

Abstract
Amyloidogenesis is the aggregation of soluble proteins into structurally conserved fibers. Amyloid fibers are distinguished by their resistance to proteinase K, tinctorial properties and β-sheet-rich secondary structure. Amyloid formation is a hallmark of many human diseases including Alzheimer’s, Huntington’s and the prion diseases. Therefore, understanding amyloidogenesis will provide insights into the development of therapeutics that target these debilitating diseases. A new class of ‘functional’ amyloids promises a unique glimpse at how nature has harnessed the amyloid fiber to accomplish important physiological tasks. Functional amyloids are produced by organisms spanning all aspects of cellular life. Herein we review amyloidogenesis, with special attention focused on the similarities and differences between the best characterized disease-associated amyloidogenic protein amyloid-β and the formation of several functional amyloids. The implications of studying functional amyloidogenesis and the strategies organisms employ to limit exposure to toxic intermediates will also be discussed.
INTRODUCTION
Amyloidogenesis is recognized as being the underlying cause of neurodegenerative diseases such as Alzheimer’s (AD), Huntington’s and Parkinson’s disease. Amyloid fibrils have biochemical and biophysical properties that distinguish them from other biological polymers. Amyloid fibers are incredibly stable, detergent insoluble, β–sheet rich structures that many proteins can form [120]. Amyloid fibers associated with neurodegenerative diseases are considered the product of a protein misfolding event. The pathology of neurodegenerative diseases defined amyloid polymerization as an aberrant process where misfolded proteins aggregate and cause disease. However, there are a number of examples where organisms can utilize either the amyloid fiber itself or intermediates formed during the amyloid polymerization process to fulfill specific biological functions [21,25,30,40, 41,64,115,132]. Unlike disease-associated amyloidogenic proteins, functional amyloid assembly is a regulated process that minimizes the cellular toxicity associated with disease-associated amyloids. There are, though, examples where organisms utilize the toxicity of the amyloid fold to carry out a function. Understanding mechanisms that promote functional amyloidogenesis will provide an unprecedented glimpse into amyloidogenic systems in general and will lead to new ideas for preventing disease-associated amyloidogenesis. Guided by this perspective we compare and contrast amyloid-β (Aβ) amyloidogenesis as it relates to AD to several systems where functional amyloidogenesis occurs presenting ideas about how these functional amyloid systems prevent the build up of amyloid associated toxicity.
ALZHEIMER’S DISEASE
AD is the most common neurodegenerative disease. More than 4 million people are afflicted with this neurodegenerative disease in the United States alone (http://www.ahaf.org/alzdis/about/adabout.htm). The clinical and neuropathological characteristics were first reported in 1906 by Alois Alzheimer. The abnormal deposits, described as both plaques and tangles, were found in the postmortem diseased brain and were later called amyloid plaques [2]. The plaques were found to be composed of long, unbranched 4–10 nanometer wide fibers when viewed with an electron microscope (Fig. 1A) [65,127]. These structures were discovered to be proteinaceous in nature and contained a uniquely stable cross-beta sheet quaternary structure. Fibers with similar structural characteristics have now been described in other neurodegenerative disorders including Parkinson’s disease and Huntington’s disease [48, 112].
Fig. 1.
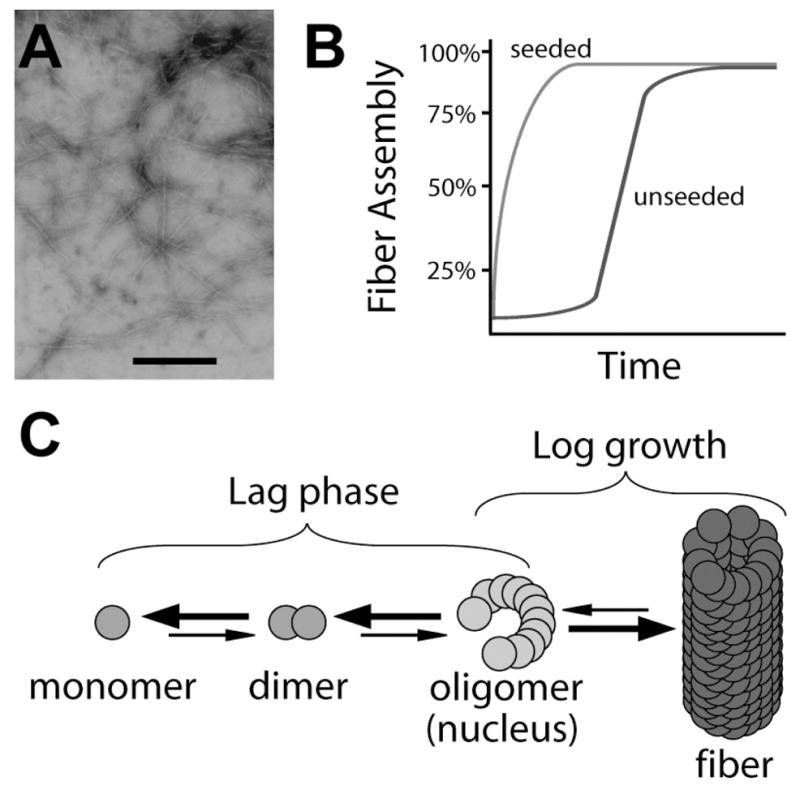
Properties of amyloid polymerization. (A) Negatively stained electron micrograph of polymerized Aβ fibers. The scale bar represents 500 nanometers. (B) A graphic representation of amyloid fiber polymerization displaying nucleus dependent kinetics (blue line). Preformed amyloid fibers can act as seeds to speed the kinetics of fiber polymerization (red line). This process eliminates the lag phase associated with nucleus formation. (C) Model of amyloid fiber polymerization. A build up of monomer occurs which leads to the formation of multimers and finally the amyloid fiber end product. Large arrows represent processes that are energetically favorable while small arrows represent energetically unfavorable processes.
The Aβ polypeptide was purified from AD associated plaques and was determined to be the major protein component of amyloid plaques [43,144]. Aβ is formed when the amyloid-β protein precursor (AβPP) is sequentially cleaved by β- and γ-secretases [49]. It is proposed that AβPP plays important physiological roles in cell adhesion, neurite outgrowth, synaptogenesis and synapse remodeling [148], however, the function of the Aβ polypeptide is currently unknown. There are two major cleavage products, Aβ40 and Aβ42 [57]. The primary sequences of Aβ40 and Aβ42 only differ in that Aβ42 has 2 additional C-terminal residues, Ile41 and Ala42. Mutations in presenilins, a central component of γ-secretase, account for most cases of familial AD. These mutations increase the production of Aβ42 in both transfected cells and transgenic mice [24]. In sporadic AD cases the apolipoprotein E (APOE) ε4 allele is the genetic risk factor most often linked to disease onset [106]. In cultured neuronal cells APOE4 enhances Aβ production by modulating AβPP processing [146]. In addition, it was reported that APOE4 also modulates the degradation and clearance of deposited Aβ [5,42,69,147].
Several lines of evidence link AβPP and misfolded Aβ to AD (for review see [13,45,50]). However, the molecular mechanism behind Aβ misfolding and how this leads to AD remains unclear. Hardy and Selkoe proposed the “Amyloid Cascade Hypothesis” in which the central event in AD development is an imbalance between Aβ production and clearance [55]. The biochemical species of Aβ that induces pathogenesis is still a hotly debated topic. Experimental evidence suggests two models: (1) the final amyloid fiber product causes neuronal damage or (2) neuronal cells are exposed to a toxic intermediates formed as Aβ polymerizes into the amyloid fiber.
In vitro self assembly of Aβ polypeptides is characterized by nucleation-dependent polymerization kinetics (Fig. 1B blue line) [61,78]. Before mature fiber aggregates are detectable, there is a time period where the Aβ polypetide polymerization appears to be stagnant. However, during this lag phase trace amounts of dimer, trimer, and eventually, nucleus (oligomer) are formed (Fig. 1C) [104,105,137]. Nucleus formation is the rate-limiting step of fibril assembly. Once a nucleus has formed, monomer addition to the growing fiber becomes thermodynamically favorable and occurs quickly (Fig. 1B and C) [61]. As with any dynamic polymerization process where different folding intermediates are present at any one time, Aβ monomer, oligomer, protofibrils (short fibrillar aggregates) and fibrils have been observed using different techniques including atomic force microscopy [108,137]. Amyloid formation inhibitors such as Congo red and curcumin potentially reduce neurotoxicity by stabilizing the monomeric state of Aβ, thus reducing the amount of oligomer intermediates formed [79,104,105,145]. Therefore neurotoxicity seems to be linked to aggregation of monomers to higher ordered structures [16,101, 102].
Amyloid laden plaques are often found in postmortem AD brains, which led to the suggestion that mature insoluble fiber aggregates of the causative agent for AD. However, statistical analyses have found only a weak correlation between the number of amyloid aggregates and the severity of AD [80,128]. In addition, high molecular weight Aβ42 aggregates do not correlate with toxicity in the Caenorhabiditis elegans AD model [27]. The formation of high molecular weight protein aggregates may be a mechanism to protect cells from cytoxicity by sequestering the toxic intermediates present formed during Aβ misregulation [3,119]. A wide range of nonfibrillar Aβ forms including dimer, trimer, oligomer, spherical aggregates and protofibrils have been reported to be cytotoxic and support this idea [26,56,60,71,75,92,108,130,138]. Different Aβ forms, including the small diffusible Aβ oligomer, high molecular weight oligomer, and fibers effect cortical neurons differently [37]. Collectively, this data suggest nonfibrillar intermediates trigger neuropathologies. Therefore, the development of nuerodegeneration could be induced by a complicated combinatory effect of several toxic Aβ conformers including the fibers themselves.
