, Graeve L. Interleukin-6-type cytokine signalling through the gp130/Jak/STAT pathway. Biochem J. 1998;334:297–314. doi: 10.1042/bj3340297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Levy DE, Darnell JE., Jr. STATs: transcriptional control and biological impact. Nat Rev Mol Cell Biol. 2002;3:651–62. doi: 10.1038/nrm909. [DOI] [PubMed] [Google Scholar]
- 49.Riedemann NC, Neff TA, Guo RF, et al. Protective effects of IL-6 blockade in sepsis are linked to reduced C5a receptor expression. J Immunol. 2003;170:503–7. doi: 10.4049/jimmunol.170.1.503. [DOI] [PubMed] [Google Scholar]
- 50.Khalil A, Tullus K, Bartfai T, Bakhiet M, Jaremko G, Brauner A. Renal cytokine responses in acute Escherichia coli pyelonephritis in IL-6-deficient mice. Clin Exp Immunol. 2000;122:200–6. doi: 10.1046/j.1365-2249.2000.01377.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dalrymple SA, Slattery R, Aud DM, Krishna M, Lucian LA, Murray R. Interleukin-6 is required for a protective immune response to systemic Escherichia coli infection. Infect Immun. 1996;64:3231–5. doi: 10.1128/iai.64.8.3231-3235.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Nadeau WJ, Pistole TG, McCormick BA. Polymorphonuclear leukocyte migration across model intestinal epithelia enhances Salmonella typhimurium killing via the epithelial derived cytokine, IL-6. Microbes Infect. 2002;4:1379–87. doi: 10.1016/s1286-4579(02)00020-5. [DOI] [PubMed] [Google Scholar]
- 53.Takeshita F, Ishii KJ, Ueda A, Ishigatsubo Y, Klinman DM. Positive and negative regulatory elements contribute to CpG oligonucleotide-mediated regulation of human IL-6 gene expression. Eur J Immunol. 2000;30:108–16. doi: 10.1002/1521-4141(200001)30:1<108::AID-IMMU108>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 54.Baccam M, Woo SY, Vinson C, Bishop GA. CD40-mediated transcriptional regulation of the IL-6 gene in B lymphocytes: involvement of NF-kappa B, AP-1, and C/EBP. J Immunol. 2003;170:3099–108. doi: 10.4049/jimmunol.170.6.3099. [DOI] [PubMed] [Google Scholar]
- 55.O'Shea JJ, Gadina M, Schreiber RD. Cytokine signaling in 2002: new surprises in the Jak/Stat pathway. Cell. 2002;109(Suppl):S121–31. doi: 10.1016/s0092-8674(02)00701-8. [DOI] [PubMed] [Google Scholar]
- 56.Xie B, Zhao J, Kitagawa M, et al. Focal adhesion kinase activates Stat1 in integrin-mediated cell migration and adhesion. J Biol Chem. 2001;276:19512–23. doi: 10.1074/jbc.M009063200. [DOI] [PubMed] [Google Scholar]
- 57.Hou SX, Zheng Z, Chen X, Perrimon N. The Jak/STAT pathway in model organisms: emerging roles in cell movement. Dev Cell. 2002;3:765–78. doi: 10.1016/s1534-5807(02)00376-3. [DOI] [PubMed] [Google Scholar]
- 58.Kira M, Sano S, Takagi S, Yoshikawa K, Takeda J, Itami S. STAT3 deficiency in keratinocytes leads to compromised cell migration through hyperphosphorylation of p130(cas). J Biol Chem. 2002;277:12931–6. doi: 10.1074/jbc.M110795200. [DOI] [PubMed] [Google Scholar]
- 59.Yahata Y, Shirakata Y, Tokumaru S, et al. Nuclear translocation of phosphorylated STAT3 is essential for vascular endothelial growth factor-induced human dermal microvascular endothelial cell migration and tube formation. J Biol Chem. 2003;278:40026–31. doi: 10.1074/jbc.M301866200. [DOI] [PubMed] [Google Scholar]
- 60.Valente AJ, Xie JF, Abramova MA, Wenzel UO, Abboud HE, Graves DT. A complex element regulates IFN-gamma-stimulated monocyte chemoattractant protein-1 gene transcription. J Immunol. 1998;161:3719–28. [PubMed] [Google Scholar]
- 61.Jaruga B, Hong F, Kim WH, Gao B. IFN-gamma/STAT1 acts as a proinflammatory signal in T cell-mediated hepatitis via induction of multiple chemokines and adhesion molecules: a critical role of IRF-1. Am J Physiol Gastrointest Liver Physiol. 2004;287:G1044–52. doi: 10.1152/ajpgi.00184.2004. [DOI] [PubMed] [Google Scholar]
- 62.Dauer DJ, Ferraro B, Song L, et al. Stat3 regulates genes common to both wound healing and cancer. Oncogene. 2005;24:3397–408. doi: 10.1038/sj.onc.1208469. [DOI] [PubMed] [Google Scholar]
- 63.Dechow TN, Pedranzini L, Leitch A, et al. Requirement of matrix metalloproteinase-9 for the transformation of human mammary epithelial cells by Stat3-C. Proc Natl Acad Sci USA. 2004;101:10602–7. doi: 10.1073/pnas.0404100101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Schumann RR, Kirschning CJ, Unbehaun A, et al. The lipopolysaccharide-binding protein is a secretory class 1 acute-phase protein whose gene is transcriptionally activated by APRF/STAT/3 and other cytokine-inducible nuclear proteins. Mol Cell Biol. 1996;16:3490–503. doi: 10.1128/mcb.16.7.3490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Klein C, Wustefeld T, Assmus U, et al. The IL-6-gp130-STAT3 pathway in hepatocytes triggers liver protection in T cell-mediated liver injury. J Clin Invest. 2005;115:860–9. doi: 10.1172/JCI200523640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Levy DE, Lee CK. What does Stat3 do? J Clin Invest. 2002;109:1143–8. doi: 10.1172/JCI15650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Hokuto I, Ikegami M, Yoshida M, et al. Stat-3 is required for pulmonary homeostasis during hyperoxia. J Clin Invest. 2004;113:28–37. doi: 10.1172/JCI200419491. [DOI] [PMC free article] [PubMed] [Google Scholar]