

Abstract
Decreased levels of the prostate-specific homeobox protein NKX3.1 are correlated with hormone-refractory and metastatic prostate cancer. Thus, it is compelling to define the NKX3.1-regulated genes that may be important for the progression of the advanced stage of the disease. In this study, we showed that vascular endothelial growth factor-C (VEGF-C) is one such target gene of NKX3.1. NKX3.1 inhibited VEGF-C expression in prostate cancer, and the loss of NKX3.1 led to increased VEGF-C expression. Histone deacetylase 1 acted as a corepressor of VEGF-C expression along with NKX3.1. Activated RalA acted in synergy with the loss of NKX3.1 for VEGF-C transcription. Patients with deletions at chromosome 8p21.1-p21.2 as a sole deletion developed lymph node metastasis. Interestingly, the higher expression of VEGF-C in prostate cancer is also correlated with lymph node metastasis. Therefore, regulation of VEGF-C expression by NKX3.1 provides a possible mechanism by which the loss of NKX3.1 protein level leads to lymphangiogenesis in the late stages of advanced prostate cancer.
Introduction
NKX3.1 is an androgen regulated, prostate-specific homeobox gene (1-3), which induces differentiation of prostate tissue and is required for normal prostate development (4-6). NKX3.1 is also important for maintaining the function of the prostate during adulthood (6). Loss of function of NKX3.1 leads to defects in prostatic protein secretion and ductal morphogenesis (5). The NKX3.1 gene is located at chromosome 8p21, which is one of the most frequently deleted regions in human prostate cancer (3, 7-9). Loss of heterozygosity of the 8p arm of the chromosome containing NKX3.1 has been observed in the early stage of prostate cancer (10-12). Therefore, it was initially considered to be a tumor suppressor gene, the loss of which was responsible for the initiation of prostate cancer (12, 13). Because of contradicting results reported by different laboratories, the role of NKX3.1 as an initiator of prostate cancer is presently inconclusive (14, 15). Interestingly, most of these studies agreed that a decreased level of NKX3.1 is correlated with advanced stage prostate cancer exhibiting extracapsular extension (10, 16, 17). Specifically, loss of NKX3.1 expression is strongly associated with hormone-refractory and metastatic prostate cancers (10). In this regard, it is important to note that advanced stage prostate cancer is usually treated with androgen deprivation therapy. Although androgen deprivation therapy may modestly prolong survival, it is palliative but not curative. The vast majority of patients who initially respond to the therapy progress to a hormone refractory, metastatic state (18-20).
Clinical studies showed that patients with deletions at 8p21.1-p21.2 as a sole deletion developed lymph node metastasis (16). Interestingly, the higher expression of vascular endothelial growth factor-C (VEGF-C) in prostate cancer is also correlated with lymph node metastasis (21-23). VEGF-C is a member of VEGF family proteins. This family of proteins is known to induce vasculogenesis, angiogenesis, and lymphangiogenesis (the formation of new lymphatic vessels; refs. 24, 25). The established function of VEGF-C is to induce lymphangiogenesis (26, 27). It is postulated that by inducing lymphangiogenesis, VEGF-C might facilitate lymph node metastasis of prostate cancer. As such, it is important to understand the mechanism of VEGF-C synthesis in the late metastatic stage of prostate cancer. Interestingly, unlike VEGF-A, VEGF-C is not induced by hypoxia, suggesting the involvement of distinct signaling events for its synthesis (28). We therefore focused on understanding whether NKX3.1 may regulate VEGF-C expression, which might provide a mechanistic explanation of the loss of NKX3.1 protein level and its correlation with metastasis in the late stages of advanced prostate cancer. We found five potential binding sites for NKX3.1 (29) in the promoter region of VEGF-C using a transcription factor binding site search algorithm. Further investigation revealed that NKX3.1 binds to the promoter region of VEGF-C and represses its transcription. Our findings suggest that loss of NKX3.1 protein in the later stage of prostate cancer facilitates the expression of VEGF-C, which in turn may promote metastasis.
Materials and Methods
Cell culture
Human prostate cancer cell lines LNCaP [American Type Culture Collection (ATCC) # CRL-1740] and PC3 (ATCC # CRL-1435) were cultured at 37.4°C in RPMI 1640 with l-glutamine (Mediatech) supplemented with penicillin/streptomycin and containing either 10% fetal bovine serum (FBS; Hyclone Laboratories) or 10% androgen-depleted Charcoal-Stripped (CS) FBS (Biomeda).
Cell transfections with NKX3.1 overexpressing plasmid and siRNA
LNCaP cells were plated in 60-mm cell culture dishes 24 h before transfection (~ 1 × 106 cells per dish). The Effectene transfection reagent kit (Qiagen) was used to transfect the NKX3.1 expression vector. pcDNA6 empty vector was used as a control for transfection experiments. All transfections were carried out according to the Qiagen Effectene transfections reagent kit protocol. In preparation for NKX3.1 siRNA transfection, LNCaP cells were cultured in 10% FBS or CS FBS for 24 h at 37.4°C. NKX3.1 Duplex I (Dharmacon RNA Technologies) was diluted to a concentration of 20 mmol/L in 1× universal buffer [20 mmol/L KCl, 6 mmol/L HEPES (pH 7.5), and 0.2 mmol/L MgCl2]. Transfection of LNCaP cells with different concentrations of NKX3.1 siRNA duplex was achieved using the DharmaFECT 3 Reagent (Dharmacon). siRNA transfection was allowed to proceed 72 h before collection of whole cell extract or total RNA.
Immunoprecipitation and Western blot assay
Immunoprecipitation was performed in 0.2 mg of cellular protein from nuclear extracts with antibodies (1 μg) directed against NKX3.1 and HDAC1 and pulled down by protein A-agarose beads (Pharmacia). For Western blot, the whole cell or nuclear extracts or the pulled down product from immunoprecipitation assay were separated by SDS-PAGE; antibodies against NKX3.1, PLCγ, HDAC1, and Histone 2B were used, followed by the secondary antibody incubation, and detected by ESL enhancer reagent from Amersham Biosciences Corp.
RNA isolation and real-time PCR
LNCaP and PC3 cells were washed twice with ice-cold PBS and lysed using the RLT buffer from the RNeasy Minkit (Qiagen). RNA was isolated according to the RNeasy Minikit protocol for animal cells, eluted into a 50 μL total volume, and stored at −70°C.
cDNA preparation
The sequences for human VEGF-C and human 36B4 (housekeeping gene) were obtained from the PubMed Gene Bank and synthesized (Integrated DNA Technology). VEGF-C Forward, 5′-AGT GTC AGG CAG CGA ACA AGA-3′. VEGF-C Reverse, CTT CCT GAG CCA GGC ATC TG-3′. 36B4 Forward, 5′-ATG CAG CAG ATC CGC ATG T-3′. 36B4 Reverse, 5′-TCA TGG TGT TCT TGC CCA TCA-3′. Real-time PCR was performed according to the SyberGreen method in a 25 μL volume using 1 μg total RNA, 12.5 μL reverse transcription-PCR Master Mix (Applied Biosystems), 0.6 μL RNase inhibitor (Applied Biosystems), and 50 nmol/L forward and reverse primers. An ABI System Sequence Detector 7700 (Applied Biosystems) was used with the following regimen of thermal cycling: stage 1, 1 cycle, 10 min at 95°C; stage 2, 40 cycles, 15 s at 95°C, 1 min 60°C.
