

Abstract
FKBP52 (FKBP59, FKBP4) is a “macro” immunophilin that, although sharing high structural and functional homologies in its amino-terminal domain with FKBP12 (FKBP1), does not have immunosuppressant activity when complexed with FK506, unlike FKBP12. To investigate the physiological function of FKBP52, we used the yeast two-hybrid system as an approach to find its potential protein partners and, from that, its cellular role. This methodology, which already has allowed us to find the FK506-binding protein (FKBP)-associated protein FAP48, also led to the detection of another FKBP-associated protein. Determination of the sequence of this protein permitted its identification as phytanoyl-CoA α-hydroxylase (PAHX), a peroxisomal enzyme that so far was unknown as an FKBP-associated protein. Inactivation of this enzyme is responsible for Refsum disease in humans. The protein also corresponds to the mouse protein LN1, which could be involved in the progress of lupus nephritis. We show here that PAHX has the physical capacity to interact with the FKBP12-like domain of FKBP52, but not with FKBP12, suggesting that it is a particular and specific target of FKBP52. Whereas the binding of calcineurin to FKBP12 is potentiated by FK506, the specific association of PAHX and FKBP52 is maintained in the presence of FK506. This observation suggests that PAHX is a serious candidate for studying the cellular signaling pathway(s) involving FKBP52 in the presence of immunosuppressant drugs.
The macrolide FK506 (tacrolimus) is a strong immunosuppressant drug widely used in organ transplantation. This pharmacological agent, like the cyclic undecapeptide cyclosporin A (CsA), also has therapeutic value in the treatment of immune and autoimmune conditions (1, 2). Both immunosuppressants are powerful biochemical tools for studying signal transduction pathways. FK506, CsA, and rapamycin—another immunosuppressant macrolide—are natural products that act by binding to endogenous, intracellular receptor proteins termed immunophilins. Based on their amino acid sequences and binding affinity for drugs, immunophilins have been divided into two distinct families: FK506-binding proteins (FKBPs), which bind FK506 and rapamycin, and cyclophilins, which bind CsA (reviewed in ref. 3).
Proteins of both immunophilin classes display a similar peptidyl–prolyl isomerase (rotamase) activity, which presumably facilitates the folding and trafficking of proteins (4, 5), though rotamase inhibition after binding of the corresponding immunosuppressant ligand is not responsible for the immunosuppressive effects of the drugs (6). Another property of both families is their ability to interact with diverse proteins involved in different signal transduction pathways. These interactions often are modulated by the binding of an immunosuppressant drug to the immunophilin.
Ranging in size from 12 to 65 kDa, several members of the FKBP family have been identified in many tissues and found in diverse subcellular compartments (reviewed in refs. 3 and 7). The principal cytoplasmic isoform of the FKBPs is FKBP12 (FKBP1, as in ref. 3), which, when liganded with an immunosuppressant drug, disrupts signal transduction responsible for T cell proliferation (8, 9). When complexed with FK506, FKBP12 inhibits calcineurin’s phosphatase activity, which is required for the Ca2+-dependent production of various growth factors including interleukin 2, whereas the rapamycin–FKBP12 complex acts by blocking the response to growth factors and progression through the cell cycle.
FKBP52 (“FKBP4,” originally FKBP59) initially was discovered as a component of inactive steroid receptor complexes (10, 11). Sequence data and biochemical analyses (12–14) have permitted the determination of a modular organization for this protein. The protein has three domains, decreasingly homologous to FKBP12 from the N-terminal end, each separated by short hydrophilic linker sequences. Only the N-terminal domain, residues 1–149 (FKBP52-I), sharing 55% homology with FKBP12, is able to bind FK506, and displays rotamase activity (15–18). Besides its immunophilin properties, FKBP52 binds ATP and GTP (19), associates with Hsp90—another component of steroid receptor heterocomplexes—via FKBP52’s three tetratricopeptide repeat unit sequences (residues 273–389) (20), and contains a calmodulin-binding site (21). FKBP52 is phosphorylated by casein kinase II (22) and has been found to have chaperone activity independent of its rotamase activity (23). However, unlike FKBP12, the FK506–FKBP52 complex does not bind calcineurin (24–26), and the protein’s physiological function remains to be established.