Despite evidence that prefibrillar aggregates may be the causative agents AD toxicity, many researchers have reported that mature Aβ fibers to be toxic to cultured neuronal cells [17,37,56,100,133]. How can this apparently conflicting data be reconciled? Recent study has demonstrated that the amyloid fiber is not a static structure. For instance, amyloid fibers formed from an SH3 domain showed very dynamic properties, in which molecules can be recycled by a dissociation and re-association mechanism within the fibril population [19]. Therefore amyloid fibrils could provide a reservoir for toxic soluble oligomers, which could trigger the pathology [50]. Under different experimental conditions amyloid fibrils may have different potentials to liberate soluble oligomers, which may be cytotoxic to the cultured neurons. Nevertheless, the ability to effectively develop therapeutics that will deter AD development is difficult due to the lack of experimental evidence defining a toxic species. Moreover, the molecular mechanism behind the initial misfolding events that convert soluble Aβ into an amyloid fiber in vivo has not been forthcoming. Perhaps exploring systems where amyloid formation occurs as a natural functional process will provide answers to these questions.
AMYLOID AS A FUNCTIONAL FOLD
Functional bacterial amyloids
Curli
The first example of a functional amyloid fiber was demonstrated in the common laboratory bacterium Escherichia coli. E. coli and other Gram-negative enteric bacteria produce a functional amyloid fiber called curli (Fig. 2A). These fibers mediate many important physiological functions for the cell. Curli fibers are the major proteinacious component of the extracellular matrix produced by bacteria during growth in biofilms. Curli also induce a potent host inflammatory response, initiate binding to host cells, and increase the ability of the bacteria to persist within the environment and the host [10,11,73,134,135,139,142]. The genetic and biochemical tools afforded by E. coli have provided an in depth look at how bacteria control the assembly of amyloid fibers [21].
Fig. 2.

Interaction between the curli subunit proteins CsgA and CsgB. (A) Negative stain electron micrograph of wild type cells producing curli. Scale bar represents 200 nanometers. (B) Model of Interbacterial Complementation. A donor cells secretes soluble CsgA that acts as a substrate for CsgB on an acceptor cells where curli biogenesis takes place. (C) A Congo red indicator plate demonstrating interbacterial complementation. The donor cells and the acceptor cells appear white until the two strains intersect. Once the two cell types intersect Congo red binding occurs demonstrating curli fiber polymerization as taken place. The arrow represents the direction in which the acceptor cells were streaked onto the plate. (D) The oligopeptide repeating units that compose the CsgA and CsgB proteins. The three dimensional structures of CsgA and CsgB are predicted to be composed of five imperfect β-strand-loop-β-strand oligopeptide repeats (R1–R5). Amino acids comprising the β-strand are located below the arrows, and amino acids predicted to comprise the loops are denoted with italicized blue letters. Bolded letters represent amino acids conserved in CsgB and CsgA at each position relative to the start of each repeating unit. Boxed letters represent amino acids conserved throughout the repeating units in both proteins.
Biosynthesis of curli fibers is dependent on two divergently transcribed operons, csgDEFG and csgBA, both of which are under the control of a complex regulatory network [6,51,109,110]. The csgBA operon encodes the minor and major curli subunit proteins, Cs-gB and CsgA, respectively. The stability and secretion of both CsgA and CsgB is dependent on the outer-membrane localized CsgG protein [21,53,77,107]. The functions of CsgF and CsgE have not been elucidated, but it is clear that CsgE plays an important role in the stability of both CsgB and CsgA, while CsgF is required for efficient curli biogenesis [21].
CsgA and CsgB are the crux of curli fibers. When incorporated into curli fibers, both CsgA and CsgB are detergent insoluble. Cells that do not express CsgB secrete CsgA into the extracellular milieu as a soluble, unpolymerized protein (Fig. 2B). Therefore, in vivo both CsgA and CsgB are required for curli biogenesis. However, CsgA and CsgB do not have to be expressed by the same cell for curli assembly to occur. In a process called interbacterial complementation, CsgA secreted from a csgB mutant, or donating cell, can be polymerized by CsgB produced on the surface of a csgA mutant or accepting cell (Fig. 2B and C) [21,52, 53,77].The ability of CsgB to convert CsgA into an insoluble fiber led it to be designated the curli nucleator protein.
CsgA and CsgB are 30% identical and 40% similar at the amino acid level, and each protein has a domain composed of five glutamine-asparagine rich oligopeptide repeats (Fig. 2D) [28,52,141]. Each glutamine-asparagine rich repeating unit is composed of roughly 20 amino acids and is predicted to form consecutive β-strand loop β-strand motifs that stack perpendicular to the axis of fiber growth (Fig. 2D). Peptides composed of the first, third and fifth oligopeptide repeats of CsgA are amyloidogenic [140]. Mature CsgA protein has been purified, and can self-assemble into curli-like amyloid fibers [21]. In vitro, purified CsgA can form amyloid in the absence of CsgB, whereas in vivo, CsgA amyloid formation is CsgB-dependent.
CsgA and Aβ in vitro polymerization share common features. First, in vitro polymerization of both proteins contains three distinct phases: a lag phase, a growth phase, and a stationary phase (Fig. 1B blue line). Second, in a process called seeding, the lag phase associated with the polymerization of CsgA and Aβ can be abrogated by the addition of preformed fibers composed of CsgA and Aβ respectively (Fig. 1B red line) [78, 140]. Lastly, a conformation specific antibody recognizes a folding intermediate formed during the lag phase of both protein’s self assembly process [63,140]. These polymerization features are characteristic of an assembly mechanism that is dependent on nucleus formation. Nucleus formation is the rate limiting step and must occur before the protein can begin self-assembly into an amyloid fiber (Fig. 1C). While the roles of nucleus formation as it relates to Aβ polymerization and AD has yet to be elucidated, the curli system is unique in that CsgB’s function is to serve as a nucleus for CsgA in vivo.
A truncated version of CsgB containing the repeating units most similar to those found in CsgA (R1–R4) was recently purified and also demonstrated the ability to self-assemble into an amyloid fiber (Fig. 2D) [53]. Similar to CsgA and Aβ polymerization, the polymerization of the truncated CsgB resembled nucleus dependent kinetics displaying a lag phase followed by a polymerization phase, followed by a stationary phase. Fibers composed of the truncated version of CsgB abrogate the CsgA’s polymerization lag phase, thus recapitulating in vivo curli biogenesis [53]. These results demonstrate that CsgB acts as a template for CsgA polymerization.
The culmination of the experimental evidence suggests E. coli has evolved an elegant strategy to control amyloid fiber biogenesis. Curli biogenesis begins when CsgB is secreted and anchored to the outer membrane where it acts as a template for newly secreted Cs-gA. In a process called nucleation CsgB converts soluble monomeric CsgA into a β–sheet-rich, detergent insoluble protein. Then, in a process called seeding, the growing fiber tip can act as a folding template for additional soluble CsgA monomers. By strictly regulating the csg operons and by separating the nucleation process and seeding process into two separate proteins, the cell ensures amyloid fiber biogenesis occurs at the right place and at the right time. This strategy decreases exposure to potentially cytotoxic folding intermediates by promoting mature amyloid fiber formation. Therefore, understanding the molecular basis of curli nucleation and polymerization may provide new insights into how the toxicity of disease-associated amyloids can be reduced.
Chaplins
The chaplins are extracellular structures produced by the Gram-positive bacterium Streptomyces coelicolor. In vivo, these amyloid fibers reduce the surface tension at the media/air interface and allow for the development of aerial hyphea [25,38]. Without the chaplins development of aerial hyphea is impaired [25,38]. Chaplin biogenesis is dependent upon the translational products of the chpA-H operon. Like Aβ and CsgA, the chaplins can assemble into β–rich insoluble fibers that bind the amyloid specific dye thioflavin T in vitro [25].
Like curli, chaplin amyloid biogenesis is temporally and positionally coordinated. Chaplin expression is dependent on the bldN developmental sigma factor, ensuring that fiber formation occurs at the proper time [38]. Furthermore, chaplin amyloid formation is localized to the extracellular space, which may limit exposure to cytotoxic intermediates [25,38].
Microcin E492 and the harpins
Two examples of bacteria utilizing the cytotoxic properties of amyloid to deter the growth of neighboring cells have been identified. Microcin E492 (also called Mcc), produced by Klebsiella pneumoniae, is a potent antibacterial bacteriocin. Mcc is most active during logarithmic growth, losing most of its cytotoxic properties in stationary phase [34,35]. Bieler and colleagues found that Mcc polymerizes into amyloid fibrils biochemically identical to Aβ and CsgA fibers. Remarkably, the polymerization of Mcc into a mature amyloid fiber coincides with a loss of Mcc antibacterial activity [12]. Thus, a pre-fiber intermediate is proposed to be the cytotoxic species of Mcc [12]. It is also interesting to note that lower concentrations of Mcc are able to induce apoptosis in some human cell lines although the mechanism of Mcc induced apoptosis is currently unknown [59].
The harpins are a second class of bacterial proteins that capitalize on the cytotoxic features of amyloid biogenesis. Produced by plant pathogens, harpins are type-III secreted proteins that induce the hypersensitive response in plants [93]. The hypersensitive response is a plant defense mechanism that slows intracellular pathogen growth by eliciting plant cell death. The hypersensitive response is similar to apoptosis in animal cells [46,47,99]. Oh et al. discovered that HpaG, a harpin produced by Xanthomonas axonopodis pv. glycines 8ra, self assembles into amyloid-like fibers. Unlike Mcc, injection of HpaG protofibrils and mature amyloid fibers into plant cells is toxic and results in cell death. Oh et al also demonstrated that a harpin from E. amylovora, HrpN, and a harpin from Pseudomonas syringae pv. syringae, HrpZ, form amyloid fibers [93]. Both of these harpins elicit the hypersensitive response. A harpin unable to induce the hypersensitive response, XopA from Xanthomonas campestris pv. vesicatoria, did not form amyloid fibers [93]. However, a gain-of-function mutant form of XopA (F48L/M52L) which does induce the hypersensitive response polymerizes into an amyloid fiber, correlating the ability to induce the hypersensitive response to the ability to form amyloid [93]. The harpins are an example of a functional amyloid fiber that is designed to be lethal.
