

Abstract
The prion protein is responsible for several fatal neurodegenerative diseases via conversion from its normal to disease-related isoform. The recombinant form of the protein is typically studied to investigate the conversion process. This constructs lacks the co- and post-translational modifications present in vivo, there the protein has two N-linked glycans and is bound to the outer leaflet of the plasma membrane via a glycosylphosphatidylinositol (GPI) anchor. The inherent flexibility and heterogeneity of the glycans, the plasticity of the GPI anchor and the localization of the protein in a membrane conspire to make experimental structural characterization of biological constructs of PrPC challenging. Yet this characterization is central to determining not only the suitability of recombinant PrPC as a model for biological forms of the protein but also the potential role of co- and post-translational modifications on the disease process. Here, we present molecular dynamics simulations of three human PrP constructs: (1) a protein-only construct modeling the recombinant form, (2) a diglycosylated and soluble construct, and (3) a diglycosylated and GPI-anchored construct bound to a lipid bilayer. We found that glycosylation and membrane anchoring do not significantly alter the structure or dynamics of PrPC, but they do appreciably modify the accessibility of the polypeptide surface PrPC. Additionally, the simulations of membrane-bound PrPC revealed likely recognition domains for the disease-initiating PrPC:PrPSc binding event and a potential mechanism for the observed inefficiency of conversion associated with differentially glycosylated PrP species.
Keywords: prion protein, protein misfolding, glycosylation, GPI anchor, lipid membrane, molecular dynamics
The prion protein (PrP) is a cell-surface glycoprotein found in mammalian tissues, particularly in neuronal cells. Therole of PrP is of yet unknown, although there is evidence that it is involved in copper metabolism (Burns et al. 2003) and signal transduction (Mouillet-Richard et al. 2000). While the biological function of PrP is of interest, even more so is the pathology of its disease-related isoform: scrapie PrP or PrPSc. The innocuous form of PrP, cellular PrP or PrPC, under certain conditions can convert to the lethal PrPSc, which can aggregate with other PrP molecules, kill glial and neuronal cells and incite a neurodegenerative process that is always fatal.
The central hypothesis in prion diseases, such as bovine spongiform encephalopathy (BSE), Creutzfeldt-Jakob disease (CJD) and fatal familial insomnia, is that it is a protein-only disease, whereby the prion protein is the only agent necessary for propagation and transmission of the disease. While recombinant (rec) PrP has been the primary construct used to study prion disease in the lab, non-protein moieties, such as the prion protein's two N-linked glycans and its glycosylphosphatidylinositol (GPI) anchor, affect the disease process in vivo (DeArmond et al. 1997; DeArmond et al. 1999). The glycans and the anchor can be found on both PrPC and the disease-related aggregate PrPSc. The N-linked glycans influence PrP expression, distribution (within regions of the brain and among different types of neuronal cells) and deposition of PrPSc plaques in vivo (DeArmond et al. 1997). While the glycans are not required for conversion of PrPC to PrPSc aggregates (Taraboulos et al. 1990; Kocisko et al. 1994; Legname et al. 2004; Tuzi et al. 2008), they play a significant and complicated role in the disease process. The complex role of glycosylation is highlighted in studies concerning homologous conversion of PrPC (conversion of PrPC by the same species of PrPSc). For one species and strain of PrPSc, unglycosylated forms of PrPC can inhibit conversion of glycosylated forms of PrPC (Nishina et al. 2006). Yet, for a different species and strain of PrPSc, the presence of unglycosylated forms of PrPC are required for conversion of glycosylated species (Nishina et al. 2006). In heterologous cases, glycosylation of host (PrPC) and donor (PrPSc) PrP can also influence the efficiency of conversion of PrPC → PrPSc. In cell-free conversion assays, for instance, glycosylated forms of PrPC convert less efficiently than their unglycosylated counterparts (when using heterologous host/donor PrP) (Priola and Lawson 2001). On the flip side, matching glycoform profiles between host PrPC and the infecting PrPSc species is likely responsible for lowering the barrier to interspecies transmission. This phenomenon has been demonstrated by the compatible glycoform profiles of PrPSc aggregates from human (variant-CJD) and bovine (BSE) prion diseases (Collinge et al. 1996). While glycosylation can modulate interspecies transmission barriers and conversion efficiencies, the sequence of the polypeptide chain remains the primary determinant of conversion (Kocisko et al. 1995; Priola and Lawson 2001; Nishina et al. 2006).
Besides the N-glycosylation, PrPC in vivo differs from rec-PrPC in that it is membrane-bound via a GPI anchor. Together, the location of PrPC (predominantly the outer-leaflet of the plasma membrane of neuronal cells) and the flexible nature of the glycans and GPI anchor have impeded structural resolution of key biological constructs of PrPC. In addition, the low-yield and heterogeneous samples resulting from the extraction of PrPC from brain tissue have made studies of biological forms of PrPC challenging. Thus, our understanding of the importance of the GPI anchor, the glycans and the plasma membrane, in the structure-function relationship of PrP remains poorly resolved. To overcome some of the current methodological obstacles, we have performed molecular dynamics (MD) simulations for atomic-resolution structural and dynamic analysis of glycosylated and membrane-bound, human prion protein (huPrPC).
We use a huPrP fragment relevant to the disease process, the most common protease resistant PrPSc fragment (residues 90-230), to investigate the role of the glycans, the GPI anchor and membrane surface on the structure and dynamics of huPrPC. Herein we describe the results of simulations of the following constructs of huPrPC: protein-only (PrPrec), diglycosylated (PrPglyco), and diglycosylated and membrane-bound (PrPgpi) (Figure 1). Each construct was studied under both converting (low pH) and non-converting, non-amyloidogenic conditions (neutral pH) to assess the influence of non-protein moieties on the early steps in the conversion of PrPC → PrPSc. The PrPrec simulations, which model rec-huPrPC, have been previously reported (DeMarco and Daggett 2007) and are included for comparative purposes. This work also builds upon several of our previous MD studies of a smaller unglycosylated fragment (residues 109-219) of Syrian hamster, human and bovine PrP (Alonso et al. 2001; Alonso et al. 2002; DeMarco and Daggett 2004).
Figure 1.
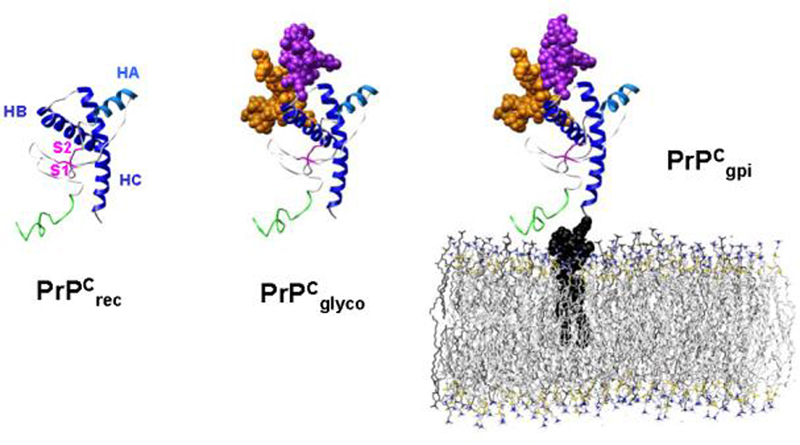
The starting structures for the PrPC simulations: PrPrec, PrPglyco and PrPgpi. The structures are colored as follows: unstructured N-terminus, residues 90-109 (green), β-strands S1 and S2 (magenta), helices HA (light blue), HB and HC (blue), glycan-1 (orange), glycan-2 (purple), GPI anchor (black), POPC bilayer (gray). Note that actual simulations contain explicit water molecules but they are not displayed here.
Materials and Methods
The systems studied include diglycosylated huPrPC (residues 90-230 and 13-residue glycans at each glycosylation site), and membrane-bound, diglycosylated huPrPC (residues 90-230, two 13-residue glycans, GPI anchor, and 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC) bilayer), referred to as PrPCglyco and PrPCgpi, respectively. In addition, previously reported simulations of protein-only huPrPC (residues 90-230), PrPrec, are discussed. The 90-230 construct was selected because it represents the most abundant fragment found in proteinase K-treated PrPSc fibrils; while it omits residues 23-89 (which includes the copper-binding domain), it contains those regions critical to PrP misfolding. All PrP models used the wild-type huPrP sequence. The starting structure for the globular region of huPrPC (residues 125-228) was taken from the rec-huPrPC NMR structure (1QLX, (Zahn et al. 2000)). The unstructured N-terminus (residues 90-124) and the two C-terminal residues (229-230) were modeled to complete the 90-230 construct. The N-terminal region was modeled in two steps: (1) residues 109-124 were modeled in based on the NMR structure of Syrian hamster PrP (structure provided by T.L. James and S. Farr-Jones, (James et al. 1997)), since both species share the same sequence over this region (note that they share 90% sequence identity over the entire 140-residue construct), (2) an unstructured sequence, residues 90-108, was then modeled onto the 109-228 construct. To limit bias introduced by modeling segments of the unstructured domain, the added sequence had no repeating secondary structure elements or designed interactions with the globular region. As previously reported, multiple simulations using this model demonstrated that the modeling did not bias the structure towards a particular conformation (DeMarco and Daggett 2007). The Cys179-Cys214 disulfide bond was left intact for both neutral and low pH simulations, in accordance with experimental evidence that the disulfide bond is present in PrPSc and is necessary for infectivity (Turk et al. 1988; Hermann and Caughey 1998).
