

Abstract
Hirano bodies are actin-rich inclusions reported most frequently in the hippocampus in association with a variety of conditions including neurodegenerative diseases and aging. We have developed a model system for formation of Hirano bodies in Dictyostelium and cultured mammalian cells to permit detailed studies of the dynamics of these structures in living cells. Model Hirano bodies are frequently observed in membrane-enclosed vesicles in mammalian cells consistent with a role of autophagy in the degradation of these structures. Clearance of Hirano bodies by an exocytotic process is supported by images from electron microscopy showing extracellular release of Hirano bodies, and observation of Hirano bodies in the culture medium of Dictyostelium and mammalian cells. An autophagosome marker protein Atg8-GFP was colocalized with model Hirano bodies in wild-type Dictyostelium cells, but not in atg5- or atg1-1 autophagy mutant strains. Induction of model Hirano bodies in Dictyostelium with a high-level expression of 34 kDa ΔEF1 from the inducible discoidin promoter resulted in larger Hirano bodies and a cessation of cell doubling. The degradation of model Hirano bodies still occurred rapidly in autophagy mutant (atg5-) Dictyostelium, suggesting that other mechanisms such as the ubiquitin-mediated proteasome pathway could contribute to the degradation of Hirano bodies. Chemical inhibition of the proteasome pathway with lactacystin significantly decreased the turnover of Hirano bodies in Dictyostelium, providing direct evidence that autophagy and the proteasome can both contribute to degradation of Hirano bodies. Short-term treatment of mammalian cells with either lactacystin or 3-methyl adenine results in higher levels of Hirano bodies and a lower level of viable cells in the cultures, supporting the conclusion that both autophagy and the proteasome contribute to degradation of Hirano bodies.
Keywords: Hirano bodies, autophagy, proteasome, actin, neuropathology, Dictyostelium, mammalian cell culture
Introduction
Hirano bodies are actin-rich inclusions reported most frequently in the hippocampus in association with a variety of conditions including neurodegenerative diseases, alcoholism and aging.1-3 These structures contain a highly ordered paracrystalline filament array that contains actin,4 actin-associated proteins5 and a broad array of proteins implicated in diverse processes in normal cells and in disease.1,6-10 Yet, the mechanisms of assembly and degradation of Hirano bodies have not been investigated, since nearly all reports of these structures utilize fixed specimens of brain tissue obtained in autopsies.
Turnover of cellular structures is accomplished by a number of distinct processes including ubiquitin-mediated proteolysis catalyzed by the proteasome11 and autophagy.12 The ubiquitin-mediated proteolysis system mediates protein degradation in the cytosolic compartment, while the autophagy pathway has the ability to promote turnover of large structures and organelles that are too large for the proteasome to enter and degrade.
We have recently reported that Hirano bodies are readily induced to form in the cellular slime mold Dictyostelium and in a variety of mammalian cells by expression of modified forms of a 34 kDa actin binding protein with activated cross-linking activity.7,10,13,14 The forms of the 34 kDa protein reported to induce formation of Hirano bodies are the carboxy-terminus (CT, amino acids 124–295) and a mutant with an alteration in the first putative EF hand (34 kDa ΔEF1). In both Dictyostelium and mammalian cells, stable lines with Hirano bodies have been established showing that these inclusions are not necessarily related to cell death. The goal of the present study is to determine the mechanism(s) contributing to degradation of Hirano bodies. Hirano bodies in cultured mammalian cells were frequently observed within membrane-bound vesicles suggesting degradation by autophagy. To investigate the role of autophagy in turnover of Hirano bodies, we utilized a combination of approaches including electron microscopy, mutant strains of Dictyostelium with defects in autophagy,15-17 and use of the inhibitors lactacystin and 3-methyl adenine to inhibit the ubiquitin mediated proteolysis and autophagy pathways, respectively. Our results reveal that both autophagy and ubiquitin-mediated degradation by the proteasome contribute to turnover of Hirano bodies.
Results
Model Hirano bodies become membrane-bound in mammalian cells
Hirano bodies are highly enriched in actin filaments, and they are expected to form in the cytoplasmic compartment. Hirano bodies in the brain have been reported in the cytoplasmic compartment, in membrane-bound vesicles, and also in the extracellular space.18-21 The localization of Hirano bodies following expression of gain of function forms of the 34 kDa actin binding protein is initially cytoplasmic as previously reported.10,13,14 We also observe that Hirano bodies are frequently present in membrane-enclosed vesicles in cultured cells. Fluorescence from a GFP fusion of the 34 kDa CT protein that induces formation of Hirano bodies is colocalized with actin filaments stained with phalloidin within a large vesicle in the perinuclear region (Fig. 1). While this localization is consistent with a membrane-bound Hirano body, this aggregation could reside above or below the vesicle. Furthermore, all actin aggregations do not have the characteristic features of Hirano bodies that are only definitively identified by electron microscopy. Therefore, we utilized electron microscopy to investigate the intracellular localization of model Hirano bodies unambiguously. In fibroblasts, model Hirano bodies are frequently sequestered within membrane-bound organelles (Fig. 2). The characteristic filament diameter and spacing that have been described for Hirano bodies in the brain and for model Hirano bodies in cultured mammalian cells10 are clearly apparent. In some cases, vesicles containing Hirano bodies appear to be engaged in exocytosis resulting in delivery of the contents of these vesicles to the extracellular space (Fig. 3). An exocytotic route for clearance of Hirano bodies from cells is also supported by the observation of Hirano bodies enriched for actin and CT-GFP in the culture medium of both Dictyostelium and cultured mammalian L cell fibroblasts (data not shown). The presence of actin-containing Hirano bodies within membrane-enclosed vesicles in cells is most easily explained by hypothesizing that Hirano bodies are substrates for macroautophagy.
Figure 1.
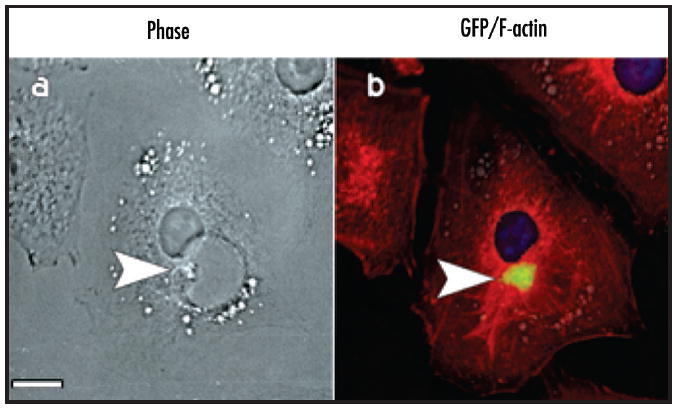
Presence of Hirano bodies in membrane-bound compartments observed by fluorescence microscopy. Stably transfected lines of mouse L cell fibroblasts expressing the CT protein and bearing Hirano bodies are shown. (a) phase contrast microscopy; (b) merged fluorescence microscope images of the same cell showing CT-GFP (green), actin filaments stained with phalloidin-TRITC (red), and nuclei stained with Hoechst dye (blue). Colocalization of CT-GFP and actin within a large vesicle in the perinuclear region of the cell is shown. Scale bar = 5 μm.