Potential bacterial amyloids
Two recently discovered bacterial structures may be composed of amyloid fibers. M. tuberculosis produces a structure that resembles E. coli curli fibers. Like curli and other amyloid fibers, the M. tuberculosis pili (MTP) are stable and non-branching fibers [1]. Also like curli, MTP bind host extracellular matrix proteins [1]. Structural and biochemical tests including circular dichroism, the ability to bind the amyloid specific dyes Congo red and thioflavin T, and resistance to proteinase K treatment will determine if these pili are in fact amyloid.
The second potential bacterial structure that might be an amyloid is the bacterial endospore. When confronted with environments limited for nutrients some species of bacteria such as Bacillus and Clostridium initiate the formation of an endospore, a unique structure that is highly resistant to heat, radiation, pH extremes, and toxic chemicals. High resolution atomic force microcopy has revealed that the spore coat of Bacillus atrophaeus is composed of fibrils similar to amyloid [103]. The inner and outer spore coats are composed of over 50 proteins and it is currently unknown which proteins form the fibril species observed and how this structure is assembled [66]. Thus further biochemical characterization of the proteins that form the spore coat, and their amyloidogenecity has not been elucidated. But it makes sense that sporulating bacteria would use the incredibly stable amyloid fiber as protective coat.
Eukaryotic functional amyloids
The yeast prion proteins: Eukaryotic functional amyloid domains
The [PSI+], [URE3], and [PIN+] phenotypes of the yeast Saccharomyces cerevisiae are defined by non-Mendelian inheritance. These phenotypes are transmitted to daughter cells via a conformationally altered amyloid version of the yeast proteins Sup35p, Ure2p, and Rnq1p, respectively [15,36,68,96,124,143]. Sup35p, Ure2p and Rnq1p all undergo a conversion to an aggregative state that can incorporate soluble protein into an insoluble amyloid aggregate [44,67,68,70,85, 97,98,116,121,122,125]. These proteins all contain a glutamine/asparagine (Q/N) rich domain that is essential for [PSI+] and [URE3] prion propagation [44,85, 121,125,126]. In both [PSI+] and [URE3] this conversion leads to the loss of wild type Sup35p and Ure2p function. The ability to confer a phenotype by converting normally soluble wild type protein into an infectious amyloid aggregate defines these proteins as prions.
[URE3]/Ure2p
Ure2p represses the genetic network needed to utilize poor nitrogen sources [29]. When yeast are provided with good nitrogen sources such as ammonia or glutamine, Ure2p binds to the positive transcriptional regulators Gln3p and Gat1p and prevents their translocation into the nucleus [7,14,18,33,54,83]. Yeast carrying the [URE3] prion have little Ure2p activity, and no phenotypic advantages have been demonstrated to correlate with the [URE3] prion. In addition, Nakayashiki et al. noted that 70 natural isolates of yeast do not carry [URE3], suggesting that the Ure2p prion state is not advantageous in a natural setting [90]. However, recent work by Shewmaker et al. demonstrated that Ure2p missing the Q/N domain had substantially reduced stability and activity [113]. Ure2p missing the Q/N domain had reduced steady state levels compared to the wild type protein suggesting the Q/N domain can act to increase Ure2p stability. These data along with the prevalence of the Q/N domain in the yeast proteome, suggest that the Q/N domain, a domain known to promote amyloidogenesis, may also function as a modular protein-stabilizing domain that also initiates protein-protein interactions.
[PSI+]/Sup35p
The Sup35p protein is an essential component of the translation termination machinery. In [psi−] yeast, Sup35p recognizes stop codons and terminates protein synthesis [123]. In [PSI+] cells wild type Sup35p is sequestered in self-assembled amyloid aggregates. Aggregated Sup35p is unable to participate in translational termination, resulting in translational read-through at wild type stop codons and C-terminally elongated proteins. As with [URE3], the [PSI+] phenotype is propagated through the community as dividing cells transmit the [PSI+] phenotype to daughter cells [31,32]. Novel work done by True and Lindquist demonstrated that the [PSI+] prion is advantageous under several growth conditions and may provide an alternative mechanism for phenotypic plasticity during environmental insult by altering the yeast proteome [131]. In addition, the Q/N domain itself has been estimated to be conserved for several hundred million years [23,62,89]. This suggests a strong selection to retain this amyloidogenic domain despite the possible detrimental effects of decreased translational termination fidelity or amyloid associated toxicity. Because some natural isolates of S. cerevisiae do not carry [PSI+], it has been proposed that the [PSI+] phenotype is not under selective pressure [90]. However, these studies are ongoing and the evolutionary impact of the [PSI+] phenotype is difficult to assess.
[PIN+]/Rnq1p
The [PIN+] phenotype is defined by the ability to induce the [PSI+] phenotype. The Rnq1p protein was discovered to induce [PSI+] [121]. Interestingly, the only known function of the Rnq1p protein is to induce the [PSI+] phenotype de novo. In addition to Rnq1p, Ure2p and New1p can also can induce [PSI+] when over-expressed [36,94]. Having a protein dedicated to the induction [PSI+] suggests that [PSI+] maybe advantageous in growth conditions where rnq1 is expressed. The study determining the prevalence of prions in natural yeast isolates found that the [PIN+] prion is present in some yeast found within the environment [90].
Regulation and coordination of yeast amyloid formation
Like Aβ and CsgA, Sup35p and Ure2p self assemble into amyloid fibers in vitro [39,116,129]. Also like CsgA and Aβ the self assembly process amyloid fiber polymerization contains a distinct lag phase that can be eliminated by the addition of preformed fibers composed of the respective protein [39,116,129,140]. Moreover, like Aβ and CsgA a conformational specific antibody reacts to an intermediate formed during Sup35 polymerization [114,116]. The curli proteins, CsgB and CsgA, are also similar to the yeast prion proteins, Sup35p, Ure2p, and Rnq1p, in that they all contain the Q/N rich domains [76,94]. In fact, the GNNQQNY peptide found within the Q/N rich domain of Sup35p forms biochemical distinct amyloid fibers [4]. This peptide has been used to examine the cross-beta structure of amyloids at high resolution using X-ray crystallography [91]. Also like CsgA and CsgB, oligopeptide repeats are found within the Q/N domain of Sup35p that aid the propagation and amyloidogenecity of the [PSI+] phenotype [76,95]. The Q/N domain of Rnq1p also contains several imperfect oligopeptide repeat sequences that are important for the propagation of [PIN+] [136]. In addition to Sup35p, Ure2p, and Rnq1p, 104 other polypeptides in the S. cerevisiae proteome contain Q/N rich domains [87]. However, their ability to form amyloid has not been demonstrated.
The conversion to the prion state for each of [PSI+], [URE3] and [PIN+] occurs at a low rate [81,143]. Even though the conversion rate to the prion state is low, yeast employ molecular chaperones called heat shock proteins to limit exposure to any toxic intermediates formed during prion propagation. Heat shock proteins, such as heat shock protein 104 (Hsp104), play an essential role in modulating the prion state. Propagation of [PSI+], [URE3], and [PIN+] all require Hsp104p as deletion of Hsp104 ‘cures’ (i.e. the prion phenotype no longer propagates to daughter cells) cells from [PSI+], [URE3] and [PIN+]. However, overexpression of Hsp104p also cures [PSI+] and overexpression of Ydj1p, a member of the Hsp40 family of proteins, cures cells of the [URE3] prion [22,88,121]. Shorter and Lindquist reconciled these seemingly contradictory findings by demonstrating that in vitro low concentrations Hsp104 catalyzed the formation of intermediates critical for Sup35p and Ure2p fiber formation, while high concentrations of Hsp104 completely abrogated the ability of both proteins to form an amyloid [114, 116]. These data suggest that Hsp104 may sequester toxic intermediates as well as decrease the time cells are exposed to such intermediates. These findings also suggests that another mechanism heat shock proteins employ to limit exposure to toxic folding intermediates is to speed the formation of an amyloid fiber to the fiber final product.
Filamentous fungi Het-S amyloid
Vegetative cell fusions occur within and between individual cells of the filamentous fungi Podospora anserina. These fusion events lead to cytoplasmic mixing and the production of a vegetative heterokaryon or multinucleated cells. The het locus controls the viability of the fused fungi, whereby heterokaryons that differ at the het locus are destroyed. This process is called heterokaryon incompatibility (For review see [111]). The het locus has two alleles, het-s and het-S. Het-S is the soluble protein product of the het-S loci while the protein product of the het-s loci, Het-s, has the ability to convert to an aggregated prion state. When fusion between a het-S cell and a het-s cell occurs the aggregated Het-s interacts with soluble Het-S and this interaction induces the incompatibility reaction. This leads to death of the heterokaryon and prevents any fusion from occurring between the two cells [30]. Maddelein et al. demonstrated that the heterokaryon incompatibility reaction is induced when cells are transformed with amyloid fibers composed of recombinant Het-s. The incompatibility reaction is not induced when a soluble version of Het-s was transformed into cells. This result provided direct experimental evidence that strengthened the protein only prion hypothesis [82]. The molecular mechanisms of Het-s-induced cell death are currently unknown.