The identification of cellular proteins that do interact with FKBP52, physically and/or functionally, is an approach for defining the cellular functions of FKBP52. Thus, the yeast two-hybrid system for detecting interacting proteins, originally described by Fields and Song (27), was carried out by using a Jurkat cell cDNA library and with the first domain of rabbit FKBP52 as bait. This methodology, which has allowed the detection of the FKBP-associated protein FAP48—interacting with both FKBP52 and FKBP12, in the absence of FK506, via the immunophilin domain (28)—also led us to the identification of another cDNA encoding a distinct protein that selectively binds FKBP52 and that is the object of this report.
MATERIALS AND METHODS
Screening.
The cDNA library from Jurkat cells and the two-hybrid screen method already have been described (28, 29). Assay of β-galactosidase activity in yeast was performed according to the standard procedure.
Sequence Analysis.
Nested deletions of the cDNA insert of the clone (clone 2) eventually found to encode incomplete PAHX (ΔPAHX) were generated by using a double-stranded nested deletion kit (Pharmacia) and then sequenced by using the Sanger dideoxy-terminal method.
Northern Blotting.
Five micrograms of poly(A)+ RNA isolated from Jurkat cells were submitted to electrophoresis through a denaturing formaldehyde agarose gel, transferred to a Hybond membrane (Amersham), and hybridized with labeled clone 2, according to the standard procedure.
5′ Rapid Amplification of cDNA Ends–PCR (RACE-PCR).
The remainder of the cDNA encoding full-length PAHX was obtained by using RACE-PCR. From 1 μg of Jurkat cell poly(A)+ RNA, the first-strand cDNA was synthesized by elongation using the Moloney murine leukemia virus RNase-H reverse transcriptase (Life Technologies, Gaithersburg, MD) with the specific antisense primer 2-Ext1: 5′-CCTGGAAATCCTGGACCTTCGTGA-3′. The PCRs were performed as described previously by using as first antisense primer the oligonucleotide 2–3′1: 5′-CGTGATCATCTTCTCACTTGGAGCA-3′, and, as second specific antisense primer, the oligonucleotide 2–3′2: 5′-GAGCATATTCGGATTTCGAAATGG-3′. After end repair using T4 DNA polymerase (Biolabs, Northbrook, IL) and phosphorylation with T4 polynucleotide kinase (Life Technologies), PCR products were subcloned into HincII-digested Puc18 vector and sequenced.
Expression Vectors.
The expression vector pGEX-1λT (Pharmacia) and pLexA containing rabbit full-length FKBP52 or FKBP52-I have been described previously (20, 28). To obtain FKBP52 as a fusion protein with glutathione S-transferase (GST), which is cleaved from FKBP52 by factor Xa, the full-length FKBP52 was excised from the vector pGEX-1λT and then inserted into the EcoRI site of pGEX-5X-1. For expressing FKBP12 as a fusion protein with GST, the FKBP12 cDNA insert was generated by PCR amplification by using as primers 5′-CGCGAATTCATGGGCGTGCAGGTGGAGAC-3′ and 5′-CGCGCTCGAGTCATTCCAGTTTTAGAAGCTCCA-3′, subsequently hydrolyzed by EcoRI and XhoI (the sites are shown in bold), and then inserted into these same restriction sites of pGEX-5X-1. Products were confirmed by sequencing.
To obtain pGEX-ΔPAHX, the cDNA insert of ΔPAHX was excised from pGAD1318 by using EcoRI and NotI restriction enzymes and inserted into pGEX-4T-2. The construction of pGAD full-length PAHX was generated from one clone obtained by rapid amplification by using forward primer 5′-CGGGGAATTCAAATGGGGCAGAGGCCGG-3′ and antisense primer 2–3′2 (as above). The forward primer has an EcoRI site, which is shown in bold. The resulting fragment was hydrolyzed by AccI and EcoRI and inserted in the two-hybrid plasmid pGAD-ΔPAHX, and the product was confirmed by sequencing.
For expression of full-length PAHX as a fusion protein with GST, PCR amplification of pGAD-PAHX was performed with forward primer 5′-CGGGGAATTCCCATGGAGCAGCTTG-3′ and the primer 2–3′2 as antisense primer. The forward primer has an EcoRI site shown in bold. After hydrolysis by EcoRI and AccI, the resulting fragment was inserted into pGEX-ΔPAHX and the product was confirmed by sequencing.
Polyclonal Antibodies.
The polyclonal antipeptide antibody 173 against FKBP52 has been described (12). The polyclonal antibody directed against the peptide 160–173 (NKPPDSGKKTSRHP) of PAHX was obtained from Eurogentec (Brussels).