Relative mRNA amounts were calculated as follows. The cycle number (CT) at which PCR amplification took place for a particular threshold value in the exponential phase for each reaction was determined by the sequence detector. Real-time PCR for the housekeeping gene, 36B4, was performed for each test sample along with VEGF-C. To normalize the value of VEGF-C for each reaction condition, the value of 36B4 at that condition was deducted and the resulting value was designated as Δ. Therefore, Δ = CT (VEGF-C sample)-CT(36B4 sample). To calculate the relative expression of the treated sample compared with the control sample, first DD was determined by deducting the D value of the control sample from the treated sample. Therefore, ΔΔ = Δ (transfected or treated sample) − Δ(empty vector or untreated sample). Finally, relative RNA amount compared with the control was calculated by using the formula, 2−DD. Average and SD for three experiments were calculated.
Detailed method for chromatin immunoprecipitation
Chromatin immunoprecipitation (ChIP) was performed using the ChIP Assay kit from Upstate Biotechnology, Inc. Briefly, 1 × 106 cells was used for each ChIP assay. Protein-DNA cross-linking was carried out by the addition of 1% formaldehyde directly to cell culture followed by incubation at 37°C for 10 min. After thorough washing of the cells with ice-cold PBS, the cells was scraped off and harvested. Cells were lysed with 400 μL of SDS-lysis buffer [1%SDS, 10 mmol/L EDTA, 50 mmol/L Tris (pH 8.1)] supplemented with protease inhibitors [1 mmol/L phenylmethylsulfonyl fluoride (PMSF), 1 mg/mL aprotinin, and 1 mg/mL pepstatin A]. Sonication was performed on ice using a sonicator (Lab-Line Ultra Tip; Lab-Line Instrument, Inc.) preset for 10-s pulse with 10-s intervals. Ten repeated sonication cycles (as previously standardized by us) were applied to achieve chromatin fragmentation of 200 to 1,000 bp range. Fragmented chromatin was diluted 10-fold in ChIP dilution buffer [0.01% SDS, 1% Triton X-100, 2 mmol/L EDTA, 16.7 mmol/L Tris (pH 8.1), and 150 mmol/L NaCl]. Diluted chromatin fragments were precleared by incubating with Protein A agarose beads under constant rotation for 2 h at 4°C. For immunoprecipitation, antibody specific for NKX3.1 and HDAC1 was used for overnight incubation with constant rotation at 4°C. Rabbit IgG was used as a control. Protein-DNA-antibody complex was pulled down by Protein A agarose–salmon sperm DNA beads. After thorough and sequential washing with low salt, high salt, and LiCl-containing buffer, the immune complex was eluted with 1% SDS and 0.1 mol/L NaHCO3. Formaldehyde-cross-links was reversed by adding 5M NaCl and heating at 65°C for 4 h. DNA fragments was recovered by ethanol precipitation after proteinase K digestion and phenol/chloroform extraction.
PCR was performed with VEGF-C promoter–specific primers that will amplify the NKX3.1 putative binding regions:
–997 Forward: AAATTATGGGTGAGGAGTTTCAAATC
–997 Reverse: TCCCCATTGGAAAAGCACTG
–1614 Forward: AACAGGCTAGATCATGGACCGA
–1614 Reverse: TCCCCAAGATGACAGTTCCAGT
–2359 Forward: ACCAAAAAGTCCTGAGCAACTATTC
–2359 Reverse: ACGCCTGTAATCCCAGCACT
The 250 to 360 bp amplicon was analyzed by 2% agarose gel electrophoresis.
Nuclear extract preparation
LNCaP cell suspension was incubated in a hypotonic buffer [10 mmol/L HEPES (pH 7.8), 10 mmol/L KCl, 2 mmol/L MgCl2, 0.1 mmol/L EDTA, 10 μg/mL aprotinin, 3 mmol/L DTT, and 0.2 mmol/L PMSF] for 15 min on ice. Nonionic detergent IGE-PAL (10%; Sigma) was then added to the cell suspension and mixed vigorously. The whole mixture was then centrifuged at 14,000 rpm in an Epppendorf centrifuge for 5 min. The pellets were again suspended in a hypertonic buffer solution [50 mmol/L HEPES (pH 7.8), 50 mmol/L KCl, 300 mmol/L NaCl, 0.1 mmol/L EDTA, 10 μg/mL aprotinin, 3 mmol/L DTT, and 0.2 mmol/L phenylmethylsulfonyl fluoride] and mixed on a rotating rack for 25 min at 4°C. The sample was then centrifuged at 14,000 rpm for 10 min, and the supernatant was collected as nuclear extract.
Retroviral infection of RalA-dominant active vector
To generate retrovirus carrying the RalA Q75L (pBabe-puro-RalA-Q75L) dominant active expression vector, 293T cells were seeded at 3 × 106 cells/100-mm plate. After 24 h, DNA transfection was performed with the EffecteneTM transfection reagent (Qiagen) and with 2 μg of targeted gene (pBabe-puro-RalA-Q75L or LacZ), 1.5 μg of pMD.MLV gag.pol, and 0.5 μg of pMD.G (encoding the cDNAs of the proteins that are required for virus packing). The medium was changed after 16 h. The retrovirus was collected 48 h after transfection and used immediately for infection or frozen at −70°C. For transducing the target gene into LNCaP, the cells were seeded at a density of 1 × 105/well in a 6-well plate. Infection was carried out by adding different volumes of retrovirus solution and fresh medium (final volume was 3 mL; ~2 × 107 plaque-forming units/mL) together with 10 μg/mL Polybrene to cells. The medium was changed after 16 h, and the cells were harvested 48 h after infection. The RalA Q75L expression vector was provided to us by Dr. Channing J Der (University of North Carolina Chapel Hill, Chapel Hill, NC). LacZ retrovirus was used as a control for cell infection experiments.
Results
Using Genomatix software, we detected five potential NKX3.1 consensus-binding sites in the promoter region of VEGF-C (Fig. 1A). This finding suggests that NKX3.1 can potentially bind the VEGF-C promoter. Interestingly, VEGF-C expression in prostate cancer is correlated with metastasis, suggesting a significant expression of VEGF-C in the late stage of invasive prostate cancer when loss of NKX3.1 expression is more pronounced (10, 16). Therefore, we hypothesized that NKX3.1 may be a transcriptional repressor of the VEGF-C gene. In this situation, loss of NKX3.1 expression would facilitate higher VEGF-C expression during the late stage of prostate cancer.
Figure 1.