A triantennary glycoform scaffold common to mouse PrPC and PrPSc (Stimson et al. 1999), and representative of vertebrate (including human) complex-type N-glycans (Varki et al. 2003), was chosen (Figure 2A). The biologically relevant PrP glycan was modeled using the Sweet online builder (Bohne et al. 1999) and then covalently attached at Asn181 and Asn197. The structure for the PrP GPI anchor was modeled based on sequence information for the PrP GPI anchor (Stahl et al. 1992) and other eukaryotic GPI anchors (Ferguson et al. 1988; Homans et al. 1989; Englund 1993; McConville and Ferguson 1993) (Figure 2B). A three-dimensional structure of the GPI anchor was also modeled using the Sweet web tool (Bohne et al. 1999).
Figure 2.
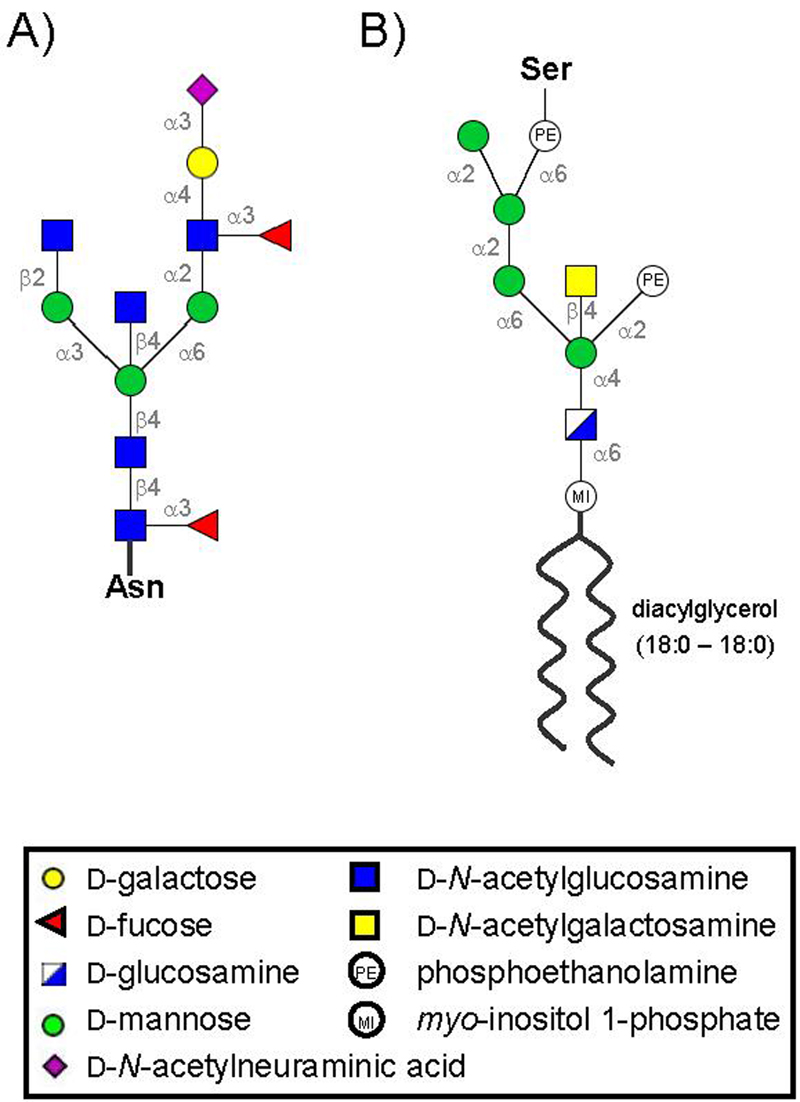
Sequence and linkage information for the models of the (A) 13-residue PrPC glycan attached at Asn-181 and Asn-197, and (B) the GPI anchor linked to Ser-230.
In membrane samples derived from rat brains, the majority of lipids (68.5%) were saturated and POPC was identified as the most abundant lipid near PrP (Brugger et al. 2004). As such, a POPC bilayer was used to model the membrane environment in the PrPgpi membrane simulations. The POPC bilayer was modeled based on a bilayer structure built for MD simulations in the Schulten group (Heller et al. 1993). To accommodate the size of the glycosylated protein, a 336-lipid bilayer was constructed. One of the POPC molecules was removed from the center of the membrane and was replaced by the lipid tail of the PrPgpi construct. Insertion of the GPI anchor was guided by matching hydrophobic and hydrophilic regions of the GPI anchor with those of the outer-leaflet of the POPC bilayer. The starting structures for the membrane simulations, with waters, included 199018 and 199026 atoms, in the neutral and low pH PrPgpi simulations, respectively.
MD simulations were performed using in lucem molecular mechanics (ilmm) software (Beck et al. 2000-2008), which employs the Levitt et al. force field (Levitt et al. 1995; Levitt et al. 1997; Beck and Daggett 2004) and the microcanonical (NVE: constant volume, energy and number of particles) ensemble. All protein and water atoms were explicitly represented. To model the low pH environment of endosomes (pH 6—4.3) (Lee et al. 1996) and in vitro conversion conditions that facilitate conversion (pH < 4) (Swietnicki et al. 1997; Hornemann and Glockshuber 1998; Jackson et al. 1999; Zou and Cashman 2002), we protonated Asp (pKa ∼ 3.9), Glu (pKa ∼ 4.3) and His (pKa ∼ 6.0) residues. The next relevant pKa is that of the terminal carbonyl groups which are protonated at ∼ pH 3.1, therefore by protonating Asp, Glu and His we achieved a pH range of approximately 4.4—3.2, which falls within the pH range of both endosomal pH and in vitro conversion conditions. Neutral pH simulations correspond to a pH range of approximately 7.9—6.1, falling between the pKa's of His side chains and the terminal amino groups. The pKa values given are only approximate, as they are highly dependent on the local environment. A theoretical investigation of pKa of His residues in PrPC demonstrates the range of pKa's possible for a single residue type along the PrP sequence (Langella et al. 2006).
For PrPglyco simulations, the protein was minimized for 1000 steps in vacuo and then solvated in a cubic box 100 × 100 × 100 Å, so that the solvent depth extended at least 20 Å from any protein atom. The total number of atoms in the PrPglyco neutral and low pH simulations was 99431 and 99427 atoms, respectively. The large box size was used to accommodate potential expansion of the glycans and unstructured N-terminus from their initial conformation. The simulations were run at 25°C, so the system density was set to the corresponding experimental density, 0.997 g/mL3 (Kell 1967), by adjusting the box volume. The PrPglyco systems were further equilibrated for 1000 steps of minimization, 2500 steps of MD of water, 500 steps of minimization, 2500 steps of MD of PrP, and a final 500 steps of minimization. For PrPgpi simulations, the system was solvated with a pre-equilibrated water box (at the desired density of 0.997 g/ml), extending at least 20 Å from any protein/carbohydrate/lipid atom, above and below the plane of the membrane. The ceramide moiety of the GPI achor was subjected to 500 steps of minimization, to remove any bad interactions since it was modeled into the membrane bilayer. The waters were minimized for 1000 steps, followed by 500 steps of minimization for waters and POPC molecules. The waters were then subjected to 5 ps of MD and 500 steps of minimization. POPCs and waters were minimized for 500 steps followed by 10 ps of MD. The entire system was then minimized for 1000 steps.
Since a majority of the existing, and likely future, experimental data were collected at room temperature, all simulations were run at 25°C to facilitate comparisons. A 10 Å force-shifted nonbonded cutoff was used and the nonbonded list was updated every 5 steps. A time step of 2 fs was used. Each simulation was carried out for 15 ns, with structures saved every 0.2 ps for analysis. The total simulation time, including all trajectories was 270 ns.