In Vitro Binding Assay.
Gel-shift assay. Overexpression of full-length PAHX and FKBP52 as fusion proteins with GST was achieved as described (30). The resulting fusion proteins were bound to glutathione (GSH)-Sepharose beads, cleaved by 7 units of thrombin (Sigma), and dialyzed overnight at 4°C against TG buffer (20 mM Tris⋅HCl/10% glycerol, pH 7.4). FKBP52 was incubated with PAHX or GST for 30 min at 25°C in a buffer containing 25 mM Hepes-KOH/50 mM KCl/2.5 mM MgCl2/5 mM βSH/1 mM EDTA/5% glycerol/0.01% Tween 20, pH 7.4. The mixtures then were analyzed by native gel gradient, 4–10% acrylamide without stacking gel, in 0.375 M Tris⋅HCl, pH 8.0; the reservoir buffer was 0.025 M Tris⋅HCl/0.0192 M glycine, pH 8.0. After transfer from polyacrylamide gel to nitrocellulose filters, the proteins were immunoblotted with 173 polyclonal antibody directed against FKBP52 by using a Vectastain ABC kit (Vector Laboratories).
GST protein-binding assay.
Forty microliters of glutathione-Sepharose beads (Pharmacia) preloaded with 500 pmol of GST-FKBP52, GST-FKBP12, or GST control protein first was blocked with 0.5% skimmed milk in binding buffer containing 20 mM Hepes-KOH/150 mM KCl/1 mM EDTA/1 mM DTT, pH 7.5. After rotating tubes overnight at 4°C, the GSH beads were washed five times with 1 ml of binding buffer, then resuspended in 100 μl of the same buffer at which increasing quantities of PAHX, at a molar ratio of 1:1 to 2:1 with respect to immobilized protein, were added. The tubes were rotated for 2.5 hr at room temperature. In FK506 experiments the GST-FKBP52 resins were preincubated previously with 1 nmol, 2.5 nmol, or 10 nmol FK506 for 1 hr at room temperature; the control sample was incubated with ethanol (a solvent for the drug). The beads then were washed three times with 1 ml binding buffer and twice with 1 ml TG buffer, and then eluted by GSH over 1 hr at room temperature. The eluted proteins were subjected to SDS/PAGE and then immunoblotted with polyclonal antibody directed against PAHX by using a Vectastain ABC kit (Vector Laboratories).
RESULTS AND DISCUSSION
Detection of the cDNA Insert Using the Two-Hybrid System.
Domain I of rabbit FKBP52 (residues 1–149) was fused to the DNA-binding domain of LexA to give pLexA-FKBP52-I, which was used as bait to screen a Jurkat cell cDNA library by the yeast two-hybrid system. The yeast strain L40, containing the two LexA responsive reporter genes His and LacZ, were used to assay the interactions. This screening allowed the detection of two cDNA clones encoding distinct proteins that interact selectively with FKBP52. One of them, which codes for the protein FAP48, has already been described (28). Here we report on the second cDNA isolated from the same screen.
The latter cDNA clone (clone 2), expressed in the L40 yeast strain containing the pLexA-FKBP52-I construct, confers on this strain the ability to grow in the absence of histidine and to have β-galactosidase activity, unlike a strain containing an extraneous bait, pLexA-lamin. This demonstrates that the clone 2 cDNA encodes for a polypeptide able to form a specific complex with FKBP52-I.
FKBP52-I is the domain responsible for the immunophilin properties and immunosuppressant binding of the whole protein. To investigate the effect of immunosuppressant drug FK506 on the complex obtained with the clone 2 and FKBP52-I, the yeast transformant pLexA-FKBP52-I/pGad-clone 2 was grown overnight in the presence or absence of increasing concentrations of FK506. Cells were collected and β-galactosidase activity was measured. As shown in Fig. 1, the β-galactosidase activity increased as a function of the amount of FK506 in the culture medium, indicating that FK506 strongly potentiates the FKBP52-I/cDNA clone 2 two-hybrid interaction and confirming the specificity of this association. This increase of β-galactosidase activity in the presence of FK506 reflects a specific action of this drug on the FKBP52-I/cDNA clone 2 two-hybrid interaction, because in yeast doubly transformed with FAP48 and FKBP52-I or FKBP52, an inhibitory effect of the FK506 could be observed on the formation of FAP48/immunophilin complexes (28).