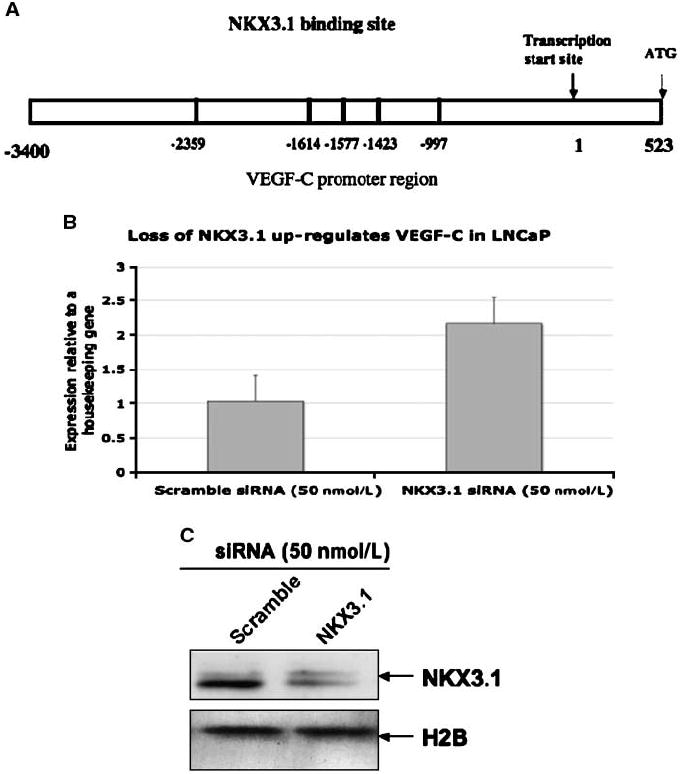
A, schematic diagram of VEGF-C promoter with potential NKX3.1 binding sites. B and C, NKX3.1 regulates VEGF-C mRNA expression in prostate cancer cell. B, LNCaP cells were transfected with NKX3.1 siRNA (50 nmol/L) or scramble siRNA (50 nmol/L). Total RNA was collected and subjected to real-time PCRusing primers for VEGF-C and 36B4 (housekeeping gene). The data presented is the mean of three individual experiments. C, LNCaP cells were transfected with the NKX3.1-siRNA as mentioned above. Nuclear extracts were subjected to Western blotting using antibody for NKX3.1.
NKX3.1 repressed VEGF-C mRNA expression in prostate cancer cells
To test whether NKX3.1 can indeed repress VEGF-C transcription, we knocked down endogenous NKX3.1 expression in LNCaP cells by siRNA specific for NKX3.1 and determined VEGF-C mRNA levels by real-time PCR (Fig. 1B). Expression of VEGF-C mRNA under each experimental condition was calculated relative to the rRNA 36B4 (details of this procedure have been discussed in the Materials and Methods section). VEGF-C mRNA levels were increased in LNCaP cells upon NKX3.1 siRNA treatment compared with scrambled siRNA, suggesting its involvement in regulating VEGF-C mRNA. Figure 1C represents the Western blot data showing efficient knockdown of NKX3.1 protein in LNCaP cells by siRNA. Next, we overexpressed increasing concentrations NKX3.1 in LNCaP cells and determined VEGF-C mRNA levels by real-time PCR. A dose-dependent decrease in VEGF-C mRNA levels was observed with increasing NKX3.1 expression in LNCaP cells (Fig. 2A). Figure 2B shows protein expression of NKX3.1 in the transfected LNCaP cells. The regulation of VEGF-C expression by NKX3.1 was further confirmed when its expression was knocked down in NKX3.1-overexpressing LNCaP cells with increasing doses of NKX3.1-targated siRNA. We detected a dose-dependent increase in VEGF-C mRNA (Fig. 2C). A dose-dependent decrease in NKX3.1 protein expression resulting from increasing siRNA concentration was confirmed by Western blots (Fig. 2D). The inhibitory effect of NKX3.1 on VEGF-C transcription was not cell line specific, as we observed similar dose-dependent regulation of VEGF-C mRNA levels in PC-3 cells (Fig. 3A). Figure 3B shows protein expression of NKX3.1 in the transfected PC3 cells. Thus, these results (Figs. 2A and 3A, C, and D) clearly point to an involvement of NKX3.1 in regulating VEGF-C expression.
Figure 2.
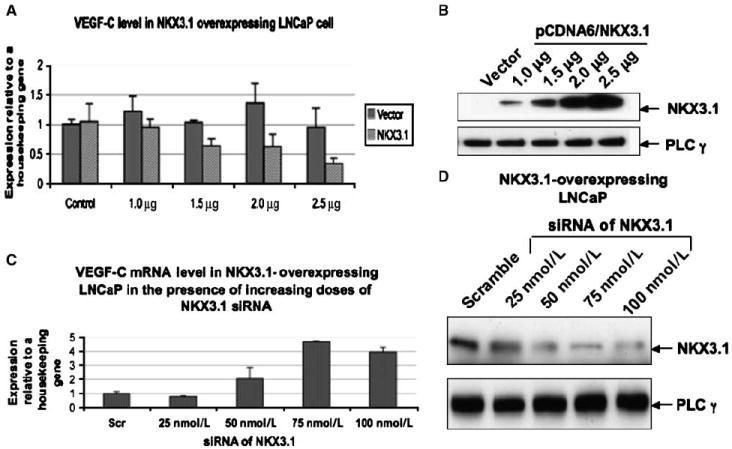
NKX3.1 overexpression inhibits VEGF-C mRNA expression in prostate cancer. A, LNCaP cells were transfected with increasing doses of NKX3.1-expressing plasmid. Total RNA was collected and subjected to real-time PCR using primers for VEGF-C and 36B4 (housekeeping gene). The data presented is the mean of three individual experiments. B, LNCaP were transfected with the NKX3.1-expressing plasmid as mentioned above. Cell lysates were subjected to Western blotting using antibody for NKX3.1. C, LNCaP cells were transfected with increasing doses of siRNA targeted to NKX3.1 and a fixed dose (2.0 mg/60-mm dish) of NKX3.1-expressing plasmid. Total RNA was collected and subjected to real-time PCR using primers for VEGF-C and 36B4 (housekeeping gene). The data presented is the mean of three individual experiments. D, LNCaP were transfected with the NKX3.1 siRNA and NKX3.1-expressing plasmid as mentioned in C. Cell lysates were subjected to Western blotting using antibody for NKX3.1.
Figure 3.

A, PC3 cells were transfected with increasing doses of NKX3.1-expressing plasmid. Total RNA was collected and subjected to real-time PCR using primers for VEGF-C and 36B4 (housekeeping gene). The data presented is the mean of three individual experiments. B, PC3 were transfected with the NKX3.1-expressing plasmid as mentioned in Fig. 2E. Cell lysates were subjected to Western blotting using antibody for NKX3.1. C and D, recruitment of NKX3.1 to VEGF-C promoter. C, ChIP assay. Cross-linked chromatin-protein complexes were isolated from LNCaP cells cultured in 10% normal serum (NS) and rum (CS, androgen deprived). Immunoprecipitation of chromatin fragments was carried out with the NKX3.1 as well as rabbit IgG (control) antibody. After reverse cross-linking, DNA fragments were isolated and the NKX3.1 binding region on the VEGF-C promoter was amplified by PCR with the VEGF-C promoter–specific primers. An amplified PCR product was detected when immunoprecipitation was carried out with the NKX3.1 antibody. D, nuclear extracts from LNCaP cells were cultured in 10% normal serum and CS, and were subjected to Western blotting using antibody against NKX3.1.