CPEB
CPEBs are highly conserved RNA-binding proteins localized at neuronal synapses that stabilize messenger RNA molecules [86]. CPEBs have been found to be important for memory retention due to their ability to activate dormant messenger RNA transcripts near neuronal synapses. These activated messages can then be translated into proteins that stabilize neuronal synapses or help create synaptic connections necessary for long term memory [117]. The CPEB protein of the sea hare, Aplaysia californica (ApCPEB), contains a Q/N rich domain. Si et al. demonstrated that ApCPEB acts as a prion in yeast and that the aggregative amyloid state of CPEB is the functional, RNA-binding, form of the protein [118]. Thus, ApCPEB is functionally active when polymerized into amyloid aggregates, whereas, the wild type functions of Ure2p and Sup35p are impaired when the proteins are aggregated. It has yet to be elucidated if CPEB folds into an amyloid in neuronal cells, but the ability of the protein to do so in yeast suggests it has the capability to do so in other cell types.
Pmel17
Melanocytes and retinal pigment epithelium are specialized cell types responsible for the production of melanin, a tyrosine based polymer that protects the mammalian eyes and epidermis from ultra-violet damage and other environmental insults. These cells types synthesize melanin within specialized membrane enclosed vesicles called melanosomes [58,72, 84]. Melanosome maturation and polymerization of melanin are dependent on insoluble fibers composed of the PMEL17 protein [8,9,20,64,74]. Fowler and colleagues demonstrated that fibers composed of PMEL17 contain the biochemical hallmarks of amyloid [40]. PMEL17 amyloid fibers function by kinetically enhancing the polymerization of melanin, presumably by acting as a scaffold for reactive melanin precursors [40]. Mammalian cells have evolved several mechanisms to reduce exposure to toxic folding intermediates produced by PMEL17 amyloid polymerization: (1) polymerizing the amyloid fiber in a membrane enclosed vesicle sequestering folding intermediates from the cytoplasm, (2) regulating the start of polymerization via proteolysis, and (3) using reaction kinetics that skew towards the stability of the mature amyloid fiber [40, 41].
CONCLUDING REMARKS
Despite over twenty years of AD research the nature of the toxic species of Aβ has yet to be conclusively identified and little is known about how Aβ polypeptide aggregation begins in vivo. Insights into these two critical phenomena will undoubtedly lead to advances in AD treatments. The functional amyloids may hold the key to understanding the molecular mechanisms of amyloid fiber toxicity and initiation of amyloid fiber polymerization because they are naturally occurring directed polymerization processes. Not only are the amyloid fiber end products in both AD and the functional amyloid systems biochemically similar, but a common intermediate has been identified for CsgA, Sup35p, and Aβ polymerization. This suggests that amyloid biogenesis occurs via a conversed mechanism. Interestingly, most of the directed amyloid synthesis pathways discussed herein polymerize to higher order aggregates/fibers in vivo and these fibers have physiological nontoxic roles in most cases. A folding intermediate of Mcc amyloid fibers is cytotoxic but not the mature amyloid fiber itself. Thus, it seems likely that the functional amyloids lend credence to the hypothesis that the fiber end product may not be toxic. The role of the chaperone Hsp104 in yeast prion propagation also supports this idea. At high concentrations the chaperone abrogates amyloid fiber polymerization, but at low concentrations Hsp104 kinetically accelerates fiber formation. These results demonstrate two mechanisms cells use to sequester the build up of toxic intermediates. Chaperones either bind the aberrant toxic intermediate, which allows the protein the opportunity to refold, or the chaperone binds to a fiber intermediate and facilitates the conversion to the amyloid form.
These functional amyloid synthesis pathways will continue to provide novel insights regarding amyloid biogenesis. The functional amyloid systems may even address pivotal questions that remain for AD progression such as, how does amyloid biogenesis begin in vivo, what is the most cytotoxic species produced during amyloid biogenesis, and what are the defining features of proteins that preclude the ability to fold into the amyloid conformation. The answers to these questions will in turn provide novel therapeutic strategies for treating disease associated amyloidosis such as AD.
Acknowledgments
We thank members of the Chapman laboratory for helpful discussions and for review of this manuscript. This work was supported by NIH AI073847-01 and by Michigan Alzheimer’s Disease Research Center award P50-A608671.
References
- 1.Alteri CJ, Xicohtencatl-Cortes J, Hess S, Caballero-Olin G, Giron JA, Friedman RL. Mycobacterium tuberculosis produces pili during human infection. Proc Natl Acad Sci USA. 2007;104:5145–5150. doi: 10.1073/pnas.0602304104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Alzheimer A. Über eine eigenartige Erkrankung der Hirnrinde. Allg Zeitschr Psychiatr Psychiatr-Gerichtl Med. 1907;64:146. [Google Scholar]
- 3.Arrasate M, Mitra S, Schweitzer ES, Segal MR, Finkbeiner S. Inclusion body formation reduces levels of mutant huntingtin and the risk of neuronal death. Nature. 2004;431:805–810. doi: 10.1038/nature02998. [DOI] [PubMed] [Google Scholar]
- 4.Balbirnie M, Grothe R, Eisenberg DS. An amyloid-forming peptide from the yeast prion Sup35 reveals a dehydrated beta-sheet structure for amyloid. Proc Natl Acad Sci USA. 2001;98:2375–2380. doi: 10.1073/pnas.041617698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bales KR, Verina T, Cummins DJ, Du Y, Dodel RC, Saura J, Fishman CE, DeLong CA, Piccardo P, Petegnief V, Ghetti B, Paul SM. Apolipoprotein E is essential for amyloid deposition in the APP(V717F) transgenic mouse model of Alzheimer’s disease. Proc Natl Acad Sci USA. 1999;96:15233–15238. doi: 10.1073/pnas.96.26.15233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Barnhart MM, Chapman MR. Curli biogenesis and function. Annu Rev Microbiol. 2006;60:131–147. doi: 10.1146/annurev.micro.60.080805.142106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Beck T, Hall MN. The TOR signalling pathway controls nuclear localization of nutrient-regulated transcription factors. Nature. 1999;402:689–692. doi: 10.1038/45287. [DOI] [PubMed] [Google Scholar]
- 8.Berson JF, Harper DC, Tenza D, Raposo G, Marks MS. Pmel17 initiates premelanosome morphogenesis within multivesicular bodies. Mol Biol Cell. 2001;12:3451–3464. doi: 10.1091/mbc.12.11.3451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Berson JF, Theos AC, Harper DC, Tenza D, Raposo G, Marks MS. Proprotein convertase cleavage liberates a fibrillogenic fragment of a resident glycoprotein to initiate melanosome biogenesis. J Cell Biol. 2003;161:521–533. doi: 10.1083/jcb.200302072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bian Z, Brauner A, Li Y, Normark S. Expression of and cytokine activation by Escherichia coli curli fibers in human sepsis. J Infect Dis. 2000;181:602–612. doi: 10.1086/315233. [DOI] [PubMed] [Google Scholar]
- 11.Bian Z, Yan ZQ, Hansson GK, Thoren P, Normark S. Activation of inducible nitric oxide synthase/nitric oxide by curli fibers leads to a fall in blood pressure during systemic Escherichia coli infection in mice. J Infect Dis. 2001;183:612–619. doi: 10.1086/318528. [DOI] [PubMed] [Google Scholar]
- 12.Bieler S, Estrada L, Lagos R, Baeza M, Castilla J, Soto C. Amyloid formation modulates the biological activity of a bacterial protein. J Biol Chem. 2005;280:26880–26885. doi: 10.1074/jbc.M502031200. [DOI] [PubMed] [Google Scholar]
- 13.Blennow K, de Leon MJ, Zetterberg H. Alzheimer’s disease. Lancet. 2006;368:387–403. doi: 10.1016/S0140-6736(06)69113-7. [DOI] [PubMed] [Google Scholar]
- 14.Blinder D, Coschigano PW, Magasanik B. Interaction of the GATA factor Gln3p with the nitrogen regulator Ure2p in Saccharomyces cerevisiae. J Bacteriol. 1996;178:4734–4736. doi: 10.1128/jb.178.15.4734-4736.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Brachmann A, Baxa U, Wickner RB. Prion generation in vitro: amyloid of Ure2p is infectious. Embo J. 2005;24:3082–3092. doi: 10.1038/sj.emboj.7600772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bucciantini M, Giannoni E, Chiti F, Baroni F, Formigli L, Zurdo J, Taddei N, Ramponi G, Dobson CM, Stefani M. Inherent toxicity of aggregates implies a common mechanism for protein misfolding diseases. Nature. 2002;416:507–511. doi: 10.1038/416507a. [DOI] [PubMed] [Google Scholar]