Figure 2.
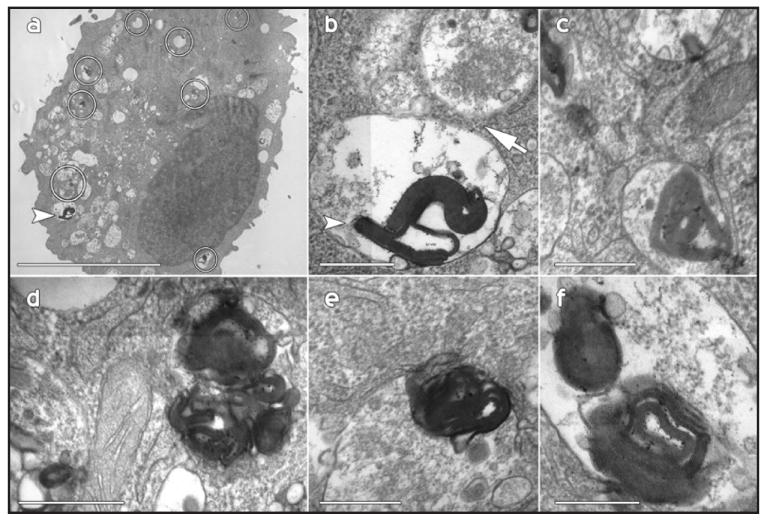
Presence of Hirano bodies in membrane-bound compartments observed by transmission electron microscopy. Stably transfected lines of mouse L cell fibroblasts expressing the CT protein and bearing Hirano bodies are shown. (a) Many of the Hirano bodies are located within vesicles (circles); (b) The filamentous structure of the inclusion marked with an arrowhead in (a) is shown at high magnification. This inclusion is in close apposition to a multivesicular body (arrow); (c–f) the small electron dense aggregations are membrane-bound and demonstrate the periodicity and organization of the Hirano bodies. Scale bar = 1.5 μm (a), 100 nm (b–f).
Figure 3.
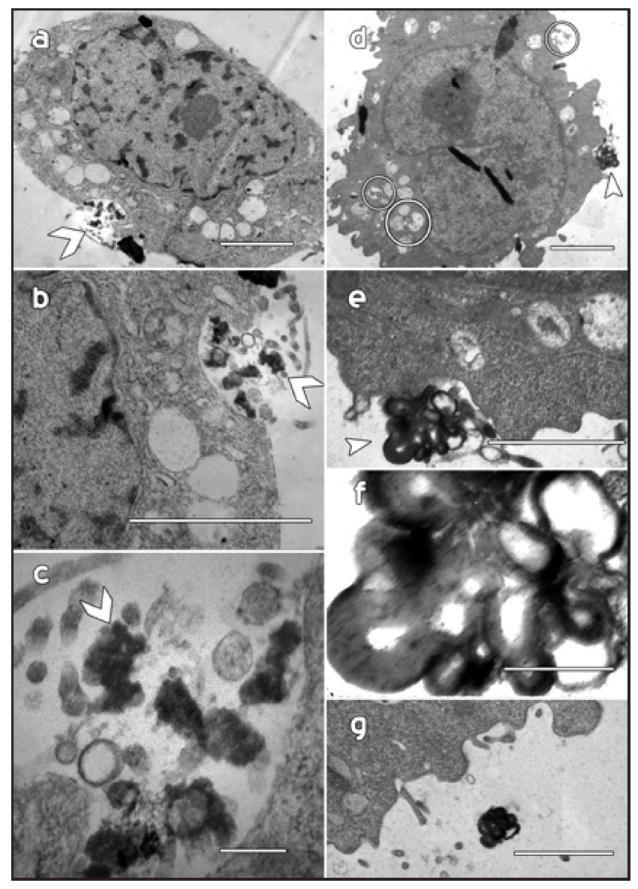
Vesicle-bound aggregations may be released from cells by exocytosis. Examples from two different cells (a–g) reveal that Hirano bodies and/or Hirano body fragments appear to undergo exocytosis. The arrowheads indicate the site of these events and the images at higher magnification allow the periodicity of the components to be more clearly resolved. (g) The same aggregation viewed in (d–f) from several serial sections later appears to be well outside of the cell. Scale bar = 1 μm (a and c), 500 nm (b, e and g), 100 nm (c and f).
Both autophagy and the proteasome contribute to degradation of model Hirano bodies
Formation of Hirano bodies in Dictyostelium was induced by expression of 34 kDa ΔEF1 from two different vectors in order to permit a high level of inducible expression using a developmentally regulated discoidin promoter,22 or a lower level of constitutive expression from an actin 15 promoter. Expression of 34 kDa ΔEF1-GFP from the discoidin promoter resulted in formation of Hirano bodies in both wild-type cells and autophagy mutant atg5- Dictyostelium (Fig. 4). By contrast, expression of GFP or wild type 34 kDa-GFP in either wild-type or autophagy mutant cells resulted in diffuse or actin cytoskeleton-like distribution, respectively, and did not induce formation of Hirano bodies (Fig. 4). Hirano body formation had already begun at 24 hours after induction of expression of 34 kDa ΔEF1-GFP, and was even more pronounced at 48 hours. The Hirano bodies in atg5- cells were much larger than the Hirano bodies in wild-type cells (Fig. 4). However, lower level constitutive expression of 34 kDa ΔEF1-GFP from the actin 15 promoter induced formation of similarly sized Hirano bodies in wild-type, atg5- and atg1-1 Dictyostelium (data not shown). Direct association of Hirano bodies and autophagy components was investigated by transfection of wild-type cells with unlabeled 34 kDa ΔEF1, and the autophagosome marker protein Atg8-GFP. Clear colocalization of Atg8-GFP and Hirano bodies stained with rhodamine phalloidin is apparent in wild-type cells (Fig. 5). However, Atg8-GFP was not colocalized with Hirano bodies in atg5- or atg1-1 cells (data not shown).
Figure 4.