Direct binding of NKX3.1 to the VEGF-C promoter region
We next tested whether NKX3.1 can bind any of the putative NKX3.1 binding sites within the VEGF-C promoter. ChIP was used to pull down DNA fragments that specifically bind to NKX3.1 followed by PCR amplification of potential NKX3.1 binding sites in the VEGF-C promoter. Interestingly, only the −997 bp region of the VEGF-C promoter was amplified. This is the NKX3.1 binding site closest to the transcription start site of the VEGF-C gene (Fig. 3C). Moreover, androgen withdrawal reduced NKX3.1 binding to VEGF-C promoter (Fig. 3C), which is consistent with our previous finding that androgens induce NKX3.1 expression in LNCaP cells (2). We also detected a decrease in NKX3.1 protein in LNCaP cells grown in the absence of androgen (Fig. 3D).
NKX3.1 requires HDAC1 for the transcriptional repression of VEGF-C
Histone acetylation is an important posttranslational modification, which is associated with transcriptional activation (30). Therefore, histone deacetylases (HDAC) are usually transcriptional repressors because of their ability to deacetylate histones (31). Previously, it was shown that NKX3.1 could associate with HDAC1 (a class 1 type HDAC; ref. 32). We hypothesized that NKX3.1 may facilitate the recruitment of HDAC to the VEGF-C promoter, which in turn could promote transcriptional repression.
To test this hypothesis, we examined the role of HDAC for NKX3.1-mediated transcriptional repression of the VEGF-C gene. We inhibited HDAC activity in LNCaP cells with a broad-spectrum HDAC inhibitor, suberoylanilide hydroxamic acid (SAHA; ref. 33). Indeed, SAHA at a concentration of 0.4 μmol/L or higher (Fig. 4A) abrogated the effects of NKX3.1 overexpression on VEGF-C mRNA expression in LNCaP cells. This experiment suggested that NKX3.1 requires HDAC activity for the transcriptional repression of VEGF-C gene. Moreover, we detected an association between endogenous NKX3.1 and HDAC1 in LNCaP cells by immunopre-cipitation-Western blot, which is consistent with a previous report that HDAC1 could associate with NKX3.1 (Fig. 4B; ref. 32). Therefore, we hypothesized that HDAC1 is recruited to the VEGF-C promoter by NKX3.1, which results in suppressed VEGF-C transcription. Indeed, by knocking down HDAC1 with siRNA in LNCaP cells, we eliminated the decrease in VEGF-C expression upon NKX3.1 overexpression, suggesting its involvement in NKX3.1-mediated transcriptional repression of VEGF-C mRNA (Fig. 4C). Efficient knocking down of HDAC1 protein in LNCaP cells by siRNA was confirmed by Western blot (Fig. 4D).
Figure 4.
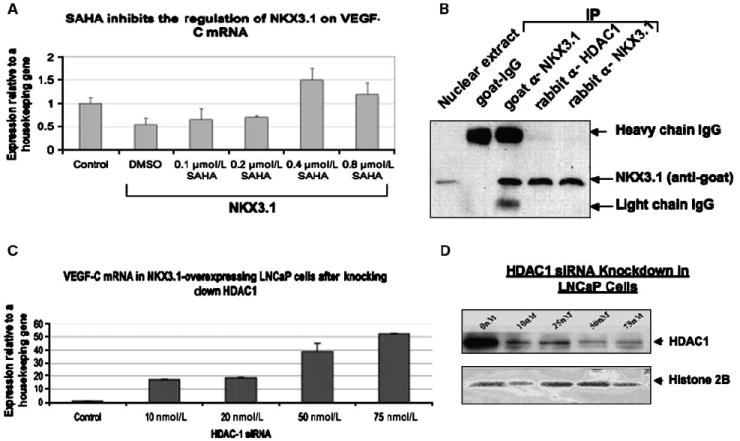
Requirement of HDAC for the transcriptional repression of VEGF-C. A, LNCaP cells were transfected with fixed dose (0.75 mg/60 mm dish) of NKX3.1-expressing plasmid. HDAC inhibitor SAHA was added to the LNCaP cells in increasing doses. Total RNA was collected and subjected to real-time PCR using primers for VEGF-C and 36B4 (housekeeping gene). The data presented is the mean of three individual experiments. B, immunoprecipitation of the nuclear extracts with anti-HDAC1 or anti-NKX3.1 antibodies followed by Western blot with anti-NKX3.1 antibody. C, LNCaP cells were transfected with increasing doses of siRNA targeted to HDAC1 and a fixed dose (0.75 mg/60-mm dish) of NKX3.1-expressing plasmid. Total RNA was collected and subjected to real-time PCR using primers for VEGF-C and 36B4 (housekeeping gene). The data presented is the mean of three individual experiments. D, LNCaP were transfected with the HDAC-1 siRNA and NKX3.1-expressing plasmid as mentioned in 4C. Cell nuclear extracts were subjected to Western blotting using antibody for HDAC1 and Histone 2B.
HDAC1 and NKX3.1 are corecruited to the VEGF-C promoter
We hypothesized that HDAC1 is recruited at the VEGF-C promoter by NKX3.1. Therefore, we wanted to determine whether we could pull down the NKX3.1 binding site (−997) of VEGF-C promoter by ChIP analysis with an anti-HDAC1 antibody. Figure 5A shows that HDAC1 was recruited to the NKX3.1 binding site of VEGF-C promoter. Importantly, knocking down NKX3.1 by siRNA inhibited HDAC1 recruitment, which indicated that HDAC1 can associate with the −997 region of VEGF-C promoter only in the presence of NKX3.1 (Fig. 5A). This result therefore suggests that HDAC1 indirectly associates with the VEGF-C promoter and that this interaction requires NKX3.1. To further confirm that NKX3.1 and HDAC1 occupy the same chromatin fragments of the VEGF-C promoter, we performed re-ChIP experiments. The first-step ChIP was performed with an anti-NKX3.1 antibody (Fig. 5B). Re-ChIP with an anti-HDAC1 antibody was performed with the cross-linked protein-DNA complex eluted from the first-step ChIP. We were able to amplify the −997 region of the VEGF-C promoter from the DNA fragments isolated from the re-ChIP with the anti-HDAC1 antibody (Fig. 5B). We also performed another re-ChIP experiment where the first-step ChIP was performed with anti-HDAC1 antibody and the re-ChIP was performed with an anti-NKX3.1 antibody (Fig. 5C). As expected, once again, we were able to amplify the −997 bp region of NKX3.1 binding site of the VEGF-C promoter (Fig. 5C). These results clearly show that NKX3.1 and HDAC1 occupy the same region of VEGF-C promoter at the same time points.
Figure 5.