- 17.Busciglio J, Lorenzo A, Yankner BA. Methodological variables in the assessment of beta amyloid neurotoxicity. Neurobiol Aging. 1992;13:609–612. doi: 10.1016/0197-4580(92)90065-6. [DOI] [PubMed] [Google Scholar]
- 18.Cardenas ME, Cutler NS, Lorenz MC, Di Como CJ, Heitman J. The TOR signaling cascade regulates gene expression in response to nutrients. Genes Dev. 1999;13:3271–3279. doi: 10.1101/gad.13.24.3271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Carulla N, Caddy GL, Hall DR, Zurdo J, Gairi M, Feliz M, Giralt E, Robinson CV, Dobson CM. Molecular recycling within amyloid fibrils. Nature. 2005;436:554–558. doi: 10.1038/nature03986. [DOI] [PubMed] [Google Scholar]
- 20.Chakraborty AK, Platt JT, Kim KK, Kwon BS, Bennett DC, Pawelek JM. Polymerization of 5,6-dihydroxyindole-2-carboxylic acid to melanin by the pmel 17/silver locus protein. Eur J Biochem. 1996;236:180–188. doi: 10.1111/j.1432-1033.1996.t01-1-00180.x. [DOI] [PubMed] [Google Scholar]
- 21.Chapman MR, Robinson LS, Pinkner JS, Roth R, Heuser J, Hammar M, Normark S, Hultgren SJ. Role of Escherichia coli curli operons in directing amyloid fiber formation. Science. 2002;295:851–855. doi: 10.1126/science.1067484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chernoff YO, Lindquist SL, Ono B, Inge-Vechtomov SG, Liebman SW. Role of the chaperone protein Hsp104 in propagation of the yeast prion-like factor [psi+] Science. 1995;268:880–884. doi: 10.1126/science.7754373. [DOI] [PubMed] [Google Scholar]
- 23.Chernoff YO, Galkin AP, Lewitin E, Chernova TA, Newnam GP, Belenkiy SM. Evolutionary conservation of prion-forming abilities of the yeast Sup35 protein. Mol Microbiol. 2000;35:865–876. doi: 10.1046/j.1365-2958.2000.01761.x. [DOI] [PubMed] [Google Scholar]
- 24.Citron M, Westaway D, Xia W, Carlson G, Diehl T, Levesque G, Johnson-Wood K, Lee M, Seubert P, Davis A, Kholodenko D, Motter R, Sherrington R, Perry B, Yao H, Strome R, Lieberburg I, Rommens J, Kim S, Schenk D, Fraser P, St George Hyslop P, Selkoe DJ. Mutant presenilins of Alzheimer’s disease increase production of 42-residue amyloid beta-protein in both transfected cells and transgenic mice. Nat Med. 1997;3:67–72. doi: 10.1038/nm0197-67. [DOI] [PubMed] [Google Scholar]
- 25.Claessen D, Rink R, de Jong W, Siebring J, de Vreugd P, Boersma FG, Dijkhuizen L, Wosten HA. A novel class of secreted hydrophobic proteins is involved in aerial hyphae formation in Streptomyces coelicolor by forming amyloid-like fibrils. Genes Dev. 2003;17:1714–1726. doi: 10.1101/gad.264303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cleary JP, Walsh DM, Hofmeister JJ, Shankar GM, Kuskowski MA, Selkoe DJ, Ashe KH. Natural oligomers of the amyloid-beta protein specifically disrupt cognitive function. Nat Neurosci. 2005;8:79–84. doi: 10.1038/nn1372. [DOI] [PubMed] [Google Scholar]
- 27.Cohen E, Bieschke J, Perciavalle RM, Kelly JW, Dillin A. Opposing activities protect against age-onset proteotoxicity. Science. 2006;313:1604–1610. doi: 10.1126/science.1124646. [DOI] [PubMed] [Google Scholar]
- 28.Collinson SK, Parker JM, Hodges RS, Kay WW. Structural predictions of AgfA, the insoluble fimbrial subunit of Salmonella thin aggregative fimbriae. J Mol Biol. 1999;290:741–756. doi: 10.1006/jmbi.1999.2882. [DOI] [PubMed] [Google Scholar]
- 29.Cooper TG. Transmitting the signal of excess nitrogen in Saccharomyces cerevisiae from the Tor proteins to the GATA factors: connecting the dots. FEMS Microbiol Rev. 2002;26:223–238. doi: 10.1111/j.1574-6976.2002.tb00612.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Coustou V, Deleu C, Saupe S, Begueret J. The protein product of the het-s heterokaryon incompatibility gene of the fungus Podospora anserina behaves as a prion analog. Proc Natl Acad Sci USA. 1997;94:9773–9778. doi: 10.1073/pnas.94.18.9773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cox BS. [PSI], a cytoplasmic suppressor of super-supression in yeast. Heredity. 1965;20:505–521. [Google Scholar]
- 32.Cox BS, Tuite MF, Mundy CJ. Reversion from suppression to nonsuppression in SUQ5 [psi+] strains of yeast: the classificaion of mutations. Genetics. 1980;95:589–609. doi: 10.1093/genetics/95.3.589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cox KH, Rai R, Distler M, Daugherty JR, Coffman JA, Cooper TG. Saccharomyces cerevisiae GATA sequences function as TATA elements during nitrogen catabolite repression and when Gln3p is excluded from the nucleus by overproduction of Ure2p. J Biol Chem. 2000;275:17611–17618. doi: 10.1074/jbc.M001648200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.de Lorenzo V, Martinez JL, Asensio C. Microcin-mediated interactions between Klebsiella pneumoniae and Escherichia coli strains. J Gen Microbiol. 1984;130:391–400. doi: 10.1099/00221287-130-2-391. [DOI] [PubMed] [Google Scholar]
- 35.de Lorenzo V. Factors affecting microcin E492 production. J Antibiot (Tokyo) 1985;38:340–345. doi: 10.7164/antibiotics.38.340. [DOI] [PubMed] [Google Scholar]
- 36.Derkatch IL, Bradley ME, Hong JY, Liebman SW. Prions affect the appearance of other prions: the story of [PIN(+)] Cell. 2001;106:171–182. doi: 10.1016/s0092-8674(01)00427-5. [DOI] [PubMed] [Google Scholar]
- 37.Deshpande A, Mina E, Glabe C, Busciglio J. Different conformations of amyloid beta induce neurotoxicity by distinct mechanisms in human cortical neurons. J Neurosci. 2006;26:6011–6018. doi: 10.1523/JNEUROSCI.1189-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Elliot MA, Karoonuthaisiri N, Huang J, Bibb MJ, Cohen SN, Kao CM, Buttner MJ. The chaplins: a family of hydrophobic cell-surface proteins involved in aerial mycelium formation in Streptomyces coelicolor. Genes Dev. 2003;17:1727–1740. doi: 10.1101/gad.264403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fay N, Inoue Y, Bousset L, Taguchi H, Melki R. Assembly of the yeast prion Ure2p into protein fibrils. Thermodynamic and kinetic characterization. J Biol Chem. 2003;278:30199–30205. doi: 10.1074/jbc.M303000200. [DOI] [PubMed] [Google Scholar]
- 40.Fowler DM, Koulov AV, Alory-Jost C, Marks MS, Balch WE, Kelly JW. Functional amyloid formation within mammalian tissue. PLoS Biol. 2006;4:e6. doi: 10.1371/journal.pbio.0040006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fowler DM, Koulov AV, Balch WE, Kelly JW. Functional amyloid – from bacteria to humans. Trends Biochem Sci. 2007;32:217–224. doi: 10.1016/j.tibs.2007.03.003. [DOI] [PubMed] [Google Scholar]
- 42.Fryer JD, Simmons K, Parsadanian M, Bales KR, Paul SM, Sullivan PM, Holtzman DM. Human apolipoprotein E4 alters the amyloid-beta 40:42 ratio and promotes the formation of cerebral amyloid angiopathy in an amyloid precursor protein transgenic model. J Neurosci. 2005;25:2803–2810. doi: 10.1523/JNEUROSCI.5170-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Glenner GG, Wong CW. Alzheimer’s disease: initial report of the purification and characterization of a novel cerebrovascular amyloid protein. Biochem Biophys Res Commun. 1984;120:885–890. doi: 10.1016/s0006-291x(84)80190-4. [DOI] [PubMed] [Google Scholar]
- 44.Glover JR, Kowal AS, Schirmer EC, Patino MM, Liu JJ, Lindquist S. Self-seeded fibers formed by Sup35, the protein determinant of [PSI+], a heritable prion-like factor of S. cerevisiae. Cell. 1997;89:811–819. doi: 10.1016/s0092-8674(00)80264-0. [DOI] [PubMed] [Google Scholar]
- 45.Goedert M, Spillantini MG. A century of Alzheimer’s disease. Science. 2006;314:777–781. doi: 10.1126/science.1132814. [DOI] [PubMed] [Google Scholar]
- 46.Greenberg JT. Programmed cell death: a way of life for plants. Proc Natl Acad Sci USA. 1996;93:12094–12097. doi: 10.1073/pnas.93.22.12094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Greenberg JT. Programmed Cell Death in Plant-Pathogen Interactions. Annu Rev Plant Physiol Plant Mol Biol. 1997;48:525–545. doi: 10.1146/annurev.arplant.48.1.525. [DOI] [PubMed] [Google Scholar]