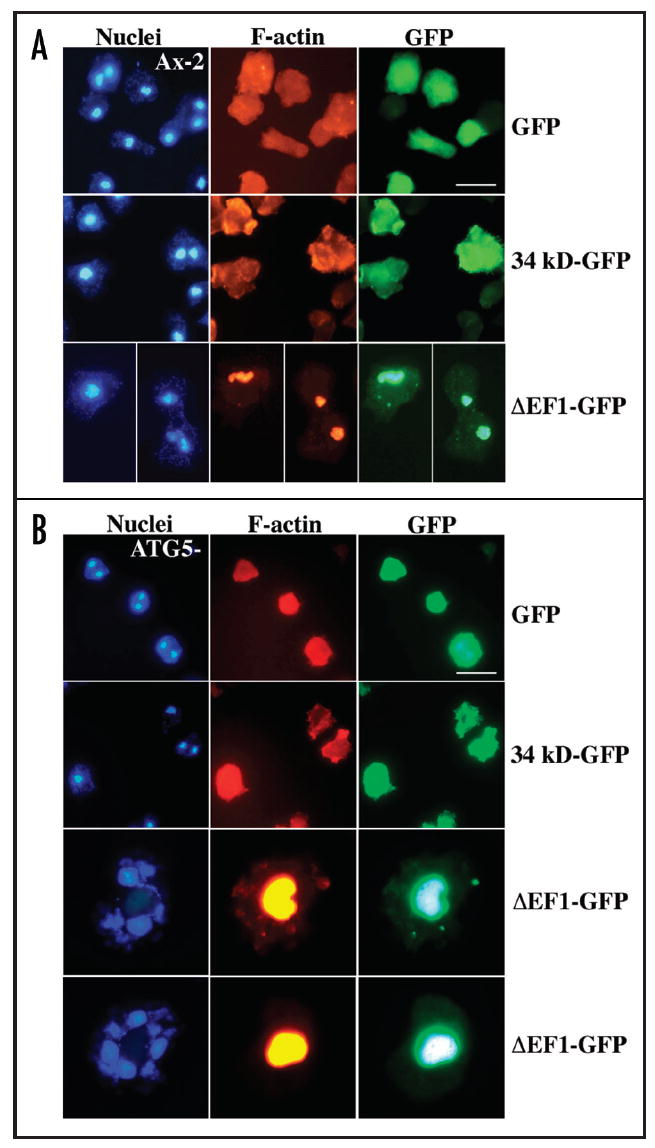
Formation of Hirano bodies after inducible expression of 34 kDa ΔEF1-GFP in wild-type and autophagy mutant Dictyostelium. Expression of GFP, 34 kDa-GFP or 34 kDa ΔEF1-GFP from the discoidin promoter in vector pVEII was induced for 48 hours by removal of folate from the growth medium of wild-type (A) and atg5- autophagy mutant (B) Dictyostelium cells. GFP and 34 kDa-GFP protein were distributed throughout the cytoplasm and in association with F-actin, respectively. In contrast, 34 kDa ΔEF1-GFP induced the formation of Hirano bodies in both strains. The Hirano bodies were larger in autophagy mutant (atg5-) cells as compared to wild-type.
Figure 5.
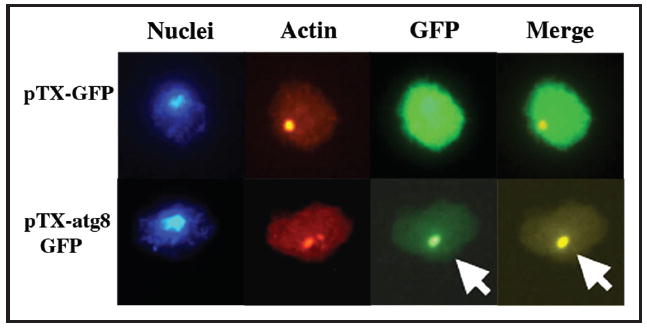
Colocalization of autophagosome marker Atg8 with Hirano bodies. Wild-type Dictyostelium constitutively expressing 34 kDa ΔEF1 protein was cotransformed with pBSR and either pTX-Atg8-GFP or pTX-GFP plasmid. Transformants were selected with blasticidin and G418. The cells were stained with Hoescht and TRITC-phalloidin. The autophagosome marker Atg8-GFP labeled punctate structures that colocalized with Hirano bodies in wild type Dictyostelium, but not in atg5- or atg1-1 autophagy mutant cells.
To assess the physiological effect of inducing formation of Hirano bodies, the growth of wild-type and atg5- cells after induction of expression of GFP, 34 kDa GFP or 34 kDa ΔEF1-GFP was measured (Fig. 6). Wild-type cells grew normally after induction of GFP, or 34 kDa GFP and showed a slight decrease in growth rate after induction of 34 kDa ΔEF1-GFP. The atg5- cells grew more slowly than wild-type in axenic culture. Expression of GFP or 34 kDa GFP has no apparent effect on the growth rate of these autophagy mutant cells. By contrast, no growth is observed following formation of Hirano bodies by induction of 34 kDa ΔEF1-GFP from the discoidin promoter in the atg5- autophagy mutant cells (Fig. 6B). Using lower-level expression of 34 kDa ΔEF1-GFP from the actin 15 promoter, major differences in growth rate of cells with Hirano bodies were not apparent (data not shown). Thus, the decreased growth rate is not due simply to the presence of Hirano bodies, but rather is due to overloading the cellular degradation machinery.
Figure 6.
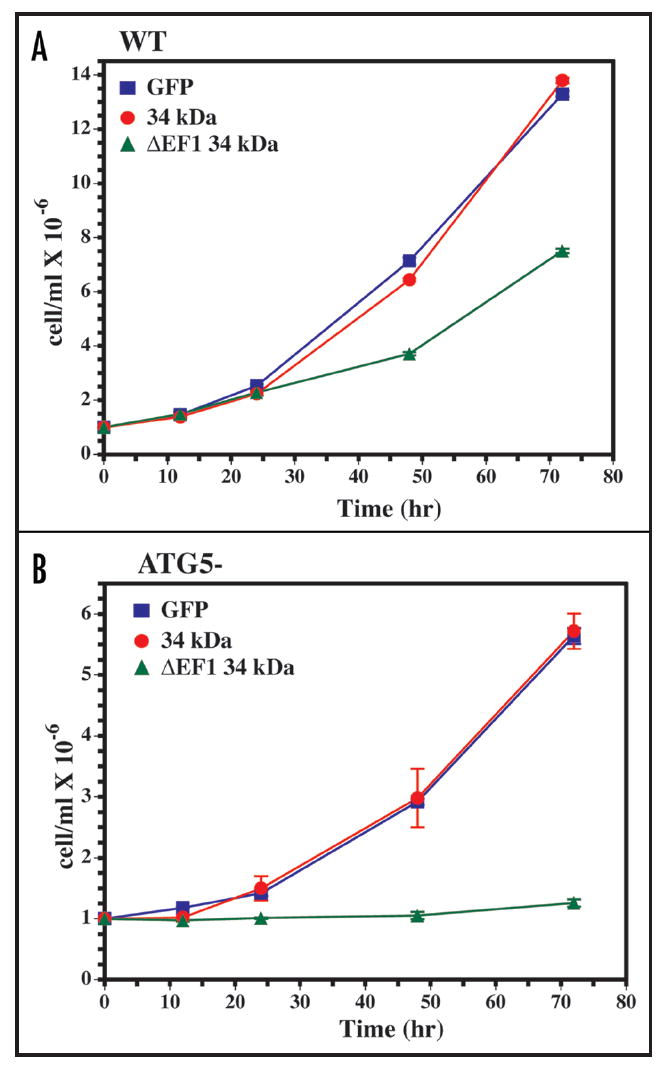
Growth arrest of autophagy mutant cells with Hirano bodies. Expression of GFP, 34 kDa GFP and 34 kDa ΔEF1-GFP was induced in wild-type (a) and atg5- (b) cells by removal of folate from the growth medium. The growth rate of autophagy mutant (atg5-) Dictyostelium with Hirano bodies was severely reduced for 72 h after induction of 34 kDa ΔEF1-GFP protein when compared to that of wild-type Dictyostelium with Hirano bodies or autophagy mutant (atg5-) Dictyostelium without Hirano bodies.