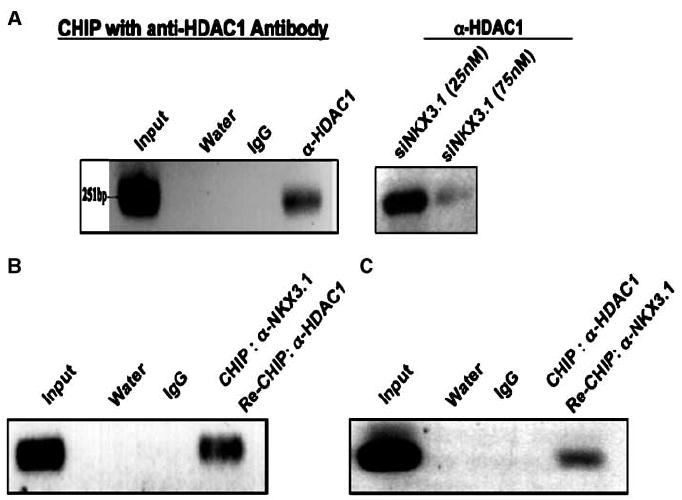
Recruitment of HDAC-1 to NKX3.1 binding site of VEGF-C promoter. A, ChIP: cross-linked chromatin-protein complexes were isolated from regular LNCaP cells or LNCaP cells transfected with different doses of siRNA against NKX3.1. Immunoprecipitation of chromatin fragments was carried out with the HDAC-1 as well as rabbit IgG (control) antibody. After reverse cross-linking, DNA fragments were isolated and the NKX3.1 binding regions (−997, −1614, and −2359 bp) on the VEGF-C promoter were amplified by PCR with the VEGF-C promoter–specific primers. Only −997 bp was positive. Re-ChIP: B, re-ChIP with anti-HDAC1 antibody as well as rabbit IgG (control) antibody was performed with the cross-linked protein-DNA complex eluted from the first step ChIP with NKX3.1 antibody as described in Fig. 3A. C, re-ChIP with anti-NKX3.1 antibody was performed with the cross-linked protein-DNA complex eluted from the first step ChIP with anti-HDAC1 antibody. Re-ChIP with rabbit IgG was used as control.
Based on the results presented in Figs. 4 and 5, it can be concluded that HDAC1 is recruited to the VEGF-C promoter by associating with NKX3.1 and thereby exerts its inhibitory effect on VEGF-C transcription.
Loss of NKX3.1 synergize with active RalA for VEGF-C expression in prostate cancer cell
In our previous publications, we showed that either activated RalA or FOXO-1 induced VEGF-C expression during androgen ablation (34, 35). A question arising from these studies was the relative contribution of NKX3.1 regulation to VEGF-C transcription in the presence of the positive regulatory signals. We hypothesized that the absence of the NKX3.1-mediated inhibitory mechanism during androgen ablation would synergize with positive stimulatory pathways to induce the up-regulation of VEGF-C mRNA. To test this hypothesis, we depleted NKX3.1 in LNCaP cells with siRNA and simultaneously transfected the cells with an activated form of RalA (Dominant active form of RalA or RalA-DA). We chose doses of RalA-DA, which had been shown previously to not induce an increase in VEGF-C mRNA (34). As a control, the same doses of RalA-DA were added to LNCaP cells with intact endogenous NKX3.1. As expected, knocking down NKX3.1 itself in LNCaP cells increased VEGF-C mRNA level compared with the control (Fig. 6A). Interestingly, addition of RalA-DA induced a further increase in VEGF-C mRNA upon knocking down NKX3.1 in LNCaP cells. However, there was no effect of RalA-DA on maintaining endogenous levels of NKX3.1 in LNCaP cells (Fig. 6A). This result clearly showed that positive stimulatory pathways, such as activated RalA and loss of NKX3.1, can synergize to induce VEGF-C transcription in prostate cancer cells.
Figure 6.
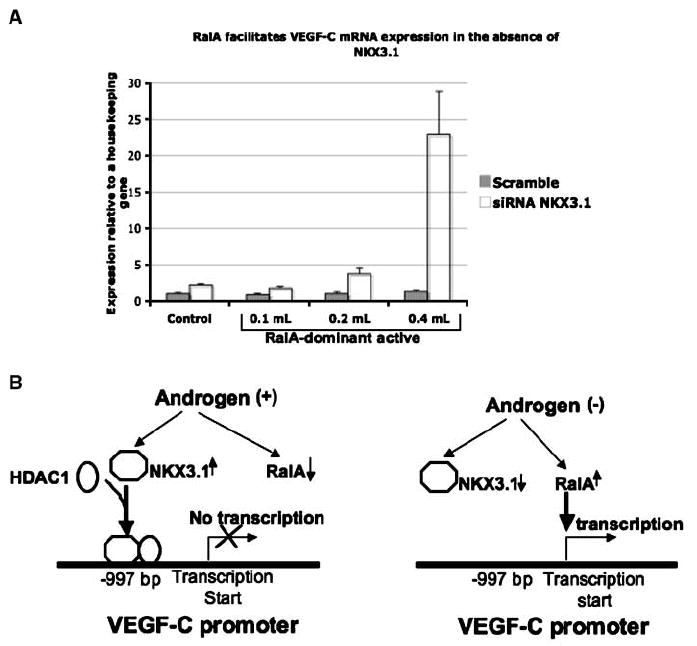
A, LNCaP cells transfected with siRNA against NKX3.1 or scramble siRNA were infected with different doses of RalA Q72L dominant active retrovirus or LacZ control retrovirus for 48 h. Total RNA was collected and subjected to real-time PCR using primers for VEGF-C and 36B4 internal control. The data presented here represents the mean of three individual experiments. B, schematic diagram detailing the molecular mechanisms of androgen-regulated VEGF-C transcription in prostate cancer.
In conclusion, our studies suggest that NKX3.1 is a transcriptional repressor of the VEGF-C gene in prostate cancer. Together with our previous reports (34, 35), these present findings elaborate the molecular mechanisms of androgen-regulated VEGF-C expression in prostate cancer cells. We propose that androgens can inhibit VEGF-C transcription by up-regulating levels of NKX3.1. Alternatively decreased levels of androgens abrogate NKX3.1-medited transcriptional repression of the VEGF-C gene, which favors positive stimulatory pathways such as RalA to induce VEGF-C transcription (Fig. 6B).
Discussion
In previous studies, we showed that VEGF-C is expressed at higher levels in prostate cancer cells in the absence of androgens and might therefore be involved in the onset of androgen refractory prostate cancer (35). In the current study, the regulation of VEGF-C by NKX3.1 was addressed because of the following reasons: (a) NKX3.1 can act as a transcriptional corepressor (29, 36); (b) the VEGF-C promoter has potential binding sites for NKX3.1 (Fig. 1A); and (c) androgen regulates NKX3.1 expression (1-3); in the absence of androgen, NKX3.1 levels are diminished and therefore are unable to inhibit VEGF-C transcription, thus explaining in part the higher expression of VEGF-C in the absence of androgen; and (d) NKX3.1 protein expression is down-regulated in hormone refractory prostate tumors (10, 37).