Secondary structure analysis was based on (ϕ,ψ) angles where only repeating structure (≥3 successive residues) was considered. Contact surfaces of a probe with a 4.0 Å radius were calculated using NACCESS software (Hubbard and Thornton 1993), using snapshots taken at 100 ps intervals from 2-15 ns (2 ns allotted for equilibration). All molecular graphics images were produced using UCSF Chimera (Pettersen et al. 2004), and portions of Figure 2 where created with GlycanBuilder (Ceroni et al. 2007).
Results
Global structural similarity
Ten different MD simulations of wild type PrPC in water were performed, six PrPrec, two PrPglyco and two PrPgpi simulations, half of which were under non-amyloidogenic (neutral pH) and the other half under amyloidogenic (low pH) conditions (Figure 1). While not presented here, duplicate simulations for both PrPglyco and PrPgpi were performed, with results similar to the initial trajectories. By approximately 2 ns, most simulations equilibrated with the α-carbons (Cα) of the structured region (residues 125-228, based on 1QLX structure) reaching an average root-mean-squared deviation (RMSD) of less than ∼3.5 Å (Figure 3A). The exception was a PrPrec low pH simulation in which conversion or misfolding was observed. Initially this trajectory equilibrated to Cα RMSD values consistent with the other simulations, but at 5 ns and again at 11 ns there were significant increases in the Cα RMSD (Figure 3A).
Figure 3.
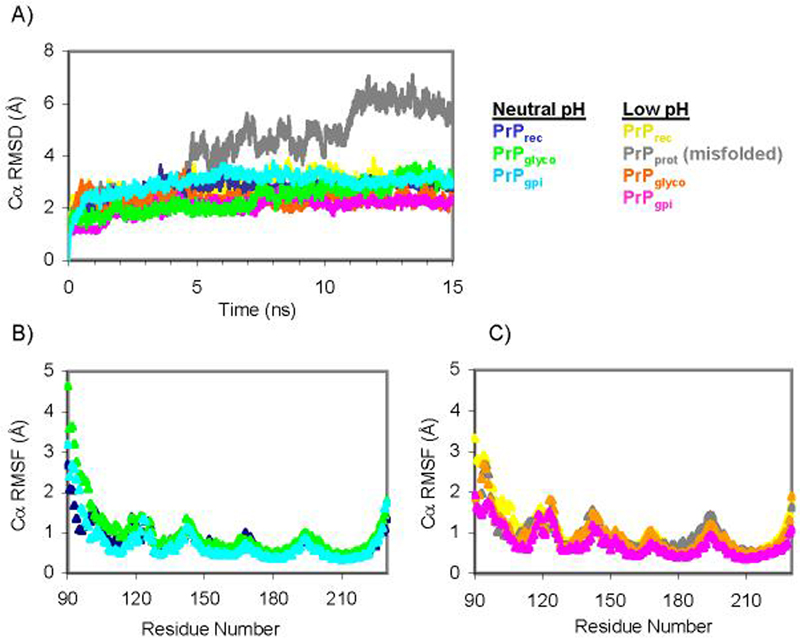
The dynamics of the different PrP constructs. (A) Cα RMSD of the globular region of PrP (residues 125-228). (B & C) Average Cα RMSF of each residue over the course of each 15 ns simulation.
Membrane parameters
The area/lipid head group of the POPC molecules in the PrPgpi simulations was 64 Å2 for both the neutral and low pH simulations at 25°C. These values are in reasonable agreement with the experimentally determined 68.3 ± 0.5 Å2 area/lipid for fully hydrated POPC bilayers in the fluid phase, measured at 30°C (Kucerka et al. 2006). In addition, the behavior of the bilayers was consistent with our simulations of aquaporin in a POPC lipid bilayer (Smolin et al. 2008).
Protein dynamics
Root-mean-squared fluctuations (RMSF) of the α-carbons from the average structure during MD were calculated to assess the dynamics of the PrP sequence. Both the neutral and low pH simulations of PrPrec, PrPglyco and PrPgpi had similar fluctuation profiles (Figure 3B). Over the structured region the average Cα RMSF for the neutral simulations were 0.69 ± 0.19 Å (PrPrec), 0.75 ± 0.21Å (PrPglyco) and 0.59 ± 0.19 Å (PrPgpi). In the structured region in the low pH simulations the corresponding average Cα RMSFs were 0.82 ± 0.21 Å (PrPrec), 0.84 ± 0.25 (PrPrec-misfolded), 0.80 ± 0.25 Å (PrPglyco) and 0.61 ± 0.18 Å (PrPgpi).
Helical Structure
The native helices, HA, HB and HC, were preserved in all simulations (Figure 4). In the absence of the GPI anchor and membrane, HC extended to the C-terminal residue; however, with the GPI-anchor attached (PrPgpi), residues ∼138-140 were unstructured.
Figure 4.
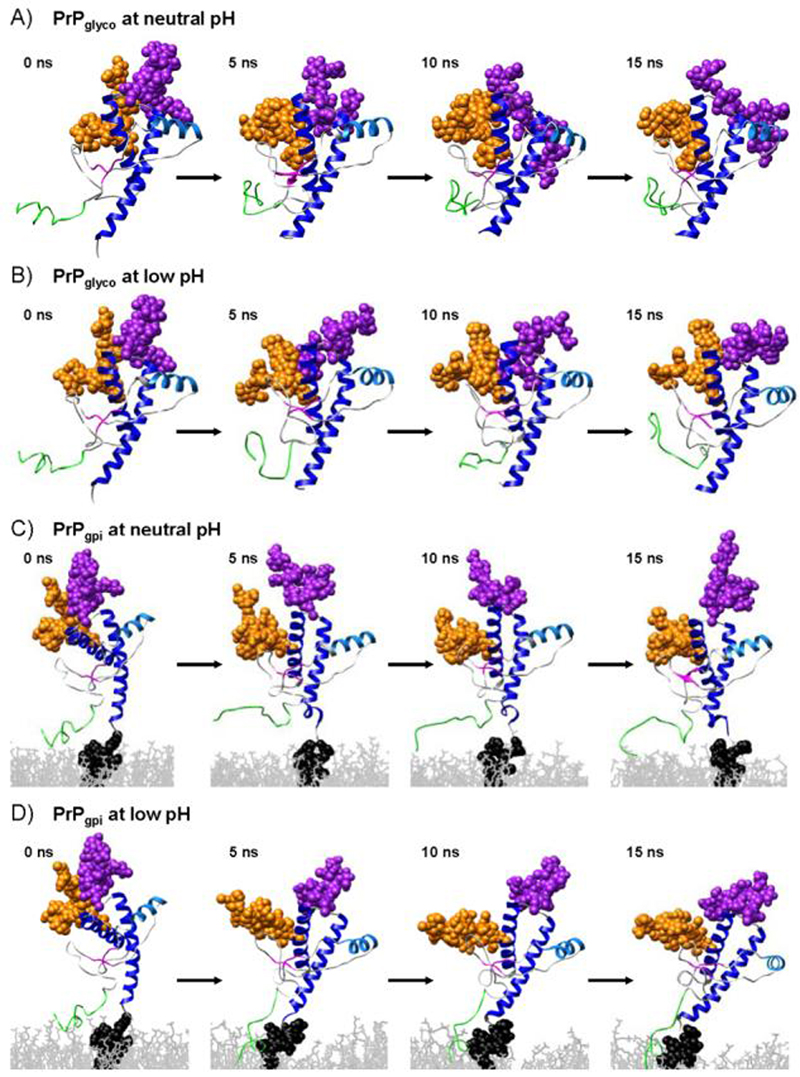
Snapshots from the PrPglyco neutral (A) and low pH (B) and the PrPgpi neutral (C) and low pH (D) simulations. Colored as in Figure 1.
Extended structure
The native β-sheet, comprised of S1 and S2, was maintained, if not extended in all simulations (Figure 4). A region that was found in all constructs (at both low and neutral pH) to be in β/extended conformation was over residues ∼135-140 (the region connecting S1 to HA). We have described formation of this strand in previous publications where we dubbed it E4, and we believe it to be an oligomerization site (Alonso et al. 2001; Alonso et al. 2002; DeMarco and Daggett 2004; DeMarco et al. 2006; DeMarco and Daggett 2007; Scouras and Daggett 2008). In the PrPglyco and PrPgpi simulations the N-terminus remained unstructured, with residues alternating between α-helical, β-strand, and other conformations; although, residues 106-110 demonstrated a propensity to form a segment of β-structure, particularly in the PrPgpi simulations.