To observe the turnover of Hirano bodies, expression of 34 kDa ΔEF1-GFP from the discoidin promoter was initiated by removal of folate from the culture medium for 24 hours, and the rate of turnover was observed following re-addition of folate to the culture medium to repress synthesis (Fig. 7). Using western blotting to directly measure the level of the 34 kDa ΔEF1-GFP protein, induction is clearly observed in wild-type cells, and decay occurs with an apparent half time of about 9 hours (Fig. 7C). In autophagy mutant cells, expression of 34 kDa ΔEF1-GFP is observed even in uninduced cells suggesting that the discoidin promoter is slightly leaky. After induction of expression in atg5- cells, the 34 kDa ΔEF1-GFP protein levels were higher than observed in wild-type, but turnover is still observed. Similar studies performed by flow cytometry to quantify the 34 kDa ΔEF1 protein, and by fluorescence microscopy to count the number of Hirano bodies directly confirm the fact that turnover still occurs in the autophagy mutant (Fig. 7A and B). The observation of efficient turnover of 34 kDa ΔEF1 in the atg5- cells suggested the involvement of a second pathway for degradation of Hirano bodies.
Figure 7.

The turnover of Hirano bodies in wild-type and autophagy mutant cells. The expression of 34 kDa ΔEF1-GFP protein was induced for 24 h and turned off by the addition of folate to the medium. Open and black bars represent wild-type and atg5- mutant cells, respectively. (A) The number of cells with Hirano bodies was counted by fluorescence microscopy. The autophagy mutant cells display more Hirano bodies than wild-type cells both prior to induction and after a 24 hour induction. (B) Flow cytometry analysis was performed to observe the turnover of 34 kDa ΔEF1-GFP in both wild type and autophagy mutant (atg5-) Dictyostelium. The mean fluorescence of the cell populations was normalized to 100% at the end of the induction. Turnover of 34 kDa ΔEF1-GFP is observed in both wild-type and autophagy mutant cells. (C) The rate of turnover of 34 kDa ΔEF1-GFP was measured by western blot using antibody directed to GFP. To control for loading of the extracts, each lane was also stained using antibody to alpha-actinin. The level of expression of 34 kDa ΔEF1-GFP protein was significantly higher in autophagy mutant (atg5-) than in wild-type Dictyostelium both prior to induction, and after 24 hours of induction. The degradation of 34 kDa ΔEF1-GFP protein was observed in wild-type and in autophagy mutant (atg5-) Dictyostelium. However, in the presence of lactacystin, turnover of the 34 kDa ΔEF1 protein was largely suppressed.
To determine the role of the proteasome in the turnover observed in autophagy mutant Dictyostelium, the proteasome inhibitor lactacystin was added to the cultures. Direct biochemical analysis showed that lactacystin blocks the activity of Dictyostelium proteasomes by 53%. Further, addition of lactacystin resulted in marked inhibition of the turnover of 34 kDa ΔEF1-GFP in atg5- mutant cells (Fig. 7C).
To confirm that both the proteasome and autophagy also contribute to turnover of Hirano bodies in mammalian cells, human H4 cells with Hirano bodies were treated with either lactacystin or 3-methyl adenine, inhibitors of the ubiquitin-mediated proteolysis system and autophagy, respectively. Treatment with either lactacystin (10 μM for 18 hours) or 3-methyl adenine (10 mM for 24 hours) had little or no effect on the shape and morphology of control H4 cells lacking Hirano bodies, but led to brighter staining for CT-GFP and larger and more prominent Hirano bodies in the drug treated as compared to control cells. In addition, treatment with lactacystin or 3-methyl adenine resulted in a decrease in the density of viable CT-GFP expressing H4 cells with Hirano bodies in the cultures, either due to cell death or an inhibition of growth (data not shown). These results support the conclusion that the proteasome and autophagy pathways contribute to turnover of Hirano bodies.
Discussion
Abnormal protein aggregation and formation of characteristic pathological structures are central features of neurodegenerative diseases. Although the physiological consequence of the formation of these abnormal inclusions remains highly controversial,23 cellular mechanisms of response to protein misfolding and aggregation, including ubiquitin-mediated proteolysis and autophagy,11,12 are focal elements of many current investigations of the mechanisms of pathogenesis and development of new therapies.24 These studies support the concept that the ability of clearance mechanisms to keep pace with accumulation of aggregates may have a major impact on disease progression.
Since the physiological role of Hirano bodies in neurodegenerative diseases and other chronic conditions is unknown, we have developed a cellular model for the formation of Hirano bodies through the expression of gain-of-function mutants of the 34 kDa actin binding protein.10,13,14 Our results help to reconcile a variety of apparently disparate observations of Hirano bodies in vivo. Hirano bodies have been reported both in the cytoplasmic space as might be expected for an actin aggregate, but have also been shown within membrane-bound compartments.19-21 Our results showing that autophagy contributes to degradation of Hirano bodies are consistent with, and serve to reconcile, these diverse localizations. In addition, our results also support the proposal that Hirano bodies which enter the degradative compartment through the autophagy pathway can be incompletely digested and released from cells by exocytosis (Fig. 3). We have also observed Hirano bodies in the growth medium of Dictyostelium and mammalian cell cultures. However, the number of Hirano bodies that we have recovered in the medium accounts for only a small fraction of all the Hirano bodies produced. Yet, the Hirano bodies may be quite unstable so that the number of Hirano bodies recovered is a minimum estimate. Thus, while the exocytosis of incompletely digested Hirano bodies can occur, the extent to which this process contributes to the clearance of Hirano bodies from cells may vary widely depending on cell type and context. This pathway for release of Hirano bodies from cells also has potential significance in vivo, since a significant fraction of the Hirano bodies in the brain are reported to reside in the extracellular space.18 In addition, the report that Hirano bodies in the brain are a byproduct of cell death and neuronal degeneration was challenged based on the lack of correlation between the position of Hirano bodies and neurons showing signs of pathology.18 Our findings that Hirano bodies form in the cytoplasm, undergo autophagy, and can be released by exocytosis following autophagy accounts for the localization of Hirano bodies in the brain either in the cytoplasm, within multivesicular bodies, or in the extracellular space.