Figure 1.

Effect of FK506 on the FKBP52-I/clone 2 (ΔPAHX) two-hybrid interaction. β-galactosidase activities were quantified by liquid assay in the strain FKBP52-I/clone2 (ΔPAHX), grown in the presence or absence of FK506. The β-galactosidase activity measured in the extract of the untreated strain was taken as 100; the values obtained for the extracts of this strain treated with drug were standardized with respect to the reference sample. Values correspond to the means of triplicates.
Structure.
The double-stranded dideoxynucleotide sequence analysis of this cDNA insert revealed an open translational frame throughout its 1,400 bp. The Northern blot carried out with 5 μg of Jurkat cell poly(A)+ RNA and using clone 2 as a probe disclosed a unique mRNA band around 1,700 bp, as shown in Fig. 2.
Figure 2.
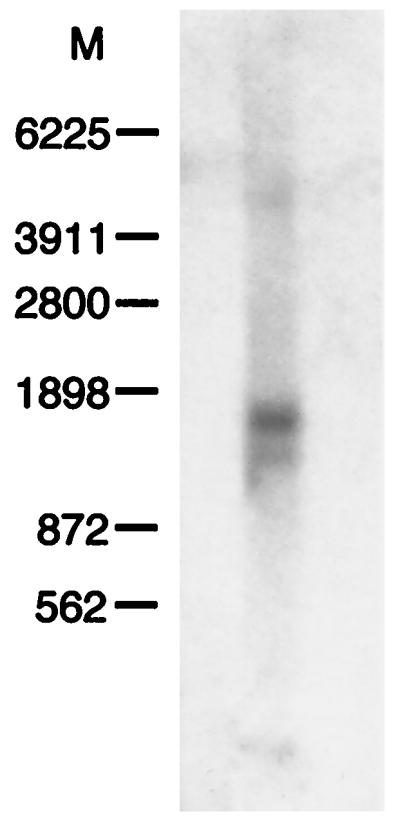
Northern blot analysis of PAHX. Poly(A)+ RNA (5 μg) isolated from Jurkat cells was electrophoresed in a 1% denaturing gel, blotted onto a Hybond membrane, and hybridized with labeled clone 2. After 1 week of exposure, a band around 1,700 bp was revealed. The position of RNA transcript molecular mass markers (Amersham Pharmacia) is shown on the left.
A 5′ RACE PCR was used to obtain the full length of this human cDNA coding sequence. Of the 20 5′ cDNA-specific clones that were analyzed, 3 had 164, 144, or 142 bp additional to clone 2. They contained an ATG flanked by sequences agreeing with the consensus for translational initiation at 44, 24, and 22 bp from their 5′ ends, respectively. Another clone, having an additional 180 bp, not only had this ATG, but also another, at 12 bp from its 5′ end, closely preceded by a TAA stop codon. This sequence has been submitted to GenBank with the accession number AF112977.
During this work searches (in April, 1997) of the GenBank and SwissProt data banks with the nucleotide and derived amino acid sequence revealed this sequence to be the human homolog (78% homology) of the mouse lupus nephritis protein 1 (LN1), which was isolated and cloned using mRNA differential displays with renal cortex mRNA from MRL/MP-lpr/lpr (MRL/lpr) mice (31). This confirmed that we had the entire coding sequence of the new FKBP-associated protein.
The complete coding sequence codes for a protein of 338 residues with a predicted molecular mass of 38,500 Da and a calculated pI of 8.66. As shown by the double-hybrid screen, the first 40 aa residues of this FKBP-associated protein are not required for the interaction with FKBP. Later searches (in December, 1997) found that the protein is identical to the recently sequenced human phytanoyl-CoA α-hydroxylase, termed PAHX or PhyH (32, 33) (the homology between LN1 and PAHX appears so far to have escaped notice). We therefore henceforth refer to the protein as PAHX.
Interaction in the Two-Hybrid System Using the Full-Length Proteins.
Additional studies were performed to determine whether FKBP52-I was as able to interact with full-length PAHX as it did with ΔPAHX (residues 41–338). The plasmid encoding full-length PAHX was fused to the activation domain of Gal4 in the pGAD1318 vector to give pGAD-PAHX and transformed in the yeast L40 strain containing pLexA-FKBP52-I. The strain obtained, although expressing both PAHX and FKBP52-I, grew very slowly without histidine and had very weak β-galactosidase activity (Fig. 3). The same results were obtained in the presence of FK506.