Glycan- & GPI anchor-protein interactions
The glycans linked to Asn181 (glycan-1) and Asn197 (glycan-2) in the neutral and low pH PrPglyco simulations had a similar number of contacts with PrP (Table 1), with an average of 96 ± 22 and 101 ± 14 glycan-protein atom-atom contacts at neutral and low pH, respectively. In the PrPgpi simulations there were slightly more glycan-protein contacts: 120 ± 12 (neutral pH) and 151 ± 27 (low pH). The increase in the number of glycan-protein contacts in PrPgpi compared to PrPglyco came from an increase in the number of contacts with glycan-1 (Figure 5). The glycans were highly flexible, occupying large regions of space (Figure 6 & 7). In addition, the plasticity of the GPI anchor permitted PrPgpi to assume several positions relative to the membrane surface (Figure 4C & D).
Table 1.
Average number of atom-atom contacts (2-15 ns) between PrP and its non-protein moieties.
| glycan-1 | glycan-2 | GPI anchor | Total | ||
|---|---|---|---|---|---|
| PrPglyco | neutral pH | 64 ± 9 | 32 ± 13 | 96 ± 22 | |
| low pH | 65 ± 9 | 35 ± 5 | 101 ± 14 | ||
| PrPgpi | neutral pH | 77 ± 8 | 43 ± 9 | 18 ± 18 | 138 ± 22 |
| low pH | 100 ± 25 | 50 ± 9 | 33 ± 11 | 184 ± 29 |
Figure 5.
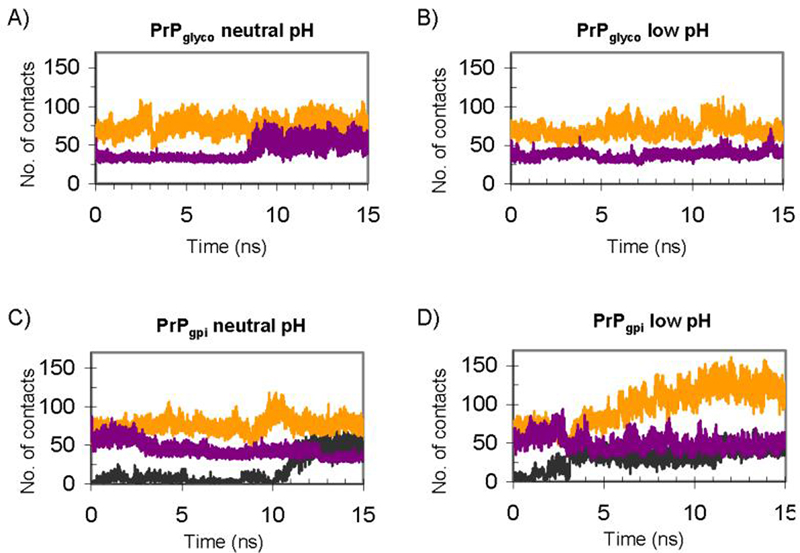
The number of glycan-protein and GPI-protein atom-atom contacts as a function of time for PrPglyco (A & B) and PrPgpi (C & D) simulations: glycan-1 (orange), glycan-2 (purple) and GPI-achor (black).
Figure 6.

Volumes occupied by glycan-1 (orange) and glycan-2 (purple) from the PrPglyco simulations. Overlay of glycans taken at 1 ns intervals from 0-15 ns, mapped onto the 15 ns structures.
Figure 7.
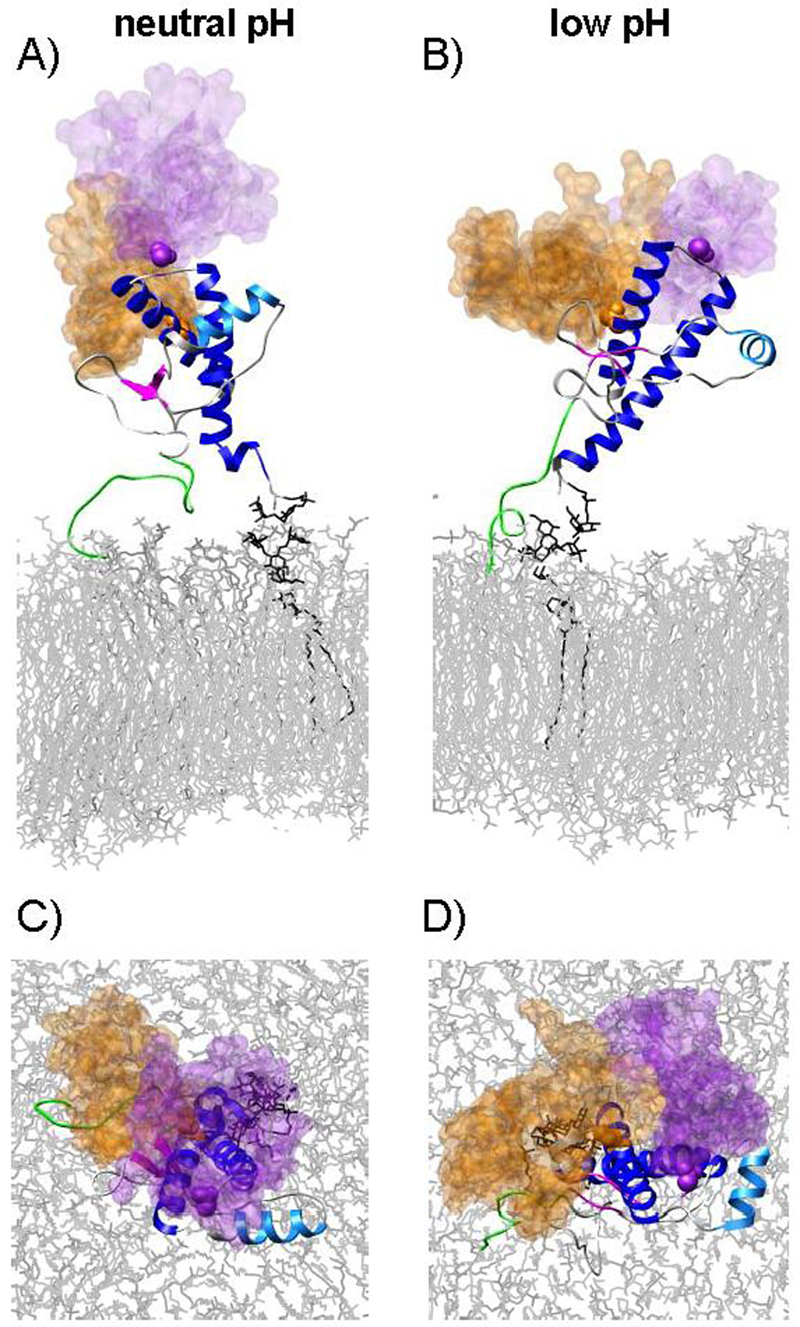
Volumes occupied by glycan-1 (orange) and glycan-2 (purple) over the course of the PrPgpi simulations. (A & B) Overlay of glycans taken at 1 ns intervals from 0-15 ns, mapped onto the 15 ns structure, and (C & D) the same image as viewed from the top of the bilayer.
For both PrPglyco and PrPgpi simulations, glycan-1 interacted with HB and the loop preceding S1 (Figure 8). For glycan-2, interactions were primarily with the loop between HB and HC (Figure 8). For PrPglyco, there was also a small contribution from transient interactions between glycan-2 and HA (Figure 8A & B). The GPI anchor interacted with protein residues proximal to its C-terminal attachment point and transiently with portions of the dynamic N-terminus (Figure 8C & D).
Figure 8.