Numerous prior studies have revealed major roles for the proteasome and autophagy in protein turnover both in healthy tissues and in association with pathology. Mice with conditional deficiencies in autophagy genes ATG5 or ATG7 develop ubiquitin-positive inclusions, increased levels of neuronal apoptosis, and behavioral disorders.25,26 These findings provide convincing evidence of the essential role of autophagy in the brain even in the absence of expression of any pathological condition or expression of disease-associated transgenes. Further, both autophagy and the proteasome contribute to the degradation of intracellular inclusions containing alpha-synuclein, polyglutamine containing proteins, superoxide dismutase and α1-antitrypsin Z.27-30 The contributions of both the proteasome and autophagy to clearance of Hirano bodies are most readily explained by positing that the soluble and aggregated forms remain in dynamic equilibrium in the cell. This proposed dynamic equilibrium between soluble and assembled components of Hirano bodies is also supported by ongoing efforts to purify Hirano bodies from Dictyostelium. Those efforts reveal that these structures are extremely labile requiring precise buffer conditions to achieve even short-term stabilization following cell lysis (Shahid-Salles S, Furukawa R and Fechheimer M, unpublished). The finding that failure to degrade Hirano bodies leads to a severe growth defect is consistent with prior studies of other inclusions showing that clearance of aggregates by autophagy reduced toxicity. Such findings support the suggestion that drugs that enhance macroautophagy, such as rapamycin, are promising therapeutic possibilities for a number of neurodegenerative diseases.24
The finding that autophagy contributes to clearance of Hirano bodies describes a mechanism for turnover of the cytoskeleton, and relates on a more general level to studies of the role of the cytoskeleton in autophagy and neurodegenerative diseases. Inhibition of anterograde or retrograde microtubule dependent transport is associated with accumulation of multivesicular bodies and enhanced formation of β-amyloid,31 inhibition of autophagic clearance of aggregates resulting in enhanced cytotoxicity,32 and failure to form aggresomes,33 respectively. In agreement with our findings with model Hirano bodies, recent studies show that both autophagy and the proteasome contribute to turnover of actin aggresomes induced by addition of jasplakinolide.34,35 The actin cytoskeleton has been implicated in a selective form of autophagy in yeast—cytoplasm to vacuole transport—but is not required for the progression of macroautophagy.36 Specifically, it has been proposed that actin may promote selective transport of cargo to the preautophagosomal structure in yeast.37,38 It is not known whether the actin cytoskeleton may promote specific degradation of cargo in higher eukaryotes, or whether there is any recognition or specificity in degradation of Hirano bodies by autophagy.
A family of lysosomal storage diseases termed the neuronal ceroid lipofuscinoses (NCL) is characterized by the accumulation within lysosomes and multivesicular bodies of granular osmiophilic deposits (GROD), curvilinear bodies (CVB) and fingerprint (FP) inclusions.39-42 The morphology of model Hirano bodies varies widely according to cell type, and the formation of Hirano bodies appears to occur by initial accumulation of smaller transient structures that then coalesce to form a larger aggregate.10 The organization of these smaller structures that coalesce to form model Hirano bodies is remarkably similar to that of the fingerprint inclusions associated with neuronal ceroid lipofuscinosis.39-42 Our finding that autophagy contributes to degradation of Hirano bodies reconciles the topological difference in subcellular localization of these structures, and suggests that the morphological and structural similarity between Hirano bodies and inclusions characteristic of NCL merit further study. It will be important to determine the relative contributions of the proteasome, autophagy and exocytosis to the clearance of Hirano bodies in diverse cell types and conditions, and to determine whether these findings provide a clue to more general mechanisms of turnover of the actin cytoskeleton in normal and pathological conditions. Finally, these studies in cultured cells must be supplemented by development of animal models in order to elucidate the physiological significance of Hirano bodies in vivo.
Materials and Methods
Formation of model Hirano bodies in Dictyostelium and in cultured mammalian cells
Formation of model Hirano bodies was induced by expression of altered forms of the Dictyostelium 34 kDa protein with activated actin binding activity. These forms are the carboxy-terminus (CT, amino acids 124–295), or the full-length protein with a mutation in the first EF hand (34 kDa ΔEF1, amino acids 1–295).13,14,43 Generation of mammalian cells with model Hirano bodies expressing the carboxy terminus of the 34 kDa protein fused to GFP (CT-GFP) was performed as previously described.7,10 Inducible formation of model Hirano bodies in Dictyostelium was performed after subcloning cDNA into the vector pVEII that contains the discoidin promoter so that expression could be suppressed or induced by addition or removal of folate, respectively, from the growth medium.22 Expression from the discoidin promoter was induced by removal of folate for a period of 24 or 48 hours. The following inserts were cloned into the BamHI site of pVEII using standard methods:44 EGFP taken from pEGFP-N1 (Clontech, Mountain View, California); 34 kDa protein fused to EGFP at the COOH terminus (34 kDa-GFP);45 34 kDa ΔEF1 or 34 kDa ΔEF1 fused to EGFP at the COOH terminus (34 kDa ΔEF1-GFP).14 Constitutive formation of Hirano bodies was performed using 34 kDa ΔEF1 expressed from the actin 15 promoter.14 Mutant Dictyostelium bearing a knockout in the autophagy gene ATG5 (atg5-) or an insertional mutation in ATG1 (atg1-1), and a vector expressing autophagosome marker protein Atg8 fused to GFP were generous gifts from Dr. Rich Kessin, Columbia University, and have been described previously.15-17 All plasmids were sequenced at the Molecular Genetics Instrumentation Facility at the University of Georgia for verification. Electroporation and selection of wild-type or mutant strains of Dictyostelium were performed as described previously.13
Analytical methods and antibodies
Protein concentrations were determined by the bicinchoninic acid (Pierce Biotechnology, Rockford, Illinois) method46 with bovine serum albumin (Sigma Chemical Co., St. Louis, Missouri) as a protein standard. Monoclonal anti-α-actinin antibody 9E6,47 and rabbit anti-GFP antibody (Molecular Probes, Eugene, Oregon) were used to probe western blots. Western blotting was performed as previously described48 except that chemiluminescent detection was performed using the Super Signal West Femto Detection (Pierce Biotechnology, Rockford, Illinois).