- 48.Guiroy DC, Miyazaki M, Multhaup G, Fischer P, Garruto RM, Beyreuther K, Masters CL, Simms G, Gibbs CJ, Jr, Gajdusek DC. Amyloid of neurofibrillary tangles of Guamanian parkinsonism-dementia and Alzheimer disease share identical amino acid sequence. Proc Natl Acad Sci USA. 1987;84:2073–2077. doi: 10.1073/pnas.84.7.2073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Haass C. Take five–BACE and the gamma-secretase quartet conduct Alzheimer’s amyloid beta-peptide generation. Embo J. 2004;23:483–488. doi: 10.1038/sj.emboj.7600061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Haass C, Selkoe DJ. Soluble protein oligomers in neurodegeneration: lessons from the Alzheimer’s amyloid beta-peptide. Nat Rev Mol Cell Biol. 2007;8:101–112. doi: 10.1038/nrm2101. [DOI] [PubMed] [Google Scholar]
- 51.Hammar M, Arnqvist A, Bian Z, Olsen A, Normark S. Expression of two csg operons is required for production of fibronectin-and congo red-binding curli polymers in Escherichia coli K-12. Mol Microbiol. 1995;18:661–670. doi: 10.1111/j.1365-2958.1995.mmi_18040661.x.. [DOI] [PubMed] [Google Scholar]
- 52.Hammar M, Bian Z, Normark S. Nucleator-dependent intercellular assembly of adhesive curli organelles in Escherichia coli. Proc Natl Acad Sci USA. 1996;93:6562–6566. doi: 10.1073/pnas.93.13.6562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hammer ND, Schmidt JC, Chapman MR. The curli nucleator protein, CsgB, contains an amyloidogenic domain that directs CsgA polymerization. Proc Natl Acad Sci USA. 2007 doi: 10.1073/pnas.0703310104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hardwick JS, Kuruvilla FG, Tong JK, Shamji AF, Schreiber SL. Rapamycin-modulated transcription defines the subset of nutrient-sensitive signaling pathways directly controlled by the Tor proteins. Proc Natl Acad Sci USA. 1999;96:14866–14870. doi: 10.1073/pnas.96.26.14866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science. 2002;297:353–356. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- 56.Hartley DM, Walsh DM, Ye CP, Diehl T, Vasquez S, Vassilev PM, Teplow DB, Selkoe DJ. Protofibrillar intermediates of amyloid beta-protein induce acute electrophysiological changes and progressive neurotoxicity in cortical neurons. J Neurosci. 1999;19:8876–8884. doi: 10.1523/JNEUROSCI.19-20-08876.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hartmann T, Bieger SC, Bruhl B, Tienari PJ, Ida N, Allsop D, Roberts GW, Masters CL, Dotti CG, Unsicker K, Beyreuther K. Distinct sites of intracellular production for Alzheimer’s disease A beta40/42 amyloid peptides. Nat Med. 1997;3:1016–1020. doi: 10.1038/nm0997-1016. [DOI] [PubMed] [Google Scholar]
- 58.Hearing VJ. The melanosome: the perfect model for cellular responses to the environment. Pigment Cell Res. 2000;13(Suppl 8):23–34. doi: 10.1034/j.1600-0749.13.s8.7.x. [DOI] [PubMed] [Google Scholar]
- 59.Hetz C, Bono MR, Barros LF, Lagos R. Microcin E492, a channel-forming bacteriocin from Klebsiella pneumoniae, induces apoptosis in some human cell lines. Proc Natl Acad Sci USA. 2002;99:2696–2701. doi: 10.1073/pnas.052709699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hoshi M, Sato M, Matsumoto S, Noguchi A, Yasutake K, Yoshida N, Sato K. Spherical aggregates of beta-amyloid (amylospheroid) show high neurotoxicity and activate tau protein kinase I/glycogen synthase kinase-3beta. Proc Natl Acad Sci USA. 2003;100:6370–6375. doi: 10.1073/pnas.1237107100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Jarrett JT, Lansbury PT., Jr Seeding “one-dimensional crystallization” of amyloid: a pathogenic mechanism in Alzheimer’s disease and scrapie? Cell. 1993;73:1055–1058. doi: 10.1016/0092-8674(93)90635-4. [DOI] [PubMed] [Google Scholar]
- 62.Jensen MA, True HL, Chernoff YO, Lindquist S. Molecular population genetics and evolution of a prion-like protein in Saccharomyces cerevisiae. Genetics. 2001;159:527–535. doi: 10.1093/genetics/159.2.527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kayed R, Head E, Thompson JL, McIntire TM, Milton SC, Cotman CW, Glabe CG. Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science. 2003;300:486–489. doi: 10.1126/science.1079469. [DOI] [PubMed] [Google Scholar]
- 64.Kelly JW, Balch WE. Amyloid as a natural product. J Cell Biol. 2003;161:461–462. doi: 10.1083/jcb.200304074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kidd M. Paired helical filaments in electron microscopy of Alzheimer’s disease. Nature. 1963;197:192–193. doi: 10.1038/197192b0. [DOI] [PubMed] [Google Scholar]
- 66.Kim H, Hahn M, Grabowski P, McPherson DC, Otte MM, Wang R, Ferguson CC, Eichenberger P, Driks A. The Bacillus subtilis spore coat protein interaction network. Mol Microbiol. 2006;59:487–502. doi: 10.1111/j.1365-2958.2005.04968.x. [DOI] [PubMed] [Google Scholar]
- 67.Kimura Y, Koitabashi S, Fujita T. Analysis of yeast prion aggregates with amyloid-staining compound in vivo. Cell Struct Funct. 2003;28:187–193. doi: 10.1247/csf.28.187. [DOI] [PubMed] [Google Scholar]
- 68.King CY, Diaz-Avalos R. Protein-only transmission of three yeast prion strains. Nature. 2004;428:319–323. doi: 10.1038/nature02391. [DOI] [PubMed] [Google Scholar]
- 69.Koistinaho M, Lin S, Wu X, Esterman M, Koger D, Hanson J, Higgs R, Liu F, Malkani S, Bales KR, Paul SM. Apolipoprotein E promotes astrocyte colocalization and degradation of deposited amyloid-beta peptides. Nat Med. 2004;10:719–726. doi: 10.1038/nm1058. [DOI] [PubMed] [Google Scholar]
- 70.Kryndushkin DS, Alexandrov IM, Ter-Avanesyan MD, Kushnirov VV. Yeast [PSI+] prion aggregates are formed by small Sup35 polymers fragmented by Hsp104. J Biol Chem. 2003;278:49636–49643. doi: 10.1074/jbc.M307996200. [DOI] [PubMed] [Google Scholar]
- 71.Lambert MP, Barlow AK, Chromy BA, Edwards C, Freed R, Liosatos M, Morgan TE, Rozovsky I, Trommer B, Viola KL, Wals P, Zhang C, Finch CE, Krafft GA, Klein WL. Diffusible, nonfibrillar ligands derived from Abeta1-42 are potent central nervous system neurotoxins. Proc Natl Acad Sci USA. 1998;95:6448–6453. doi: 10.1073/pnas.95.11.6448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Land EJ, Ramsden CA, Riley PA. Quinone chemistry and melanogenesis. Methods Enzymol. 2004;378:88–109. doi: 10.1016/S0076-6879(04)78005-2. [DOI] [PubMed] [Google Scholar]
- 73.Lawley TD, Chan K, Thompson LJ, Kim CC, Govoni GR, Monack DM. Genome-wide screen for Salmonella genes required for long-term systemic infection of the mouse. PLoS Pathog. 2006;2:e11. doi: 10.1371/journal.ppat.0020011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Lee ZH, Hou L, Moellmann G, Kuklinska E, Antol K, Fraser M, Halaban R, Kwon BS. Characterization and subcellular localization of human Pmel 17/silver, a 110-kDa (pre)melanosomal membrane protein associated with 5,6,-dihydroxyindole-2-carboxylic acid (DHICA) converting activity. J Invest Dermatol. 1996;106:605–610. doi: 10.1111/1523-1747.ep12345163. [DOI] [PubMed] [Google Scholar]
- 75.Lesne S, Koh MT, Kotilinek L, Kayed R, Glabe CG, Yang A, Gallagher M, Ashe KH. A specific amyloid-beta protein assembly in the brain impairs memory. Nature. 2006;440:352–357. doi: 10.1038/nature04533. [DOI] [PubMed] [Google Scholar]
- 76.Liu JJ, Lindquist S. Oligopeptide-repeat expansions modulate ’protein-only’ inheritance in yeast. Nature. 1999;400:573–576. doi: 10.1038/23048. [DOI] [PubMed] [Google Scholar]
- 77.Loferer H, Hammar M, Normark S. Availability of the fibre subunit CsgA and the nucleator protein CsgB during assembly of fibronectin-binding curli is limited by the intracellular concentration of the novel lipoprotein CsgG. Mol Microbiol. 1997;26:11–23. doi: 10.1046/j.1365-2958.1997.5231883.x. [DOI] [PubMed] [Google Scholar]
- 78.Lomakin A, Chung DS, Benedek GB, Kirschner DA, Teplow DB. On the nucleation and growth of amyloid beta-protein fibrils: detection of nuclei and quantitation of rate constants. Proc Natl Acad Sci USA. 1996;93:1125–1129. doi: 10.1073/pnas.93.3.1125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Lorenzo A, Yankner BA. Beta-amyloid neurotoxicity requires fibril formation and is inhibited by congo red. Proc Natl Acad Sci USA. 1994;91:12243–12247. doi: 10.1073/pnas.91.25.12243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Lue LF, Kuo YM, Roher AE, Brachova L, Shen Y, Sue L, Beach T, Kurth JH, Rydel RE, Rogers J. Soluble amyloid beta peptide concentration as a predictor of synaptic change in Alzheimer’s disease. Am J Pathol. 1999;155:853–862. doi: 10.1016/s0002-9440(10)65184-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Lund PM, Cox BS. Reversion analysis of [psi−] mutations in Saccharomyces cerevisiae. Genet Res. 1981;37:173–182. doi: 10.1017/s0016672300020140. [DOI] [PubMed] [Google Scholar]