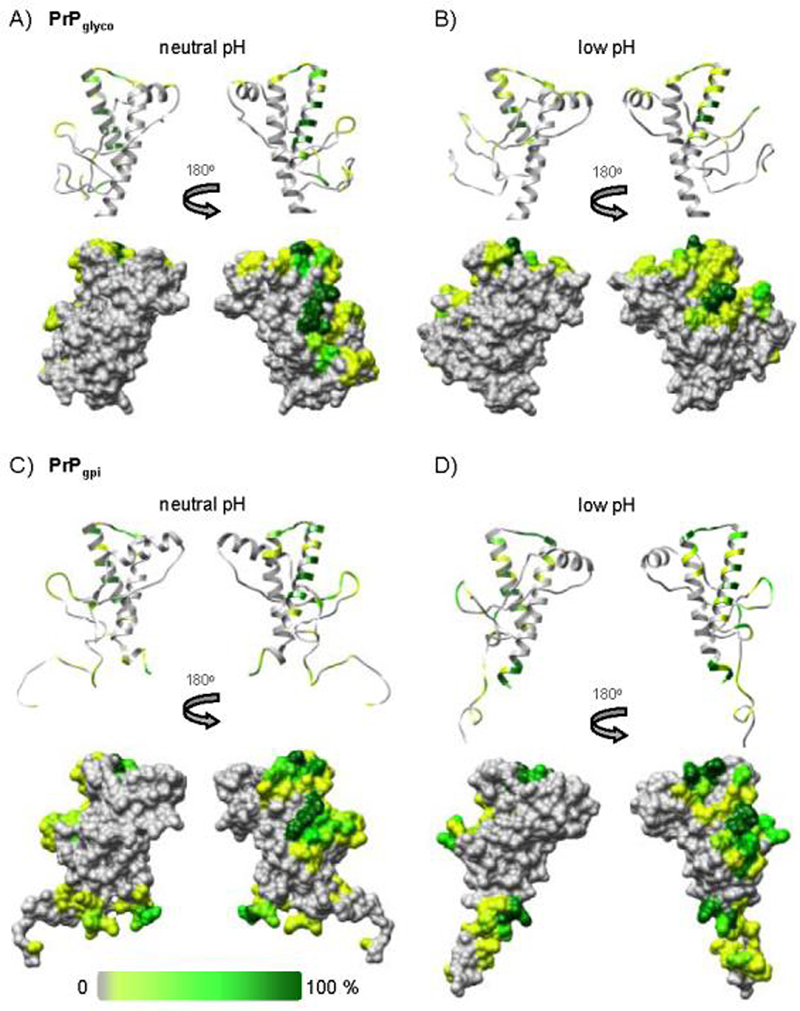
The percentage of time (over the course of the simulations) that glycan and/or GPI anchor atoms were in contact with the polypeptide of PrPglyco (A & B), and PrPgpi (C & D); mapped onto the corresponding final structures from simulation.
PrPC surfaces remaining accessible upon glycosylation and membrane-anchoring
We have evaluated the accessible surface area of the various constructs to determine the effect of the glycans and membrane on potential self-association surfaces and to compare with experimental studies of anti-PrP antibodies. Typically, a probe with a 1.4 Å radius is used to calculate solvent accessible surface area; here we employed a larger probe size (4.0 Å radius) to approximate surface accessibility to larger receptors, such as peptides and proteins. By calculating the contact area with and without modifications present, surfaces unaltered by the presence of the glycans, GPI anchor, and lipid bilayer were identified for PrPgpi (Figure 9). The “HA face” of the protein, remained accessible, forming a continuous surface (Figure 9A and C). Additionally, on the other face of the protein (Figure 9B & D), HC and the base of HB remained exposed. In the PrPgpi simulations, the proximity of the unstructured N-terminus to the membrane affected its accessibility. However, these interactions were transient (Figure 8) and non-specific (based on examination and analysis of snapshots from the simulations), and therefore the N-terminus should be accessible to antibodies or other molecules. For comparison, epitopes from antibodies that bind both cell-surface and rec-PrPC were mapped onto the 15 ns neutral pH PrPgpi structure (Figure 9E), and they correspond to the ‘unaffected’ surfaces of PrPgpi. The following antibodies and their epitopes were considered: R10 (res. 96-105), D18 (res. 134-157), 6H4 (res. 144-152) and R1/R2 (res. 225-230) (Leclerc et al. 2003). Similarly, peptides derived from the PrP sequence that are known to bind to PrPC and inhibit conversion to PrPSc (Horiuchi et al. 2001), map to exposed surfaces of PrPgpi (Figure 9G). The antibody and peptide surfaces combined (Figure 9E & G) yield a very similar pattern, a negative image, to the surface of PrPgpi not obstructed by the glycans, GPI anchor or membrane (Figure 9A-C). A rec-PrPC-selective epitope (res. 152-163) (Leclerc et al. 2003) also mapped to the exposed HA-face of the protein (Figure 9F), which is inconsistent with the inability of the antibody to bind cell-surface PrPC. However, both glycans interact with regions neighboring the epitope (Figure 7 & 8) and they could sterically prevent the antibody from binding to this epitope in the cell-surface form. In support of this hypothesis, an unglycosylated mouse-hamster PrPC chimera with the hamster-specific amino acid at residue 155, converts twice as readily as its glycosylated counterpart (using a hamster PrPSc seed) (Priola and Lawson 2001). Removal of the glycans likely exposes this residue, which is an important determinant of the mouse-hamster species barrier (Figure 9H) (Kocisko et al. 1995; Priola et al. 2001). Both the rec-PrPC-selective epitope and the mouse-hamster conversion assays are consistent with the results from the PrPglyco and PrPgpi simulations, which show that the glycans can obstruct the protein's surface near residue 155 (a region implicated in the PrPC:PrPSc binding event). It should be noted that due to the modest probe size used to calculate surface accessibility, in comparison to the size of an antibody, a larger probe would help to further refine the predicted antibody-accessible surfaces.
Figure 9.
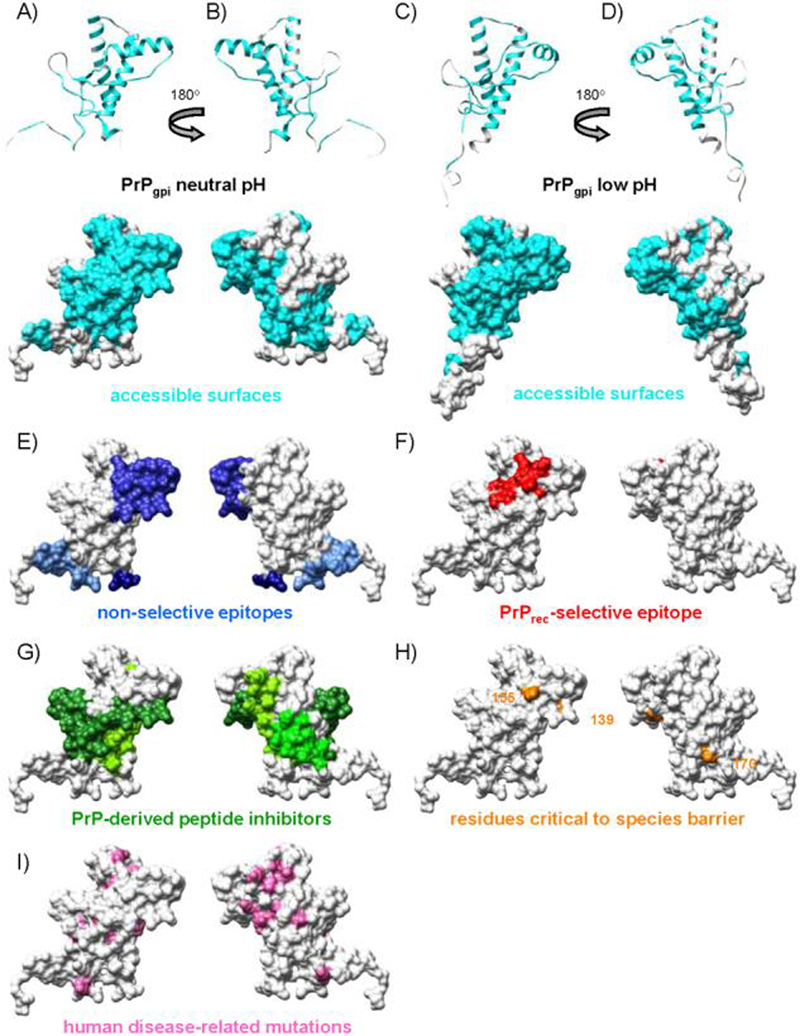
Surface properties of the polypeptide from cell-surface PrPC relative to its recombinant analogue. To approximate accessibility of the surface to receptor molecules, a probe with a 4.0 Å radius was used to calculate surface accessibility. (A-D) Accessible PrP surfaces not affected by the presence of the glycans, GPI anchor or lipid bilayer (cyan) for the neutral (A & B) and low pH (C&D) simulations of PrPgpi. (E) Experimentally determined antibody epitopes accessible in both recombinant and cell-surface PrPC (blue). (F) Experimentally determined epitope accessible in recombinant but not cell-surface PrPC (red). (G) Residues (109-141, 166-179, 200-223) that correspond to peptides that bind PrPC and inhibit conversion (green). (H) Residues critical to species barrier (orange). (I) Human disease-related mutations and the M129V polymorphism (pink).