Growth
Axenic strains of Dictyostelium were routinely maintained in axenic cultures with shaking at 150 rpm in HL-5 medium49 at 20°C. To assess growth rates, cells were inoculated at a starting concentration of 1 × 106 cells/ml into axenic HL-5 culture medium with shaking at 150 rpm at 20°C. Each datum point represents the average for samples obtained daily in duplicate from three flasks and counted on a standard hemocytometer. Growth rates of wild-type and atg5- cells expressing GFP, 34 kDa-GFP or 34 kDa ΔEF1-GFP were measured.
Localization of Hirano bodies and autophagosomes in Dictyostelium
Dictyostelium cells were fixed for 25 min in 3.7% formaldehyde in 17 mM phosphate buffer, pH 6.1, permeabilized in acetone at -20°C for 2 min, and stained as described previously.50 Rhodamine-labeled or Oregon green-labeled phalloidin (Molecular Probes, Eugene, Oregon) was used to localize F-actin, and monoclonal antibody B2C47 followed by a rhodamine-labeled or a fluorescein-labeled secondary antibody (Sigma Chemical Co., St. Louis, Missouri) was used to localize the 34 kDa protein. Colocalization of Hirano bodies and autophagosomes was performed with Hirano bodies induced by expression of unlabelled 34 kDa ΔEF1 protein driven by the actin 15 promoter, and transformed with autophagy protein Atg8 fused to GFP.15,16 Hirano bodies were stained with rhodamine phalloidin as described above, and compared to the localization of fluorescence from Atg8-GFP. Nuclei were stained using Hoechst 33342 to label DNA. Methods for light and electron microscopy of model Hirano bodies in mammalian cells have been described previously.10
Study of the rate of protein turnover
For studies of the degradation of the 34 kDa ΔEF1 protein, expression was induced from the discoidin promoter in wild-type or autophagy mutant Dictyostelium cells by removal of folate from the media for 24 hours, and the level of the protein was assessed at varying times from 0–24 hours after readdition of folate. The decay of the 34 kDa ΔEF1 protein after re-addition of folate was measured by one of three methods. First, the number of Hirano bodies was counted by fluorescence microscopy using the GFP fluorescence to identify these condensed structures. Second, the rate of turnover of GFP fluorescence was measured by flow cytometry using the Becton Dickinson FACSCalibur with data collected in list mode, and analysis was performed using FloJo (Tree Star Inc., Stanford University, Palo Alto, California). Third, the rate of turnover of GFP was measured by western blot using rabbit anti-GFP antibody. In experiments to test the role of the proteasome in turnover, lactacystin (Calbiochem, San Diego, California) was added to the cell cultures at a concentration of 10 μM. Similarly, 3-methyladenine was added to cultures at a concentration of 10 mM to inhibit autophagy.
Verification of the inhibition of proteasome activity by lactacystin in Dictyostelium
The extent of inhibition of lactacystin on Dictyostelium proteasomes was determined using methods described previously.51 Briefly, 200 ml of DH-1 Dictyostelium was grown to a density of 5 × 106 cells/ml. The cells were harvested by sedimentation and washed three times with 17 mM Sorensen’s buffer, pH 6.1. Cells were resuspended in one volume of homogenization buffer (50 mM Tris, pH 7.5, 0.25 M sucrose, 5 mM MgCl2, 2 mM ATP, 1 mM DTT, 0.5 mM EDTA, 0.025% digitonin) and sonicated for 4 minutes in a bath sonicator, followed by 75 pulses with 30% duty cycle and tip power 4 (Branson sonifier, cell disruptor 200, Danbury, Connecticut). This resulted in approximately 95% cell lysis determined by light microscopy. The lysate was clarified for 15 minutes at 13,000 Xg, 4°C. The supernatant was sedimented for 2 hours at 240,000 Xg, 4°C. The resultant pellet was resuspended in one volume of proteasome assay buffer (50 mM Tris, pH 7.5, 40 mM KCl, 0.5 mM MgCl2, 0.5 mM ATP, 1 mM DTT, 0.5 mg/ml BSA). 50 μl of partially purified proteasomes was utilized in a 200 μl assay. Proteasome activity was determined by the cleavage of 600 μM Boc-LSTR (Sigma Aldrich Chemical Co., St. Louis, Missouri), measuring the resultant 7-amino-4-methylcourmarin (amc) fluorescence with 380 nm excitation and 460 nm emission wavelengths (Perkin Elmer LS5, Waltham, Massachusetts). Standard curves were determined with amc (Sigma Aldrich Chemical Co., St. Louis, Missouri). Measurements were determined in triplicate of partially purified proteasomes, and in absence or presence of either 10 μM lactacystin or 20 μM epoxomicin (Sigma Aldrich Chemical Co., St. Louis, Missouri) as a function of time. Epoxomicin was used to define the fraction of the activity in the partially purified preparation that was due to the action of proteasomes.
Acknowledgments
We thank Dr. Rich Kessin and the Dictyostelium stock center for the Atg8-GFP plasmid, and atg5- and atg1-1 Dictyostelium. We are grateful to the UGA Flow Cytometry Facility for assistance with the flow cytometer, and the Center for Advanced Ultrastructure Research for confocal and transmission electron microscopy, respectively. This work was supported by NSF (MCB 98-08748), the Alzheimer’s Association (IIRG-00-2436), and NIH R01—NS 04645101 to M.F. and R.F.