In this present study, we first showed that NKX3.1 is able to repress VEGF-C mRNA in prostate cancer cells, which suggests its involvement as a repressor of VEGF-C gene transcription (Figs. 1B, 2, and 3A). We then showed by ChIP assay that NKX3.1 indeed binds at one of the potential binding sites of VEGF-C promoter (−997 bp from the transcription start site; Fig. 3B). Our hypothesis that NKX3.1 was low in the absence of androgen and, therefore, could not inhibit VEGF-C transcription, was further supported by the observation of less binding of NKX3.1 to the VEGF-C promoter when cells were cultured in androgen-depleted medium (Fig. 3B). We also observed that SAHA, a generic HDAC inhibitor, could override transcriptional repression by NKX3.1 (Fig. 4A). We then showed that HDAC1, a specific type I HDAC, associates with NKX3.1 and also was responsible for repressing VEGF-C mRNA (Fig. 4B and C). From ChIP assays, we concluded that HDAC1 is recruited to the same region of the VEGF-C promoter where HDAC1 binds and that this recruitment is dependent on NKX3.1 (Fig. 5A). Finally, by re-ChIP assays, we conclusively proved that HDAC1 was corecruited to the VEGF-C promoter with NKX3.1 (Fig. 5B and C). These observations suggest a mechanism of transcriptional repression of VEGF-C by NKX3.1 whereby NKX3.1 specifically binds −997 bp region of VEGF-C promoter and recruits HDAC1. HDAC1 by deacetylating the histones, promotes a transcriptionally inactive chromatin domain at the VEGF-C promoter and, thus inhibits VEGF-C gene transcription.
HDAC inhibitors are known to stimulate antiproliferative factors and apoptosis both in vitro and in vivo. These observations have generated considerable interest in HDAC inhibitors as a potential form of cancer treatment. Based on structural homologies, there are four major classes (class I–IV) of HDAC inhibitors (31). Despite the rapid incorporation of this class of compounds into clinical trials, the precise mechanism of action of each of the HDAC isoforms is not completely known. Our present study suggests that inhibiting HDAC1 (a class 1 HDAC inhibitor) in prostate cancer cells will facilitate VEGF-C synthesis and therefore should favor prostate cancer progression. SAHA being a broad-spectrum HDAC inhibitor also inhibits HDAC1 and therefore would not be the ideal therapeutic choice in advanced prostate cancer. Alternatively, targeting specific HDAC isoforms other than HDAC1 may be a better approach at this stage of the disease. Like HDAC1, other members of class I HDACs (HDAC 2, 3, and 8) have similar antiproliferative and apoptotic effects, and should be considered as therapeutic targets (31). Class II inhibitors also show significant anticancer functions. A recent finding suggests that inhibition of class II HDAC6 leads to acetylation and disruption of the chaperone function of heat-shock protein 90 (HSP90; ref. 38). HSP90 is crucial for androgen receptor function, and therefore inhibiting HDAC6 may be important in prostate cancer treatment. Other studies suggest a general therapeutic advantage in treating solid tumors by combining SAHA with ionizing radiation. It has been shown recently that a class II HDAC, HDAC4, interacts with 53BP1 (p53 binding protein) and facilitates the repair of damaged DNA (39). Inhibiting HDAC4 enhances sensitivity to the effects of ionizing radiation in cancer cells similar to SAHA. Interestingly, nuclear accumulation of HDAC4 also coincides with the loss of androgen sensitivity in hormone refractory prostate cancer cells (40). Therefore, we would suggest that isoform-specific HDAC inhibitors might inhibit progression of prostate cancer, as does SAHA for a wide range of hematologic and solid tumors. These isoform-specific inhibitors may offer greater therapeutic advantages compared with broad-spectrum HDACs such as SAHA, as they should modulate only the disease-focused genes and have a reduced possibility for developing resistance to HDAC inhibitors (31). In this respect, it should be noted that increased thioredoxins, a scavenger for reactive oxygen species, in prostate cancer cells have been shown to be responsible for resistance toward SAHA (41). Our results along with other published data therefore suggest that although SAHA has shown positive clinical responses in many cancers, it might not be effective in all cancer types and in all stages. In some specific cancer type, it may even induce expression of genes that facilitate cancer progression and/ or drug resistance. Therefore, inhibitors for specific HDAC isoforms may be the better choice in this scenario.
It is also important to note that androgen ablation induces positive stimulatory pathways such as activation of RalA for VEGF-C transcription (34, 35). Therefore, we propose that prolonged withdrawal of androgen favors high levels of VEGF-C transcription by decreasing the transcriptional inhibition of NKX3.1 and also by activating positive regulatory pathways such as RalA. The activator stimulus should be more pronounced without the inhibitory signals. This was indeed the case as we observed a significant increase in VEGF-C expression by activated RalA only when endogenous NKX3.1 was depleted in the LNCaP cells (Fig. 6A). Similar synergism with NKX3.1 was observed in Nkx3.1-Pten mutant mice, where inactivation of Pten synergizes with loss of Nkx3.1 during prostate cancer progression (42, 43). Interestingly, prostate epithelial cells from these mutant mice can proliferate and survive in the absence of androgens and develop an androgen refractory phenotype (43, 44).
Basic and clinical studies involving tissue culture or animal models underscore the importance of NKX3.1 in prostate cancer (42-50). However, few NKX3.1 target genes in prostate cancer, which lead to cancer-specific phenotypes, are known. In this study, we identified VEGF-C as one such target gene of NKX3.1 that might be important for the metastatic progression of prostate cancer. Previous reports have correlated loss of NKX3.1 with lymph node metastasis in human prostate cancer (7). Our study therefore provides a mechanistic explanation linking NKX3.1 to lymph node metastasis via VEGF-C.
Acknowledgments
Grant support: Research Scholar Grant from American Cancer Society (RSG-070944-01-CSM), Career Development project in prostate spore grant (Mayo Clinic; 1 PSOCA91956-3), and New Investigator award grant from US Army Medical Research and Material Command (2B1636; K. Datta); NIH grants CA 121277, CA 91956, CA 125747, and the TJ Martell Foundation (FNDT Martell/#1-8; D.J. Tindall); and a SPORE developmental project grant and an Eagle’s grant (Eagle Foundation for Cancer Research; M.H. Muders).
We thank Dr. Scott Dehm for his helpful discussion.
Footnotes
Disclosure of Potential Conflicts of Interest No potential conflicts of interest were disclosed.