- 82.Maddelein ML, Dos Reis S, Duvezin-Caubet S, Coulary-Salin B, Saupe SJ. Amyloid aggregates of the HET-s prion protein are infectious. Proc Natl Acad Sci USA. 2002;99:7402–7407. doi: 10.1073/pnas.072199199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Magasanik B, Kaiser CA. Nitrogen regulation in Sac-charomyces cerevisiae. Gene. 2002;290:1–18. doi: 10.1016/s0378-1119(02)00558-9. [DOI] [PubMed] [Google Scholar]
- 84.Marks MS, Seabra MC. The melanosome: membrane dynamics in black and white. Nat Rev Mol Cell Biol. 2001;2:738–748. doi: 10.1038/35096009. [DOI] [PubMed] [Google Scholar]
- 85.Masison DC, Wickner RB. Prion-inducing domain of yeast Ure2p and protease resistance of Ure2p in prion-containing cells. Science. 1995;270:93–95. doi: 10.1126/science.270.5233.93. [DOI] [PubMed] [Google Scholar]
- 86.Mendez R, Richter JD. Translational control by CPEB: a means to the end. Nat Rev Mol Cell Biol. 2001;2:521–529. doi: 10.1038/35080081. [DOI] [PubMed] [Google Scholar]
- 87.Michelitsch MD, Weissman JS. A census of glutamine/asparagine-rich regions: implications for their conserved function and the prediction of novel prions. Proc Natl Acad Sci USA. 2000;97:11910–11915. doi: 10.1073/pnas.97.22.11910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Moriyama H, Edskes HK, Wickner RB. [URE3] prion propagation in Saccharomyces cerevisiae: requirement for chaperone Hsp104 and curing by overexpressed chaperone Ydj1p. Mol Cell Biol. 2000;20:8916–8922. doi: 10.1128/mcb.20.23.8916-8922.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Nakayashiki T, Ebihara K, Bannai H, Nakamura Y. Yeast [PSI+] “prions” that are crosstransmissible and susceptible beyond a species barrier through a quasi-prion state. Mol Cell. 2001;7:1121–1130. doi: 10.1016/s1097-2765(01)00259-3. [DOI] [PubMed] [Google Scholar]
- 90.Nakayashiki T, Kurtzman CP, Edskes HK, Wickner RB. Yeast prions [URE3] and [PSI+] are diseases. Proc Natl Acad Sci USA. 2005;102:10575–10580. doi: 10.1073/pnas.0504882102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Nelson R, Sawaya MR, Balbirnie M, Madsen AO, Riekel C, Grothe R, Eisenberg D. Structure of the cross-beta spine of amyloid-like fibrils. Nature. 2005;435:773–778. doi: 10.1038/nature03680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Nilsberth C, Westlind-Danielsson A, Eckman CB, Condron MM, Axelman K, Forsell C, Stenh C, Luthman J, Teplow DB, Younkin SG, Naslund J, Lannfelt L. The ‘Arctic’ APP mutation (E693G) causes Alzheimer’s disease by enhanced Abeta protofibril formation. Nat Neurosci. 2001;4:887–893. doi: 10.1038/nn0901-887. [DOI] [PubMed] [Google Scholar]
- 93.Oh J, Kim JG, Jeon E, Yoo CH, Moon JS, Rhee S, Hwang I. Amyloidogenesis of type III-dependent harpins from plant pathogenic bacteria. J Biol Chem. 2007;282:13601–13609. doi: 10.1074/jbc.M602576200. [DOI] [PubMed] [Google Scholar]
- 94.Osherovich LZ, Weissman JS. Multiple Gln/Asn-rich prion domains confer susceptibility to induction of the yeast [PSI(+)] prion. Cell. 2001;106:183–194. doi: 10.1016/s0092-8674(01)00440-8. [DOI] [PubMed] [Google Scholar]
- 95.Osherovich LZ, Cox BS, Tuite MF, Weissman JS. Dissection and design of yeast prions. PLoS Biol. 2004;2:E86. doi: 10.1371/journal.pbio.0020086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Patel BK, Liebman SW. “Prion-proof” for [PIN+]: infection with in vitro-made amyloid aggregates of Rnq1p-(132–405) induces [PIN+] J Mol Biol. 2007;365:773–782. doi: 10.1016/j.jmb.2006.10.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Patino MM, Liu JJ, Glover JR, Lindquist S. Support for the prion hypothesis for inheritance of a phenotypic trait in yeast. Science. 1996;273:622–626. doi: 10.1126/science.273.5275.622. [DOI] [PubMed] [Google Scholar]
- 98.Paushkin SV, Kushnirov VV, Smirnov VN, Ter-Avanesyan MD. Propagation of the yeast prion-like [psi+] determinant is mediated by oligomerization of the SUP35-encoded polypeptide chain release factor. Embo J. 1996;15:3127–3134. [PMC free article] [PubMed] [Google Scholar]
- 99.Pennell RI, Lamb C. Programmed Cell Death in Plants. Plant Cell. 1997;9:1157–1168. doi: 10.1105/tpc.9.7.1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Petkova AT, Leapman RD, Guo Z, Yau WM, Mattson MP, Tycko R. Self-propagating, molecular-level polymorphism in Alzheimer’s beta-amyloid fibrils. Science. 2005;307:262–265. doi: 10.1126/science.1105850. [DOI] [PubMed] [Google Scholar]
- 101.Pike CJ, Walencewicz AJ, Glabe CG, Cotman CW. In vitro aging of beta-amyloid protein causes peptide aggregation and neurotoxicity. Brain Res. 1991;563:311–314. doi: 10.1016/0006-8993(91)91553-d. [DOI] [PubMed] [Google Scholar]
- 102.Pike CJ, Burdick D, Walencewicz AJ, Glabe CG, Cotman CW. Neurodegeneration induced by beta-amyloid peptides in vitro: the role of peptide assembly state. J Neurosci. 1993;13:1676–1687. doi: 10.1523/JNEUROSCI.13-04-01676.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Plomp M, Leighton TJ, Wheeler KE, Hill HD, Malkin AJ. In vitro high-resolution structural dynamics of single germinating bacterial spores. Proc Natl Acad Sci USA. 2007;104:9644–9649. doi: 10.1073/pnas.0610626104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Podlisny MB, Ostaszewski BL, Squazzo SL, Koo EH, Rydell RE, Teplow DB, Selkoe DJ. Aggregation of secreted amyloid beta-protein into sodium dodecyl sulfate-stable oligomers in cell culture. J Biol Chem. 1995;270:9564–9570. doi: 10.1074/jbc.270.16.9564. [DOI] [PubMed] [Google Scholar]
- 105.Podlisny MB, Walsh DM, Amarante P, Ostaszewski BL, Stimson ER, Maggio JE, Teplow DB, Selkoe DJ. Oligomerization of endogenous and synthetic amyloid beta-protein at nanomolar levels in cell culture and stabilization of monomer by Congo red. Biochemistry. 1998;37:3602–3611. doi: 10.1021/bi972029u. [DOI] [PubMed] [Google Scholar]
- 106.Raber J, Huang Y, Ashford JW. ApoE genotype accounts for the vast majority of AD risk and AD pathology. Neurobiol Aging. 2004;25:641–650. doi: 10.1016/j.neurobiolaging.2003.12.023. [DOI] [PubMed] [Google Scholar]
- 107.Robinson LS, Ashman EM, Hultgren SJ, Chapman MR. Secretion of curli fibre subunits is mediated by the outer membrane-localized CsgG protein. Mol Microbiol. 2006;59:870–881. doi: 10.1111/j.1365-2958.2005.04997.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Roher AE, Chaney MO, Kuo YM, Webster SD, Stine WB, Haverkamp LJ, Woods AS, Cotter RJ, Tuohy JM, Krafft GA, Bonnell BS, Emmerling MR. Morphology and toxicity of Abeta-(1–42) dimer derived from neuritic and vascular amyloid deposits of Alzheimer’s disease. J Biol Chem. 1996;271:20631–20635. doi: 10.1074/jbc.271.34.20631. [DOI] [PubMed] [Google Scholar]
- 109.Romling U, Sierralta WD, Eriksson K, Normark S. Multicellular and aggregative behaviour of Salmonella typhimurium strains is controlled by mutations in the agfD promoter. Mol Microbiol. 1998;28:249–264. doi: 10.1046/j.1365-2958.1998.00791.x. [DOI] [PubMed] [Google Scholar]
- 110.Romling U, Bokranz W, Rabsch W, Zogaj X, Nimtz M, Tschape H. Occurrence and regulation of the multicellular morphotype in Salmonella serovars important in human disease. Int J Med Microbiol. 2003;293:273–285. doi: 10.1078/1438-4221-00268. [DOI] [PubMed] [Google Scholar]
- 111.Saupe SJ. Molecular genetics of heterokaryon incompatibility in filamentous ascomycetes. Microbiol Mol Biol Rev. 2000;64:489–502. doi: 10.1128/mmbr.64.3.489-502.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Scherzinger E, Lurz R, Turmaine M, Mangiarini L, Hollenbach B, Hasenbank R, Bates GP, Davies SW, Lehrach H, Wanker EE. Huntingtin-encoded polyglutamine expansions form amyloid-like protein aggregates in vitro and in vivo. Cell. 1997;90:549–558. doi: 10.1016/s0092-8674(00)80514-0. [DOI] [PubMed] [Google Scholar]
- 113.Shewmaker F, Mull L, Nakayashiki T, Masison DC, Wickner RB. Ure2p Function Is Enhanced by Its Prion Domain in Saccharomyces cerevisiae. Genetics. 2007;176:1557–1565. doi: 10.1534/genetics.107.074153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Shorter J, Lindquist S. Hsp104 catalyzes formation and elimination of self-replicating Sup35 prion conformers. Science. 2004;304:1793–1797. doi: 10.1126/science.1098007. [DOI] [PubMed] [Google Scholar]
- 115.Shorter J, Lindquist S. Prions as adaptive conduits of memory and inheritance. Nat Rev Genet. 2005;6:435–450. doi: 10.1038/nrg1616. [DOI] [PubMed] [Google Scholar]
- 116.Shorter J, Lindquist S. Destruction or potentiation of different prions catalyzed by similar Hsp104 remodeling activities. Mol Cell. 2006;23:425–438. doi: 10.1016/j.molcel.2006.05.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Si K, Giustetto M, Etkin A, Hsu R, Janisiewicz AM, Miniaci MC, Kim JH, Zhu H, Kandel ER. A neuronal isoform of CPEB regulates local protein synthesis and stabilizes synapse-specific long-term facilitation in aplysia. Cell. 2003;115:893–904. doi: 10.1016/s0092-8674(03)01021-3. [DOI] [PubMed] [Google Scholar]