References
- 1.Hirano A. Hirano bodies and related neuronal inclusions. Neuropathol Appl Neurobiol. 1994;20:3–11. doi: 10.1111/j.1365-2990.1994.tb00951.x. [DOI] [PubMed] [Google Scholar]
- 2.Lass R, Hagel C. Hirano bodies and chronic alcoholism. Neuropath Appl Neurobiol. 1994;20:12–21. doi: 10.1111/j.1365-2990.1994.tb00952.x. [DOI] [PubMed] [Google Scholar]
- 3.Gibson PH, Tomlinson BE. Numbers of Hirano bodies in the hippocampus of normal and demented people with Alzheimer’s disease. J Neurol Sci. 1977;33:199–206. doi: 10.1016/0022-510x(77)90193-9. [DOI] [PubMed] [Google Scholar]
- 4.Goldman JE. The association of actin with Hirano bodies. J Neuropathol Exp Neurol. 1983;42:146–52. doi: 10.1097/00005072-198303000-00004. [DOI] [PubMed] [Google Scholar]
- 5.Galloway PG, Perry G, Gambetti P. Hirano body filaments contain actin and actin-associated proteins. J Neuropathol Exp Neurol. 1987;46:185–99. doi: 10.1097/00005072-198703000-00006. [DOI] [PubMed] [Google Scholar]
- 6.Munoz DG, Wang D, Greenberg BD. Hirano bodies accumulate C-terminal sequences of beta-amyloid precursor protein (beta-APP) epitopes. J Neuropathol Exp Neurol. 1993;52:14–21. doi: 10.1097/00005072-199301000-00003. [DOI] [PubMed] [Google Scholar]
- 7.Ha S, Furukawa R, Fechheimer M. Association of the carboxyl-terminal domain of the amyloid-β precursor protein with Hirano bodies reduces activation of transcription and initiation of apoptosis. 2008 Submitted for publication. [Google Scholar]
- 8.Galloway PG, Perry G, Kosik KS, Gambetti P. Hirano bodies contain tau protein. Brain Res. 1987;403:337–40. doi: 10.1016/0006-8993(87)90071-0. [DOI] [PubMed] [Google Scholar]
- 9.Peterson C, Kress Y, Vallee R, Goldman JE. High molecular weight microtubule-associated proteins bind to actin lattices (Hirano bodies) Acta Neuropathol. 1988;77:168–74. doi: 10.1007/BF00687427. [DOI] [PubMed] [Google Scholar]
- 10.Davis RC, Furukawa R, Fechheimer M. A cell culture model for investigation of Hirano bodies. Acta Neuropathol. 2008;115:205–17. doi: 10.1007/s00401-007-0275-9. [DOI] [PubMed] [Google Scholar]
- 11.Goldberg AL. Functions of the proteasome: From protein degradation and immune surveillance to cancer therapy. Biochem Soc Trans. 2007;35:12–7. doi: 10.1042/BST0350012. [DOI] [PubMed] [Google Scholar]
- 12.Mizushima N, Levine B, Cuervo AM, Klionsky DJ. Autophagy fights disease through cellular self-digestion. Nature. 2008;451:1069–75. doi: 10.1038/nature06639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Maselli AG, Davis R, Furukawa R, Fechheimer M. Formation of Hirano bodies in Dictyostelium and mammalian cells induced by expression of a modified form of an actin cross-linking protein. J Cell Sci. 2002;115:1939–52. doi: 10.1242/jcs.115.9.1939. [DOI] [PubMed] [Google Scholar]
- 14.Maselli AG, Furukawa R, Thomson SAM, Davis RC, Fechheimer M. Formation of Hirano bodies induced by expression of an actin cross-linking protein with a gain of function mutation. Eucaryot Cell. 2003;2:778–87. doi: 10.1128/EC.2.4.778-787.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Otto GP, Wu MY, Kazgan N, Anderson OR, Kessin RH. Macroautophagy is required for multicellular development of the social amoeba Dictyostelium discoideum. J Biol Chem. 2003;278:17636–45. doi: 10.1074/jbc.M212467200. [DOI] [PubMed] [Google Scholar]
- 16.Otto GP, Wu MY, Kazgan N, Anderson OR, Kessin RH. Macroautophagy is required for multicellular development of the social amoeba Dictyostelium discoideum. J Biol Chem. 2004;278:17636–45. doi: 10.1074/jbc.M212467200. [DOI] [PubMed] [Google Scholar]
- 17.Tekinay T, Wu MY, Otto GP, Anderson OR, Kessin RH. Function of the Dictyostelium discoideum Atg1 kinase during autophagy and development. Eukaryot Cell. 2006;5:1797–806. doi: 10.1128/EC.00342-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gibson PH. Light and electron microscopic observations on the relationship between Hirano bodies, neuron and glial perikarya in the human hippocampus. Acta Neuropathol. 1978;42:165–71. doi: 10.1007/BF00690353. [DOI] [PubMed] [Google Scholar]
- 19.Schochet SS, Jr, McCormick WF. Ultrastructure of Hirano bodies. Acta Neuropathol. 1972;21:50–60. doi: 10.1007/BF00687999. [DOI] [PubMed] [Google Scholar]
- 20.Izumiyama N, Ohtsubo K, Tachikawa T, Nakamura H. Elucidation of three-dimensional ultrastructure of Hirano bodies by the quick-freeze, deep-etch and replica method. Acta Neuropathol. 1991;81:248–54. doi: 10.1007/BF00305865. [DOI] [PubMed] [Google Scholar]
- 21.Tomonaga M. Ultrastructure of Hirano bodies. Acta Neuropathol. 1974;28:365–6. doi: 10.1007/BF00685292. [DOI] [PubMed] [Google Scholar]
- 22.Blusch J, Morandini P, Nellen W. Transcriptional regulation by folate: Inducible gene expression in Dictyostelium transformants during growth and early development. Nucl Acids Res. 1992;20:6235–8. doi: 10.1093/nar/20.23.6235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kirkitadze MD, Bitan G, Teplow DB. Paradigm shifts in Alzheimer’s disease and other neurodegenerative disorders: the emerging role of oligomeric assemblies. J Neurosci Res. 2002;69:567–77. doi: 10.1002/jnr.10328. [DOI] [PubMed] [Google Scholar]
- 24.Rubinsztein DC. The roles of intracellular protein-degradation pathways in neurodegeneration. Nature. 2006;443:780–6. doi: 10.1038/nature05291. [DOI] [PubMed] [Google Scholar]
- 25.Hara T, Nakamura K, Matsui M, Yamamoto A, Nakahara Y, Suzuki-Migishima R, Yokoyama M, Mishima K, Saito I, Okano H, Mizushima N. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature. 2006;441:885–9. doi: 10.1038/nature04724. [DOI] [PubMed] [Google Scholar]
- 26.Komatsu M, Waguri S, Chiba T, Murata S, Iwata J-i, Tanida I, Ueno T, Koike M, Uchiyama Y, Kominami E, Tanaka K. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature. 2006;441:880–4. doi: 10.1038/nature04723. [DOI] [PubMed] [Google Scholar]