References
- 1.Bieberich CJ, Fujita K, He WW, Jay G. Prostate-specific and androgen-dependent expression of a novel homeobox gene. J Biol Chem. 1996;271:31779–82. doi: 10.1074/jbc.271.50.31779. [DOI] [PubMed] [Google Scholar]
- 2.Prescott JL, Blok L, Tindall DJ. Isolation and androgen regulation of the human homeobox cDNA, NKX3.1. Prostate. 1998;35:71–80. doi: 10.1002/(sici)1097-0045(19980401)35:1<71::aid-pros10>3.0.co;2-h. [DOI] [PubMed] [Google Scholar]
- 3.He WW, Sciavolino PJ, Wing J, et al. A novel human prostate-specific, androgen-regulated homeobox gene (NKX3.1) that maps to 8p21, a region frequently deleted in prostate cancer. Genomics. 1997;43:69–77. doi: 10.1006/geno.1997.4715. [DOI] [PubMed] [Google Scholar]
- 4.Tanaka M, Lyons GE, Izumo S. Expression of the Nkx3.1 homobox gene during pre and postnatal development. Mech Dev. 1999;85:179–82. doi: 10.1016/s0925-4773(99)00084-2. [DOI] [PubMed] [Google Scholar]
- 5.Bhatia-Gaur R, Donjacour AA, Sciavolino PJ, et al. Roles for Nkx3.1 in prostate development and cancer. Genes Dev. 1999;13:966–77. doi: 10.1101/gad.13.8.966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shen MM, Abate-Shen C. Roles of the Nkx3.1 homeobox gene in prostate organogenesis and carcinogenesis. Dev Dyn. 2003;228:767–78. doi: 10.1002/dvdy.10397. [DOI] [PubMed] [Google Scholar]
- 7.Bova GS, Carter BS, Bussemakers MJ, et al. Homozygous deletion and frequent allelic loss of chromosome 8p22 loci in human prostate cancer. Cancer Res. 1993;53:3869–73. [PubMed] [Google Scholar]
- 8.MacGrogan D, Levy A, Bostwick D, Wagner M, Wells D, Bookstein R. Loss of chromosome arm 8p loci in prostate cancer: mapping by quantitative allelic imbalance. Genes Chromosomes Cancer. 1994;10:151–9. doi: 10.1002/gcc.2870100302. [DOI] [PubMed] [Google Scholar]
- 9.Suzuki H, Emi M, Komiya A, et al. Localization of a tumor suppressor gene associated with progression of human prostate cancer within a 1.2 Mb region of 8p22–21.3. Genes Chromosomes Cancer. 1995;13:168–74. doi: 10.1002/gcc.2870130306. [DOI] [PubMed] [Google Scholar]
- 10.Bowen C, Bubendorf L, Voeller HJ, et al. Loss of NKX3.1 expression in human prostate cancers correlates with tumor progression. Cancer Res. 2000;60:6111–5. [PubMed] [Google Scholar]
- 11.Bethel CR, Faith D, Li X, et al. Decreased NKX3.1 protein expression in focal prostatic atrophy, prostatic intraepithelial neoplasia, and adenocarcinoma: association with gleason score and chromosome 8p deletion. Cancer Res. 2006;66:10683–90. doi: 10.1158/0008-5472.CAN-06-0963. [DOI] [PubMed] [Google Scholar]
- 12.Magee JA, Abdulkadir SA, Milbrandt J. Haploinsufficiency at the Nkx3.1 locus. A paradigm for stochastic, dosage-sensitive gene regulation during tumor initiation. Cancer Cell. 2003;3:273–83. doi: 10.1016/s1535-6108(03)00047-3. [DOI] [PubMed] [Google Scholar]
- 13.Kim MJ, Bhatia-Gaur R, Banach-Petrosky WA, et al. Nkx3.1 mutant mice recapitulate early stages of prostate carcinogenesis. Cancer Res. 2002;62:2999–3004. [PubMed] [Google Scholar]
- 14.Xu LL, Srikantan V, Sesterhenn IA, et al. Expression profile of an androgen regulated prostate specific homeobox gene NKX3.1 in primary prostate cancer. J Urol. 2000;163:972–9. [PubMed] [Google Scholar]
- 15.Ornstein DK, Cinquanta M, Weiler S, et al. Expression studies and mutational analysis of the androgen regulated homeobox gene NKX3.1 in benign and malignant prostate epithelium. J Urol. 2001;165:1329–34. [PubMed] [Google Scholar]
- 16.Oba K, Matsuyama H, Yoshihiro S, et al. Two putative tumor suppressor genes on chromosome arm 8p may play different roles in prostate cancer. Cancer Genet Cytogenet. 2001;124:20–6. doi: 10.1016/s0165-4608(00)00248-x. [DOI] [PubMed] [Google Scholar]
- 17.Korkmaz CG, Korkmaz KS, Manola J, et al. Analysis of androgen regulated homeobox gene NKX3.1 during prostate carcinogenesis. J Urol. 2004;172:1134–9. doi: 10.1097/01.ju.0000136526.78535.b8. [DOI] [PubMed] [Google Scholar]
- 18.Dehm SM, Tindall DJ. Androgen receptor structural and functional elements: role and regulation in prostate cancer. Mol Endocrinol. 2007;21:2855–63. doi: 10.1210/me.2007-0223. [DOI] [PubMed] [Google Scholar]
- 19.Dehm SM, Tindall DJ. Molecular regulation of androgen action in prostate cancer. J Cell Biochem. 2006;99:333–44. doi: 10.1002/jcb.20794. [DOI] [PubMed] [Google Scholar]
- 20.Debes JD, Tindall DJ. Mechanisms of androgen-refractory prostate cancer. N Engl J Med. 2004;351:1488–90. doi: 10.1056/NEJMp048178. [DOI] [PubMed] [Google Scholar]
- 21.Jennbacken K, Vallbo C, Wang W, Damber JE. Expression of vascular endothelial growth factor C (VEGF-C) and VEGF receptor-3 in human prostate cancer is associated with regional lymph node metastasis. Prostate. 2005;65:110–6. doi: 10.1002/pros.20276. [DOI] [PubMed] [Google Scholar]
- 22.Tsurusaki T, Kanda S, Sakai H, et al. Vascular endothelial growth factor-C expression in human prostatic carcinoma and its relationship to lymph node metastasis. Br J Cancer. 1999;80:309–13. doi: 10.1038/sj.bjc.6690356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zeng Y, Opeskin K, Horvath LG, Sutherland RL, Williams ED. Lymphatic vessel density and lymph node metastasis in prostate cancer. Prostate. 2005;65:222–30. doi: 10.1002/pros.20288. [DOI] [PubMed] [Google Scholar]
- 24.Dvorak HF, Brown LF, Detmar M, Dvorak AM. Vascular permeability factor/vascular endothelial growth factor, microvascular hyperpermeability, and angiogenesis. Am J Pathol. 1995;146:1029–39. [PMC free article] [PubMed] [Google Scholar]
- 25.Folkman J. Tumor angiogenesis: therapeutic implications. N Engl J Med. 1971;285:1182–6. doi: 10.1056/NEJM197111182852108. [DOI] [PubMed] [Google Scholar]