Discussion
Investigations into the mechanism of pathogenesis of PrP have been strongly weighted towards the use of recombinant PrP versus its in vivo counterpart, which is diglycosylated and membrane-bound via a GPI anchor. While the diglycosylated and membrane-bound form of PrPC is the predominant biological species, recombinant PrP is favored for structural studies for several reasons, most notably because of the difficulty in isolating and purifying the necessary amounts of PrPC from tissue. The combination of circular dichroism (CD) (Eberl et al. 2004; Hornemann et al. 2004) and 1H-NMR spectroscopies (Hornemann et al. 2004), as well as antibody binding studies (Leclerc et al. 2003), indicate that recombinant (unglycosylated and soluble) PrPC and glycosylated (both membrane-bound and in solution) share similar structural characteristics, making rec-PrPC a suitable model for biological forms of PrPC. However, in addition to polypeptide structure, MD simulations permitted further assessments of the compatibility of synthetic versus biological forms of PrP, including the relative stability/dynamics of the polypeptide chain, its interaction with non-protein moieties and the issues of accessibility induced by the presence of the glycans, GPI anchor and the proximity to the membrane surface.
The MD simulations presented here of a model of the recombinant form of huPrPC (PrPrec), soluble glycosylated huPrPC (PrPglyco) and glycosylated and GPI-anchored huPrPC (PrPgpi), also indicate that glycosylation and attachment to a membrane do not alter the structure of PrPC (Figure 4). Over the globular region of PrP, PrPgpi and PrPglyco simulations maintained the same structured domain as the NMR structure of recombinant PrPC (Zahn et al. 2000), in agreement with circular dichroism and NMR studies of glycosylated (with a partial GPI anchor) versus recombinant forms of bovine PrPC (Hornemann et al. 2004) and CD spectra for liposome-coupled murine rec-PrPC (Eberl et al. 2004). Antibody binding studies of recombinant versus cell-surface PrPC allowed for further validation of the trajectories. Using a set of antibodies that bind unfolded HA and folded HA, it was determined that more than 90% of the population of cell-surface PrPC maintains HA (Leclerc et al. 2003). In the MD simulations, HA also remains helical (Figure 4). In agreement with cross-competition antibody binding studies for PrPgpi-like constructs (Leclerc et al. 2003), the unstructured N-terminus does not interact with the C-terminal globular domain in a manner that would inhibit antibody binding to either region. Additionally, for PrPgpi the N-terminus makes non-specific transient interactions with the POPC bilayer, which is consistent with peptides from this region having a low affinity for large unilamellar POPC vesicles (Henriques et al. 2008) and with soluble rec-huPrP not interacting appreciably with POPC vesicles (Morillas et al. 1999). In addition to structure, PrPrec, PrPglyco and PrPgpi shared similar dynamic profiles over both the unstructured N-terminus and structured C-terminal domain (Figure 3).
Both experiment and MD simulations suggest that biological and recombinant forms of PrPC share a similar structure; however, the conversion efficiencies of these species can be markedly different. Glycosylation of PrPC has been shown in some cases to reduce the conversion efficiency of PrPC → PrPSc in (Taraboulos et al. 1990; Lehmann and Harris 1997; Priola and Lawson 2001), while, in the case of in vivo experiments, it is unknown whether this effect could be directly attributed to glycosylation or aberrant trafficking of the tunicamycin-treated or mutant unglycosylated-PrPC. Unlike previous low pH simulations of recombinant forms of PrPC (both for the protease-K resistant fragment (DeMarco and Daggett 2007) and the smaller 109-219 fragment (Alonso et al. 2001; Alonso et al. 2002; DeMarco and Daggett 2004)), neither glycosylated construct (PrPglyco, PrPgpi) populated misfolded species. Unfortunately, since there are only a few simulations of glycosylated constructs at low pH, it is not possible to determine whether the lack of observed misfolding at low pH is due to the glycans or due to limited sampling, but we note that this degree of sampling is sufficient to see conversion of the unglycosylated constructs. In the case of the glycosylated protein, spontaneous conversion is discouraged via glycan-1 interactions with the loop leading to S1 and glycan-2 interactions with HA (Figure 8A & B), both of which limit the mobility of HA. Simulations have revealed that displacement of HA is needed for conversion of huPrPrec (DeMarco and Daggett 2007), as well as for conversion of other PrP species (Alonso et al. 2001; Alonso et al. 2002; DeMarco and Daggett 2004; Scouras and Daggett 2008). So, limiting the flexibility of this region in the PrPC state appears to affect the ability to misfold (Figure 10).
Figure 10.
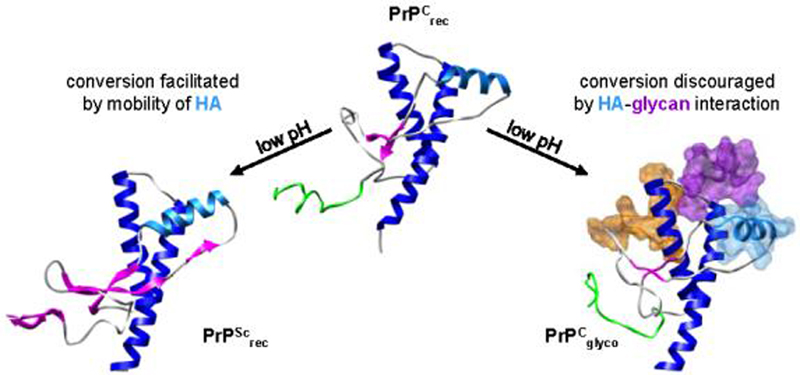
Conversion inefficiencies associated with glycosylation may be due in part to interactions between HA (light blue) and glycan-2 (purple). As previously demonstrated, the conversion of huPrPCrec → PrPScrec is facilitated by the mobility of HA (DeMarco and Daggett 2007).
Besides the internal structure and mobility of the protein-portion of PrP, the glycans may serve a more obvious constraint to conversion by limiting the accessible surface for PrPC:PrPSc interactions. Surfaces of PrPgpi unaffected by the presence of the non-protein moieties matched well to epitopes from non-selective antibodies (antibodies that bind both cell-surface and rec-PrPC) (Figure 9E) and PrP-derived inhibitory peptides that bind PrPC (Figure 9G). Taken together the antibody, PrP-derived peptides and simulation results identify potential PrPC binding surfaces that PrPSc aggregates could exploit to convert PrPC into the pathogenic isoform within the membrane (Caughey 2001; Baron et al. 2002; Baron and Caughey 2003). These regions include the unstructured N-terminus, the large surface that includes HA and the C-terminal third of HC. Also of interest were the surfaces obstructed by the glycans, GPI anchor or membrane, since it has relevance for the suitability of rec-PrPC to model biological forms of cell-surface PrPC. An antibody that bound to rec-PrPC but not to cell surface PrPC identified an epitope (res. 152-163) that is somehow obstructed in cell-surface PrPC (Figure 9F). The MD simulations indicate that the flexible nature of the glycans allows them to protect regions near this epitope (Figure 7C & D) and that they are likely responsible for the selectivity of the antibody. In support of this conclusion, are studies that indicate that unglycosylated PrPC converts more readily than glycosylated PrPC by revealing a key determinant of transmissibility, residue 155 (Priola and Lawson 2001), which is found within the rec-PrPC-selective epitope (Figure 9H). As for disease-related huPrP mutations, many reside on obstructed surfaces or are buried in the core of the protein and therefore likely affect conversion/aggregation processes beyond the initial step of PrPC binding to PrPSc aggregates (Figure 9I).
MD simulations of PrPgpi, PrPglyco and PrPrec, in combination with experimental results, indicate that glycosylation does not markedly affect the structure and dynamics of the polypeptide chain, relative to rec-PrPC. However, based on the MD simulations we hypothesize that the glycans may modulate the early steps in conversion, and resulting structure, of the misfolded species by stabilizing the globular domain via interactions with the HA-face. As previously demonstrated, mobility of the HA segment facilitates conversion (Figure 10). Even under misfolding conditions, this motion can be inhibited by glycan-2; helping glycosylated forms maintain native PrPC structure, particularly near the unstructured N-terminus (Figure 10). Although potentially inhibited, conversion of glycosylated PrPC can proceed. Similar to the subtle differences in PrPSc structure induced by sequence variation (Alonso et al. 2002; DeMarco and Daggett 2007; Scouras and Daggett 2008), steric interference by the glycans could force the molecule to misfold into a slightly different structure. This could contribute to the observed differences in PrPSc-binding efficiencies associated with glycosylated and unglycosylated species of PrPC (Priola and Lawson 2001), and ultimately affect the morphology of the resultant PrPSc fibril.
Similar to glycosylation, attachment of PrPC to the membrane surface via a GPI anchor does not significantly alter the structure or dynamics of PrPC, and while anchoring brings the unstructured N-terminus in close proximity to the membrane surface, there are limited interactions with the bilayer. The proximity to the membrane and bulk of the glycans do, however, limit accessibility to certain surfaces. Antibodies that target the N-terminus or HA-face are likely to have better selectivity and affinity for PrPC in vivo than those targeted to regions near HB. These exposed surfaces in PrPCgpi also reflect possible recognition domains with infectious PrPSc aggregates.