- 27.Kabuta T, Suzuki Y, Waka K. Degradation of amyotrophic lateral sclerosis-linked mutant Cu,Zn-superoxide dismutase proteins by macroautophagy. J Biol Chem. 2006;281:30524–33. doi: 10.1074/jbc.M603337200. [DOI] [PubMed] [Google Scholar]
- 28.Ravikumar B, Duden B, Rubinsztein DC. Aggregate-prone proteins with polyglutamine and polyalanine expansions are degraded by autophagy. Human Mol Genet. 2002;11:1107–17. doi: 10.1093/hmg/11.9.1107. [DOI] [PubMed] [Google Scholar]
- 29.Webb JL, Ravikumar B, Atkins J, Skepper JN, Rubinsztein DC. Alpha-synuclein is degraded by both autophagy and the proteasome. J Biol Chem. 2003;278:25009–13. doi: 10.1074/jbc.M300227200. [DOI] [PubMed] [Google Scholar]
- 30.Kamimoto T, Shoji S, Hidvegi T, Mizushima N, Umebayashi K, Perlmutter DH, Yoshimori T. Intracellular inclusions containing mutant alpha1-antitrypsin Z are propagated in the absence of autophagic activity. J Biol Chem. 2006;281:4467–76. doi: 10.1074/jbc.M509409200. [DOI] [PubMed] [Google Scholar]
- 31.Stokin GB, Lillo C, Falzone TL, Brusch RG, Rockenstein E, Mount SL, Raman R, Davies P, Masliah E, Williams DS, Goldstein LSB. Axonopathy and transport deficits early in the pathogenesis of Alzheimer’s disease. Science. 2005;307:1282–8. doi: 10.1126/science.1105681. [DOI] [PubMed] [Google Scholar]
- 32.Ravikumar B, Acevedo-Arozena A, Imarisio S, Berger Z, Vacher C, O’Kane CJ, Brown SDM, Rubinsztein DC. Dynein mutations impair autophagic clearance of aggregate-prone proteins. Nat Genetics. 2005;37:771–6. doi: 10.1038/ng1591. [DOI] [PubMed] [Google Scholar]
- 33.Johnston JA, Ward CL, Kopito RR. Aggresomes: a cellular response to misfolded proteins. J Cell Biol. 1998;143:1883–98. doi: 10.1083/jcb.143.7.1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lázaro-Diéguez F, Aguado C, Mato E, Sánchez-Ruiz Y, Esteban I, Alberch J, Knecht E, Egea G. Dynamics of an F-actin aggresome generated by the actin-stabilizing toxin jasplakinolide. J Cell Sci. 2008;121:1415–25. doi: 10.1242/jcs.017665. [DOI] [PubMed] [Google Scholar]
- 35.Lázaro-Diéguez F, Knecht E, Egea G. Clearance of a Hirano body-like F-actin aggresome generated by jasplakinolide. Autophagy. 2008;4:717–20. doi: 10.4161/auto.6345. [DOI] [PubMed] [Google Scholar]
- 36.Reggiori F, Monastyrska I, Shintani T, Klionsky DJ. The actin cytoskeleton is required for selective types of autophagy, but nonspecific autophagy, in the yeast Saccharomyces cerevisiae. Mol Biol Cell. 2005;16:5843–56. doi: 10.1091/mbc.E05-07-0629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Monastyrska I, Shintani T, Klionsky DJ, Reggiori F. Atg11 directs autophagosome cargoes to the PAS along actin cables. Autophagy. 2006;2:119–21. doi: 10.4161/auto.2.2.2298. [DOI] [PubMed] [Google Scholar]
- 38.Monastyrska I, He C, Geng J, Hoppe AD, Klionsky DJ. Arp2 links autophagic machinery with the actin cytoskeleton. Mol Biol Cell. 2008;19:1962–75. doi: 10.1091/mbc.E07-09-0892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Goebel HH. The neuronal ceroid-lipofuscinoses. Semin Pediatr Neurol. 1996;3:270–8. doi: 10.1016/s1071-9091(96)80031-3. [DOI] [PubMed] [Google Scholar]
- 40.Mole SE, Williams RE, Goebel HH. Correlations between genotype, ultrastructural morphology and clinical phenotype in the neuronal ceroid lipofuscinoses. Neurogenetics. 2005;6:107–26. doi: 10.1007/s10048-005-0218-3. [DOI] [PubMed] [Google Scholar]
- 41.Anderson GW, Smith VV, Brooke I, Malone M, Sebire NJ. Diagnosis of neuronal ceroid lipofuscinosis (Batten disease) by electron microscopy in peripheral blood specimens. Ultrastruct Pathol. 2006;30:373–8. doi: 10.1080/01913120500406566. [DOI] [PubMed] [Google Scholar]
- 42.Haltia M. The neuronal ceroid lipofuscinoses: From past to present. Biochim Biophys Acta. 2006;1762:850–6. doi: 10.1016/j.bbadis.2006.06.010. [DOI] [PubMed] [Google Scholar]
- 43.Lim RWL, Furukawa R, Fechheimer M. Evidence of intramolecular regulation of the Dictyostelium discoideum 34,000 dalton F-actin bundling protein. Biochemistry. 1999;38:16323–32. doi: 10.1021/bi991100o. [DOI] [PubMed] [Google Scholar]
- 44.Maniatis T, Fritsch EF, Sambrook J. Molecular Cloning: A Laboratory Manual. Cold Spring Harbor: Cold Spring Harbor Press; 1982. [Google Scholar]
- 45.Furukawa R, Maselli AG, Thomson SAM, Lim RW-L, Stokes JV, Fechheimer M. Calcium Regulation of actin cross-linking is important for function of the actin cytoskeleton in Dictyostelium. J Cell Sci. 2003;116:187–96. doi: 10.1242/jcs.00220. [DOI] [PubMed] [Google Scholar]
- 46.Smith PK, Krohn RI, Hermanson GT, Mallia AK, Gartner FH, Provenzano MD, Fujimoto EK, Goeke NM, Olson BJ, Klenk DC. Measurement of protein using bicinchoninic acid. Anal Biochem. 1985;150:76–85. doi: 10.1016/0003-2697(85)90442-7. [DOI] [PubMed] [Google Scholar]
- 47.Furukawa R, Butz S, Fleischmann E, Fechheimer M. The Dictyostelium discoideum 30,000 dalton protein contributes to phagocytosis. Protoplasma. 1992;169:18–27. [Google Scholar]
- 48.Towbin H, Staehelin T, Gordon J. Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: procedure and some applications. Proc Natl Acad Sci USA. 1979;76:4350–4. doi: 10.1073/pnas.76.9.4350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Loomis WF. Sensitivity of Dictyostelium discoideum to nucleic acid analogues. Exp Cell Res. 1971;64:484–6. doi: 10.1016/0014-4827(71)90107-8. [DOI] [PubMed] [Google Scholar]
- 50.Fechheimer M. The Dictyostelium discoideum 30,000-dalton protein is an actin filament-bundling protein that is selectively present in filopodia. J Cell Biol. 1987;104:1539–51. doi: 10.1083/jcb.104.6.1539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kisselev AF, Goldberg AL. Monitoring activity and inhibition of 26S proteasomes with fluorogenic peptide substrates. Meth Enzymol. 2005;398:364–78. doi: 10.1016/S0076-6879(05)98030-0. [DOI] [PubMed] [Google Scholar]