- 26.Karpanen T, Egeblad M, Karkkainen MJ, et al. Vascular endothelial growth factor C promotes tumor lymphangiogenesis and intralymphatic tumor growth. Cancer Res. 2001;61:1786–90. [PubMed] [Google Scholar]
- 27.Skobe M, Hawighorst T, Jackson DG, et al. Induction of tumor lymphangiogenesis by VEGF-C promotes breast cancer metastasis. Nat Med. 2001;7:192–8. doi: 10.1038/84643. [DOI] [PubMed] [Google Scholar]
- 28.Enholm B, Paavonen K, Ristimaki A, et al. Comparison of VEGF, VEGF-B, VEGF-C and Ang-1 mRNA regulation by serum, growth factors, oncoproteins and hypoxia. Oncogene. 1997;14:2475–83. doi: 10.1038/sj.onc.1201090. [DOI] [PubMed] [Google Scholar]
- 29.Steadman DJ, Giuffrida D, Gelmann EP. DNA-binding sequence of the human prostate-specific homeodomain protein NKX3.1. Nucleic Acids Res. 2000;28:2389–95. doi: 10.1093/nar/28.12.2389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shahbazian MD, Grunstein M. Functions of site-specific histone acetylation and deacetylation. Annu Rev Biochem. 2007;76:75–100. doi: 10.1146/annurev.biochem.76.052705.162114. [DOI] [PubMed] [Google Scholar]
- 31.Karagiannis TC, El-Osta A. Will broad-spectrum histone deacetylase inhibitors be superseded by more specific compounds? Leukemia. 2007;21:61–5. doi: 10.1038/sj.leu.2404464. [DOI] [PubMed] [Google Scholar]
- 32.Lei Q, Jiao J, Xin L, et al. NKX3.1 stabilizes p53, inhibits AKT activation, and blocks prostate cancer initiation caused by PTEN loss. Cancer Cell. 2006;9:367–78. doi: 10.1016/j.ccr.2006.03.031. [DOI] [PubMed] [Google Scholar]
- 33.Finnin MS, Donigian JR, Cohen A, et al. Structures of a histone deacetylase homologue bound to the TSA and SAHA inhibitors. Nature. 1999;401:188–93. doi: 10.1038/43710. [DOI] [PubMed] [Google Scholar]
- 34.Rinaldo F, Li J, Wang E, Muders M, Datta K. RalA regulates vascular endothelial growth factor-C (VEGF-C) synthesis in prostate cancer cells during androgen ablation. Oncogene. 2007;26:1731–8. doi: 10.1038/sj.onc.1209971. [DOI] [PubMed] [Google Scholar]
- 35.Li J, Wang E, Rinaldo F, Datta K. Upregulation of VEGF-C by androgen depletion: the involvement of IGF-IR-FOXO pathway. Oncogene. 2005;24:5510–20. doi: 10.1038/sj.onc.1208693. [DOI] [PubMed] [Google Scholar]
- 36.Abdulkadir SA. Mechanisms of prostate tumorigenesis: roles for transcription factors Nkx3.1 and Egr1. Ann N Y Acad Sci. 2005;1059:33–40. doi: 10.1196/annals.1339.018. [DOI] [PubMed] [Google Scholar]
- 37.Lee EC, Tenniswood MP. Emergence of metastatic hormone-refractory disease in prostate cancer after anti-androgen therapy. J Cell Biochem. 2004;91:662–70. doi: 10.1002/jcb.20040. [DOI] [PubMed] [Google Scholar]
- 38.Murphy PJ, Morishima Y, Kovacs JJ, Yao TP, Pratt WB. Regulation of the dynamics of hsp90 action on the glucocorticoid receptor by acetylation/deacetylation of the chaperone. J Biol Chem. 2005;280:33792–9. doi: 10.1074/jbc.M506997200. [DOI] [PubMed] [Google Scholar]
- 39.Kao GD, McKenna WG, Guenther MG, Muschel RJ, Lazar MA, Yen TJ. Histone deacetylase 4 interacts with 53BP1 to mediate the DNA damage response. J Cell Biol. 2003;160:1017–27. doi: 10.1083/jcb.200209065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Halkidou K, Cook S, Leung HY, Neal DE, Robson CN. Nuclear accumulation of histone deacetylase 4 (HDAC4) coincides with the loss of androgen sensitivity in hormone refractory cancer of the prostate. Eur Urol. 2004;45:382–9. doi: 10.1016/j.eururo.2003.10.005. author reply 9. [DOI] [PubMed] [Google Scholar]
- 41.Xu W, Ngo L, Perez G, Dokmanovic M, Marks PA. Intrinsic apoptotic and thioredoxin pathways in human prostate cancer cell response to histone deacetylase inhibitor. Proc Natl Acad Sci U S A. 2006;103:15540–5. doi: 10.1073/pnas.0607518103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Abate-Shen C, Banach-Petrosky WA, Sun X, et al. Nkx3.1; Pten mutant mice develop invasive prostate adenocarcinoma and lymph node metastases. Cancer Res. 2003;63:3886–90. [PubMed] [Google Scholar]
- 43.Gao H, Ouyang X, Banach-Petrosky WA, Shen MM, Abate-Shen C. Emergence of androgen independence at early stages of prostate cancer progression in Nkx3.1; Pten mice. Cancer Res. 2006;66:7929–33. doi: 10.1158/0008-5472.CAN-06-1637. [DOI] [PubMed] [Google Scholar]
- 44.Banach-Petrosky W, Jessen WJ, Ouyang X, et al. Prolonged exposure to reduced levels of androgen accelerates prostate cancer progression in Nkx3.1; Pten mutant mice. Cancer Res. 2007;67:9089–96. doi: 10.1158/0008-5472.CAN-07-2887. [DOI] [PubMed] [Google Scholar]
- 45.Abate-Shen C, Shen MM. Molecular genetics of prostate cancer. Genes Dev. 2000;14:2410–34. doi: 10.1101/gad.819500. [DOI] [PubMed] [Google Scholar]
- 46.Abdulkadir SA, Magee JA, Peters TJ, et al. Conditional loss of Nkx3.1 in adult mice induces prostatic intraepithelial neoplasia. Mol Cell Biol. 2002;22:1495–503. doi: 10.1128/mcb.22.5.1495-1503.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Asatiani E, Huang WX, Wang A, et al. Deletion, methylation, and expression of the NKX3.1 suppressor gene in primary human prostate cancer. Cancer Res. 2005;65:1164–73. doi: 10.1158/0008-5472.CAN-04-2688. [DOI] [PubMed] [Google Scholar]
- 48.Bethel CR, Bieberich CJ. Loss of Nkx3.1 expression in the transgenic adenocarcinoma of mouse prostate model. Prostate. 2007;67:1740–50. doi: 10.1002/pros.20579. [DOI] [PubMed] [Google Scholar]
- 49.Jiang AL, Hu XY, Zhang PJ, et al. Up-regulation of NKX3.1 expression and inhibition of LNCaP cell proliferation induced by an inhibitory element decoy. Acta Biochim Biophys Sin (Shanghai) 2005;37:335–40. doi: 10.1111/j.1745-7270.2005.00047.x. [DOI] [PubMed] [Google Scholar]
- 50.Jiang AL, Zhang JY, Young C, et al. Molecular cloning and characterization of human homeobox gene Nkx3.1 promoter. Acta Biochim Biophys Sin (Shanghai) 2004;36:64–7. doi: 10.1093/abbs/36.1.64. [DOI] [PubMed] [Google Scholar]