Acknowledgements
We are grateful for financial support provided by the National Institutes of Health (GM 81407 to V. D.) and the Hope Barnes Fellowship to M. L. D.
Abbreviations
- GPI
glycosylphosphatidylinositol
- MD
molecular dynamics
- POPC
1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine
- PrP
prion protein
- PrPC
cellular prion protein
- PrPSc
infectious and/or misfolded form of the prion protein
- PrPrec
protein-only portion of PrP
- PrPglyco
diglycosylated (soluble) prion protein
- PrPgpi
diglycosylated and GPI-anchored prion protein
References
- Alonso DOV, An C, Daggett V. Simulations of biomolecules: characterization of the early steps in the pH-induced conformational conversion of the hamster, bovine and human forms of the prion protein. Philos. Trans. R. Soc. Lond. B. Biol. Sci. 2002;360:1165–1178. doi: 10.1098/rsta.2002.0986. [DOI] [PubMed] [Google Scholar]
- Alonso DOV, DeArmond SJ, Cohen FE, Daggett V. Mapping the early steps in the pH-induced conformational conversion of the prion protein. Proc. Natl. Acad. Sci. U. S. A. 2001;98:2985–2989. doi: 10.1073/pnas.061555898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baron GS, Caughey B. Effect of glycosylphosphatidylinositol anchor-dependent and-independent prion protein association with model raft membranes on conversion to the protease-resistant isoform. J. Biol. Chem. 2003;278:14883–14892. doi: 10.1074/jbc.M210840200. [DOI] [PubMed] [Google Scholar]
- Baron GS, Wehrly K, Dorward DW, Chesebro B, Caughey B. Conversion of raft associated prion protein to the protease-resistant state requires insertion of PrP-res (PrPSc) into contiguous membranes. EMBO J. 2002;21:1031–1040. doi: 10.1093/emboj/21.5.1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beck DAC, Daggett V. Methods for molecular dynamics simulations of protein folding/unfolding in solution. Methods. 2004;34:112–120. doi: 10.1016/j.ymeth.2004.03.008. [DOI] [PubMed] [Google Scholar]
- Beck DAC, Alonso DOV, Daggett V. In lucem molecular mechanics, Computer Program. University of Washington; 2000-2008. [Google Scholar]
- Bohne A, Lang E, von der Lieth CW. SWEET - WWW-based rapid 3D construction of oligo- and polysaccharides. Bioinformatics. 1999;15:767–768. doi: 10.1093/bioinformatics/15.9.767. [DOI] [PubMed] [Google Scholar]
- Brugger B, Graham C, Leibrecht I, Mombelli E, Jen A, Wieland F, Morris R. The membrane domains occupied by glycosylphosphatidylinositol-anchored prion protein and Thy-1 differ in lipid composition. J. Biol. Chem. 2004;279:7530–7536. doi: 10.1074/jbc.M310207200. [DOI] [PubMed] [Google Scholar]
- Burns CS, Aronoff-Spencer E, Legname G, Prusiner SB, Antholine WE, Gerfen GJ, Peisach J, Millhauser GL. Copper coordination in the full-length, recombinant prion protein. Biochemistry. 2003;42:6794–6803. doi: 10.1021/bi027138+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caughey B. Interactions between prion protein isoforms: the kiss of death? Trends Biochem. Sci. 2001;26:235–242. doi: 10.1016/s0968-0004(01)01792-3. [DOI] [PubMed] [Google Scholar]
- Ceroni A, Dell A, Haslam S. The GlycanBuilder: a fast, intuitive and flexible software tool for building and displaying glycan structures. Source Code for Biology and Medicine. 2007;2:3. doi: 10.1186/1751-0473-2-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collinge J, Sidle KC, Meads J, Ironside J, Hill AF. Molecular analysis of prion strain variation and the aetiology of 'new variant' CJD. Nature. 1996;383:685–690. doi: 10.1038/383685a0. [DOI] [PubMed] [Google Scholar]
- DeArmond SJ, Qiu Y, Sanchez H, Spilman PR, Ninchak-Casey A, Alonso D, Daggett V. PrPC glycoform heterogeneity as a function of brain region: Implications for selective targeting of neurons by prion strains. J. Neuropathol. Exp. Neurol. 1999;58:1000–1009. doi: 10.1097/00005072-199909000-00010. [DOI] [PubMed] [Google Scholar]
- DeArmond SJ, Sanchez H, Yehiely F, Qiu Y, Ninchak-Casey A, Daggett V, Camerino AP, Cayetano J, Rogers M, Groth D, Torchia M, Tremblay P, Scott MR, Cohen FE, Prusiner SB. Selective neuronal targeting in prion disease. Neuron. 1997;19:1337–1348. doi: 10.1016/s0896-6273(00)80424-9. [DOI] [PubMed] [Google Scholar]
- DeMarco ML, Daggett V. From conversion to aggregation: Protofibril formation of the prion protein. Proc. Natl. Acad. Sci. U. S. A. 2004;101:2293–2298. doi: 10.1073/pnas.0307178101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeMarco ML, Daggett V. Molecular Mechanism for Low pH Triggered Misfolding of the Human Prion Protein. Biochemistry. 2007;46:3045–3054. doi: 10.1021/bi0619066. [DOI] [PubMed] [Google Scholar]
- DeMarco ML, Silveira JR, Caughey B, Daggett V. Structural Properties of Prion Protein Protofibrils and Fibrils: An Experimental Assessment of Atomic Models. Biochemistry. 2006;445:15573–15582. doi: 10.1021/bi0612723. [DOI] [PubMed] [Google Scholar]
- Eberl H, Tittmann P, Glockshuber R. Characterization of recombinant, membrane-attached full-length prion protein. J. Biol. Chem. 2004;279:25058–25065. doi: 10.1074/jbc.M400952200. [DOI] [PubMed] [Google Scholar]
- Englund PT. The structure and biosynthesis of glycosyl phosphatidylinositol protein anchors. Annu. Rev. Biochem. 1993;62:121–138. doi: 10.1146/annurev.bi.62.070193.001005. [DOI] [PubMed] [Google Scholar]
- Ferguson MA, Homans SW, Dwek RA, Rademacher TW. The glycosylphosphatidylinositol membrane anchor of Trypanosoma brucei variant surface glycoprotein. Biochem. Soc. Trans. 1988;16:265–268. doi: 10.1042/bst0160265. [DOI] [PubMed] [Google Scholar]
- Heller H, Schaefer M, Schulten K. Molecular-Dynamics Simulation of a Bilayer of 200 Lipids in the Gel and in the Liquid-Crystal Phases. J. Phys. Chem. 1993;97:8343–8360. [Google Scholar]
- Henriques ST, Pattenden LK, Aguilar MI, Castanho MA. PrP(106-126) does not interact with membranes under physiological conditions. Biophys J. 2008 doi: 10.1529/biophysj.108.131458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hermann LM, Caughey B. The importance of the disulfide bond in prion protein conversion. Neuroreport. 1998;9:2457–2461. doi: 10.1097/00001756-199808030-00006. [DOI] [PubMed] [Google Scholar]
- Homans SW, Edge CJ, Ferguson MA, Dwek RA, Rademacher TW. Solution structure of the glycosylphosphatidylinositol membrane anchor glycan of Trypanosoma brucei variant surface glycoprotein. Biochemistry. 1989;28:2881–2887. doi: 10.1021/bi00433a020. [DOI] [PubMed] [Google Scholar]
- Horiuchi M, Baron GS, Xiong LW, Caughey B. Inhibition of interactions and interconversions of prion protein isoforms by peptide fragments from the C-terminal folded domain. J. Biol. Chem. 2001;276:15489–15497. doi: 10.1074/jbc.M100288200. [DOI] [PubMed] [Google Scholar]
- Hornemann S, Glockshuber R. A scrapie-like unfolding intermediate of the prion protein domain PrP(121-231) induced by acidic pH. Proc. Natl. Acad. Sci. U. S. A. 1998;95:6010–6014. doi: 10.1073/pnas.95.11.6010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hornemann S, Schorn C, Wuthrich K. NMR structure of the bovine prion protein isolated from healthy calf brains. EMBO Report. 2004;5:1159–1164. doi: 10.1038/sj.embor.7400297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubbard SJ, Thornton JM. ‘NACCESS’, Computer Program, Department of Biochemistry and Molecular Biology. University College London; 1993. [Google Scholar]
- Jackson GS, Hill SF, Joseph C, Hosszu L, Power A, Waltho JP, Clarke AR, Collinge J. Multiple folding pathways for heterologously expressed human prion protein. Biochim. Biophys. Acta. 1999;1431:1–13. doi: 10.1016/s0167-4838(99)00038-2. [DOI] [PubMed] [Google Scholar]