- 118.Si K, Lindquist S, Kandel ER. A neuronal isoform of the aplysia CPEB has prion-like properties. Cell. 2003;115:879–891. doi: 10.1016/s0092-8674(03)01020-1. [DOI] [PubMed] [Google Scholar]
- 119.Slow EJ, Graham RK, Osmand AP, Devon RS, Lu G, Deng Y, Pearson J, Vaid K, Bissada N, Wetzel R, Leavitt BR, Hayden MR. Absence of behavioral abnormalities and neurodegeneration in vivo despite widespread neuronal huntingtin inclusions. Proc Natl Acad Sci USA. 2005;102:11402–11407. doi: 10.1073/pnas.0503634102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Smith JF, Knowles TP, Dobson CM, Macphee CE, Welland ME. Characterization of the nanoscale properties of individual amyloid fibrils. Proc Natl Acad Sci USA. 2006;103:15806–15811. doi: 10.1073/pnas.0604035103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Sondheimer N, Lindquist S. Rnq1: an epigenetic modifier of protein function in yeast. Mol Cell. 2000;5:163–172. doi: 10.1016/s1097-2765(00)80412-8. [DOI] [PubMed] [Google Scholar]
- 122.Speransky VV, Taylor KL, Edskes HK, Wickner RB, Steven AC. Prion filament networks in [URE3] cells of Saccharomyces cerevisiae. J Cell Biol. 2001;153:1327–1336. doi: 10.1083/jcb.153.6.1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Stansfield I, Jones KM, Kushnirov VV, Dagkesaman-skaya AR, Poznyakovski AI, Paushkin SV, Nierras CR, Cox BS, Ter-Avanesyan MD, Tuite MF. The products of the SUP45 (eRF1) and SUP35 genes interact to mediate translation termination in Saccharomyces cerevisiae. Embo J. 1995;14:4365–4373. doi: 10.1002/j.1460-2075.1995.tb00111.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Tanaka M, Chien P, Naber N, Cooke R, Weissman JS. Conformational variations in an infectious protein determine prion strain differences. Nature. 2004;428:323–328. doi: 10.1038/nature02392. [DOI] [PubMed] [Google Scholar]
- 125.Taylor KL, Cheng N, Williams RW, Steven AC, Wickner RB. Prion domain initiation of amyloid formation in vitro from native Ure2p. Science. 1999;283:1339–1343. doi: 10.1126/science.283.5406.1339. [DOI] [PubMed] [Google Scholar]
- 126.Ter-Avanesyan MD, Dagkesamanskaya AR, Kushnirov VV, Smirnov VN. The SUP35 omnipotent suppressor gene is involved in the maintenance of the non-Mendelian determinant [psi+] in the yeast Saccharomyces cerevisiae. Genetics. 1994;137:671–676. doi: 10.1093/genetics/137.3.671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Terry RD, Gonatas NK, Weiss M. Ultrastructural Studies In Alzheimer’s Presenile Dementia. Am J Pathol. 1964;44:269–297. [PMC free article] [PubMed] [Google Scholar]
- 128.Terry RD, Masliah E, Salmon DP, Butters N, DeTeresa R, Hill R, Hansen LA, Katzman R. Physical basis of cognitive alterations in Alzheimer’s disease: synapse loss is the major correlate of cognitive impairment. Ann Neurol. 1991;30:572–580. doi: 10.1002/ana.410300410. [DOI] [PubMed] [Google Scholar]
- 129.Thual C, Komar AA, Bousset L, Fernandez-Bellot E, Cullin C, Melki R. Structural characterization of Saccharomyces cerevisiae prion-like protein Ure2. J Biol Chem. 1999;274:13666–13674. doi: 10.1074/jbc.274.19.13666. [DOI] [PubMed] [Google Scholar]
- 130.Townsend M, Shankar GM, Mehta T, Walsh DM, Selkoe DJ. Effects of secreted oligomers of amyloid beta-protein on hippocampal synaptic plasticity: a potent role for trimers. J Physiol. 2006;572:477–492. doi: 10.1113/jphysiol.2005.103754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.True HL, Lindquist SL. A yeast prion provides a mechanism for genetic variation and phenotypic diversity. Nature. 2000;407:477–483. doi: 10.1038/35035005. [DOI] [PubMed] [Google Scholar]
- 132.True HL, Berlin I, Lindquist SL. Epigenetic regulation of translation reveals hidden genetic variation to produce complex traits. Nature. 2004;431:184–187. doi: 10.1038/nature02885. [DOI] [PubMed] [Google Scholar]
- 133.Tsai J, Grutzendler J, Duff K, Gan WB. Fibrillar amyloid deposition leads to local synaptic abnormalities and breakage of neuronal branches. Nat Neurosci. 2004;7:1181–1183. doi: 10.1038/nn1335. [DOI] [PubMed] [Google Scholar]
- 134.Uhlich GA, Cooke PH, Solomon EB. Analyses of the red-dry-rough phenotype of an Escherichia coli O157:H7 strain and its role in biofilm formation and resistance to antibacterial agents. Appl Environ Microbiol. 2006;72:2564–2572. doi: 10.1128/AEM.72.4.2564-2572.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Vidal O, Longin R, Prigent-Combaret C, Dorel C, Hooreman M, Lejeune P. Isolation of an Escherichia coli K-12 mutant strain able to form biofilms on inert surfaces: involvement of a new ompR allele that increases curli expression. J Bacteriol. 1998;180:2442–2449. doi: 10.1128/jb.180.9.2442-2449.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Vitrenko YA, Pavon ME, Stone SI, Liebman SW. Propagation of the [PIN+] prion by fragments of Rnq1 fused to GFP. Curr Genet. 2007;51:309–319. doi: 10.1007/s00294-007-0127-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Walsh DM, Hartley DM, Kusumoto Y, Fezoui Y, Condron MM, Lomakin A, Benedek GB, Selkoe DJ, Teplow DB. Amyloid beta-protein fibrillogenesis. Structure and biological activity of protofibrillar intermediates. J Biol Chem. 1999;274:25945–25952. doi: 10.1074/jbc.274.36.25945. [DOI] [PubMed] [Google Scholar]
- 138.Walsh DM, Klyubin I, Fadeeva JV, Cullen WK, Anwyl R, Wolfe MS, Rowan MJ, Selkoe DJ. Naturally secreted oligomers of amyloid beta protein potently inhibit hippocampal long-term potentiation in vivo. Nature. 2002;416:535–539. doi: 10.1038/416535a. [DOI] [PubMed] [Google Scholar]
- 139.Wang X, Rochon M, Lamprokostopoulou A, Lunsdorf H, Nimtz M, Romling U. Impact of biofilm matrix components on interaction of commensal Escherichia coli with the gastrointestinal cell line HT-29. Cell Mol Life Sci. 2006;63:2352–2363. doi: 10.1007/s00018-006-6222-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Wang X, Smith DR, Jones JW, Chapman MR. In vitro polymerization of a functional Escherichia coli amyloid protein. J Biol Chem. 2007;282:3713–3719. doi: 10.1074/jbc.M609228200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.White AP, Collinson SK, Banser PA, Gibson DL, Paetzel M, Strynadka NC, Kay WW. Structure and characterization of AgfB from Salmonella enteritidis thin aggregative fimbriae. J Mol Biol. 2001;311:735–749. doi: 10.1006/jmbi.2001.4876. [DOI] [PubMed] [Google Scholar]
- 142.White AP, Gibson DL, Kim W, Kay WW, Surette MG. Thin aggregative fimbriae and cellulose enhance long-term survival and persistence of Salmonella. J Bacteriol. 2006;188:3219–3227. doi: 10.1128/JB.188.9.3219-3227.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Wickner RB. [URE3] as an altered URE2 protein: evidence for a prion analog in Saccharomyces cerevisiae. Science. 1994;264:566–569. doi: 10.1126/science.7909170. [DOI] [PubMed] [Google Scholar]
- 144.Wischik CM, Novak M, Edwards PC, Klug A, Tichelaar W, Crowther RA. Structural characterization of the core of the paired helical filament of Alzheimer disease. Proc Natl Acad Sci USA. 1988;85:4884–4888. doi: 10.1073/pnas.85.13.4884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Yang F, Lim GP, Begum AN, Ubeda OJ, Simmons MR, Ambegaokar SS, Chen PP, Kayed R, Glabe CG, Frautschy SA, Cole GM. Curcumin inhibits formation of amyloid beta oligomers and fibrils, binds plaques, and reduces amyloid in vivo. J Biol Chem. 2005;280:5892–5901. doi: 10.1074/jbc.M404751200. [DOI] [PubMed] [Google Scholar]
- 146.Ye S, Huang Y, Mullendorff K, Dong L, Giedt G, Meng EC, Cohen FE, Kuntz ID, Weisgraber KH, Mahley RW. Apolipoprotein (apo) E4 enhances amyloid beta peptide production in cultured neuronal cells: apoE structure as a potential therapeutic target. Proc Natl Acad Sci USA. 2005;102:18700–18705. doi: 10.1073/pnas.0508693102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Zerbinatti CV, Wahrle SE, Kim H, Cam JA, Bales K, Paul SM, Holtzman DM, Bu G. Apolipoprotein E and low density lipoprotein receptor-related protein facilitate intraneuronal Abeta42 accumulation in amyloid model mice. J Biol Chem. 2006;281:36180–36186. doi: 10.1074/jbc.M604436200. [DOI] [PubMed] [Google Scholar]
- 148.Zheng H, Koo EH. The amyloid precursor protein: beyond amyloid. Mol Neurodegener. 2006;1:5. doi: 10.1186/1750-1326-1-5. [DOI] [PMC free article] [PubMed] [Google Scholar]