- James TL, Liu H, Ulyanov NB, FarrJones S, Zhang H, Donne DG, Kaneko K, Groth D, Mehlhorn I, Prusiner SB, Cohen FE. Solution structure of a 142-residue recombinant prion protein corresponding to the infectious fragment of the scrapie isoform. Proc. Natl. Acad. Sci. U. S. A. 1997;94:10086–10091. doi: 10.1073/pnas.94.19.10086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kell GS. Precise Representation of Volume Properties of Water at 1 Atmosphere. J. Chem. Eng. Data. 1967;12:66. [Google Scholar]
- Kocisko DA, Priola SA, Raymond GJ, Chesebro B, Lansbury PT, Caughey B. Species specificity in the cell-free conversion of prion protein to protease-resistant forms: a model for the scrapie species barrier. Proc. Natl. Acad. Sci. U. S. A. 1995;92:3923–3927. doi: 10.1073/pnas.92.9.3923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kocisko DA, Come JH, Priola SA, Chesebro B, Raymond GJ, Lansbury PT, Caughey B. Cell-Free Formation of Protease-Resistant Prion Protein. Nature. 1994;370:471–474. doi: 10.1038/370471a0. [DOI] [PubMed] [Google Scholar]
- Kucerka N, Tristram-Nagle S, Nagle JF. Structure of fully hydrated fluid phase lipid bilayers with monounsaturated chains. J. Membr. Biol. 2006;208:193–202. doi: 10.1007/s00232-005-7006-8. [DOI] [PubMed] [Google Scholar]
- Langella E, Improta R, Crescenzi O, Barone V. Assessing the acid-base and conformational properties of histidine residues in human prion protein (125-228) by means of pKa calculations and molecular dynamics simulations. Proteins. 2006;64:167–177. doi: 10.1002/prot.20979. [DOI] [PubMed] [Google Scholar]
- Leclerc E, Peretz D, Ball H, Solforosi L, Legname G, Safar J, Serban A, Prusiner SB, Burton DR, Williamson RA. Conformation of PrPC on the cell surface as probed by antibodies. J. Mol. Biol. 2003;326:475–483. doi: 10.1016/s0022-2836(02)01365-7. [DOI] [PubMed] [Google Scholar]
- Lee RJ, Wang S, Low PS. Measurement of endosome pH following folate receptor-mediated endocytosis. Biochim. Biophys. Acta. 1996;1312:237–242. doi: 10.1016/0167-4889(96)00041-9. [DOI] [PubMed] [Google Scholar]
- Legname G, Baskakov IV, Nguyen HO, Riesner D, Cohen FE, DeArmond SJ, Prusiner SB. Synthetic mammalian prions. Science. 2004;305:673–676. doi: 10.1126/science.1100195. [DOI] [PubMed] [Google Scholar]
- Lehmann S, Harris DA. Blockade of Glycosylation Promotes Acquistion of Scrapie-like Properties by the Prion Protein in Cultured Cells. J. Biol. Chem. 1997;272:21479–21487. doi: 10.1074/jbc.272.34.21479. [DOI] [PubMed] [Google Scholar]
- Levitt M, Hirshberg M, Sharon R, Daggett V. Potential-Energy Function and Parameters for Simulations of the Molecular-Dynamics of Proteins and Nucleic-Acids in Solution. Comput. Phys. Commun. 1995;91:215–231. [Google Scholar]
- Levitt M, Hirshberg M, Sharon R, Laidig KE, Daggett V. Calibration and testing of a water model for simulation of the molecular dynamics of proteins and nucleic acids in solution. J. Phys. Chem. B. 1997;101:5051–5061. [Google Scholar]
- McConville MJ, Ferguson MA. The structure, biosynthesis and function of glycosylated phosphatidylinositols in the parasitic protozoa and higher eukaryotes. Biochem. J. 1993;294(Pt 2):305–324. doi: 10.1042/bj2940305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morillas M, Swietnicki W, Gambetti P, Surewicz WK. Membrane environment alters the conformational structure of the recombinant human prion protein. J. Biol. Chem. 1999;274:36859–36865. doi: 10.1074/jbc.274.52.36859. [DOI] [PubMed] [Google Scholar]
- Mouillet-Richard S, Ermonval M, Chebassier C, Laplanche JL, Lehmann S, Launay JM, Kellermann O. Signal transduction through prion protein. Science. 2000;289:1925–1928. doi: 10.1126/science.289.5486.1925. [DOI] [PubMed] [Google Scholar]
- Nishina KA, Deleault NR, Mahal SP, Baskakov I, Luhrs T, Riek R, Supattapone S. The Stoichiometry of Host PrPC Glycoforms Modulates the Efficiency of PrPSc Formation in Vitro. Biochemistry. 2006;45:14129–14139. doi: 10.1021/bi061526k. [DOI] [PubMed] [Google Scholar]
- Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. UCSF chimera - A visualization system for exploratory research and analysis. J. Comput. Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- Priola SA, Lawson VA. Glycosylation influences cross-species formation of protease-resistant prion protein. EMBO J. 2001;20:6692–6699. doi: 10.1093/emboj/20.23.6692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Priola SA, Chabry J, Chan K. Efficient conversion of normal prion protein (PrP) by abnormal hamster PrP is determined by homology at amino acid residue 155. J. Virol. 2001;75:4673–4680. doi: 10.1128/JVI.75.10.4673-4680.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scouras AD, Daggett V. Species variation in PrPSc protofibril models. J. Mater. Sci. 2008;43:3625–3637. [Google Scholar]
- Smolin N, Li B, Beck DAC, Daggett V. Side-chain dynamics are critical for water permeation through aquaporin-1. Biophys. J. 2008;95:1089–1098. doi: 10.1529/biophysj.107.125187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stahl N, Baldwin MA, Hecker R, Pan KM, Burlingame AL, Prusiner SB. Glycosylinositol phospholipid anchors of the scrapie and cellular prion proteins contain sialic acid. Biochemistry. 1992;31:5043–5053. doi: 10.1021/bi00136a600. [DOI] [PubMed] [Google Scholar]
- Stimson E, Hope J, Chong A, Burlingame AL. Site-specific characterization of the N-linked glycans of murine prion protein by high-performance liquid chromatography electrospray mass spectrometry and exoglycosidase digestions. Biochemistry. 1999;38:4885–4895. doi: 10.1021/bi982330q. [DOI] [PubMed] [Google Scholar]
- Swietnicki W, Petersen R, Gambetti P, Surewicz WK. pH-dependent stability and conformation of the recombinant human prion protein PrP(90-231) J. Biol. Chem. 1997;272:27517–27520. doi: 10.1074/jbc.272.44.27517. [DOI] [PubMed] [Google Scholar]
- Taraboulos A, Rogers M, Borchelt DR, McKinley M,P, Scott M, Serban D, Prusiner SB. Acquisition of protease resistance by prion proteins in scrapie-infected cells does not require asparagine-linked glycosylation. Proc. Natl. Acad. Sci. U. S. A. 1990;87:8262–8266. doi: 10.1073/pnas.87.21.8262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turk E, Teplow DB, Hood LE, Prusiner SB. Purification and Properties of the Cellular and Scrapie Hamster Prion Proteins. Eur. J. Biochem. 1988;176:21–30. doi: 10.1111/j.1432-1033.1988.tb14246.x. [DOI] [PubMed] [Google Scholar]
- Tuzi NL, Cancellotti E, Baybutt H, Blackford L, Bradford B, Plinston C, Coghill A, Hart P, Piccardo P, Barron RM, Manson JC. Host PrP Glycosylation: A Major Factor Determining the Outcome of Prion Infection. PLoS Biology. 2008;6:e100. doi: 10.1371/journal.pbio.0060100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varki A, Cummings R, Esko J, Freeze H, Hart G, Marth J. The Essentials of Glycobiology. Cold Spring Harbor Laboratory Press; New York: 2003. [PubMed] [Google Scholar]
- Zahn R, Liu AZ, Luhrs T, Riek R, von Schroetter C, Garcia FL, Billeter M, Calzolai L, Wider G, Wuthrich K. NMR solution structure of the human prion protein. Proc. Natl. Acad. Sci. U. S. A. 2000;97:145–150. doi: 10.1073/pnas.97.1.145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou WQ, Cashman NR. Acidic pH and detergents enhance in vitro conversion of human brain PrPC to a PrPSc-like form. J. Biol. Chem. 2002;277:43942–43947. doi: 10.1074/jbc.M203611200. [DOI] [PubMed] [Google Scholar]