Figure 3.

Interaction of ΔPAHX, or PAHX with FKBP52, and FKBP52-I in the yeast two-hybrid system. β-Galactosidase activities were quantified by liquid assay in the strains FKBP52-I/ΔPAHX, FKBP52-I/PAHX, FKBP52/ΔPAHX, and FKBP52/PAHX.
The same experiment was carried out with the yeast strain containing full-length FKBP52, expressed as a fusion protein with the DNA-binding domain of LexA (pLexA-FKBP52), and pGAD-PAHX. No growth was detected in the absence of histidine, nor was there any β-galactosidase activity in the absence (Fig. 3) or presence of FK506.
The observation of increased interaction in the strain pLexA-FKBP52-I/pGAD-ΔPAHX (residues 41–338) as a function of the increased presence of the immunosuppressor FK506 is good evidence that detection of the protein interaction was not an artifact and suggests that the lack of signal in the strain pLexA-FKBP52/pGAD-PAHX is insufficient evidence alone to refute an interaction. In fact, this result does not necessarily preclude an interaction because the hybrid proteins may not be stable or, in this experimental system, the extra domains of the complete proteins may sterically hinder the DNA-binding domain of LexA and the Gal4 activation domain, preventing detection of an interaction. Alternatively, other endogenous yeast FKBPs may be competing with the FKBP52 bait protein. This uncertainty prompted us to undertake in vitro binding assays.
In Vitro Studies of the Interaction Between PAHX and FKBP52.
FKBP52 and PAHX were expressed as fusion proteins with GST in Escherichia coli, and the fusion proteins were purified on a GSH-Sepharose column and cleaved by thrombin. PAGE under nondenaturing conditions, after incubation of the purified FKBP52 and PAHX together, showed a band of modified electrophoretic mobility. Western blotting using the anti-FKBP52 antibody demonstrated the presence of FKBP52 in this band (Fig. 4). Parallel incubation with GST did not modify the migration of either protein, thereby demonstrating not only that FKBP52 and PAHX interact, but that the interaction is direct, not involving an intermediary molecule.
Figure 4.

Analysis of the FKBP52/PAHX complex in vitro using a gel-shift assay. FKBP52 alone or incubated with GST or PAHX, as indicated, was subjected to nondenaturing PAGE followed by Western blotting using the polyclonal antibody 173 directed against FKBP52.
In addition, FKBP52 protein affinity beads were used to capture PAHX. For this purpose, FKBP52 was subcloned into vector pGEX-5X-1 and expressed as a fusion protein with GST, which can be cleaved from the FKBP52 by factor Xa and not by thrombin. The affinity beads were generated by binding the GST-FKBP52 fusion protein to GSH-Sepharose. The affinity beads then were incubated with protein PAHX, previously expressed in E. coli as a fusion protein with GST and cleaved by thrombin. After incubation and extensive washing of the beads, the retained proteins were eluted with GSH and analyzed by Western blotting using the anti-PAHX antibody (Fig. 5A). As shown in the figure, analysis of the protein adsorbed on the GSH beads revealed the presence of PAHX exclusively on the GST-FKBP52 resin. The GST resin alone was unable to retain PAHX, confirming the specificity of this interaction.
Figure 5.

In vitro binding assay of PAHX with immunophilin. (A) Human PAHX specifically binds FKBP52 in vitro. In vitro binding assays were carried out by incubation of 500 pmol GST-FKBP52 resin (lanes 1–3), 500 pmol GST-FKBP12 resin (lanes 4–6), or 500 pmol GST resin as control (lanes 7–9) in the presence of increasing quantities of PAHX. The molar ratio of PAHX added to different resins is indicated under each lane. After processing the resin as described in Material and Methods, the presence of PAHX retained on the GSH beads was analyzed by Western blotting by using a polyclonal antibody directed against PAHX. Recombinant purified PAHX is loaded as control in lane 10. (B) The PAHX/immunophilin complex formation is not modulated by FK506 in vitro. (Upper) GSH beads loaded with 500 pmol of GST-FKBP52 were treated with increasing quantities of FK506 as indicated under each lane, before incubation with 500 pmol of PAHX. Bound proteins were subjected to Western blotting analysis by using a polyclonal antibody directed against PAHX. Lane 1, no PAHX was added to the resin. (Lower) GSH beads loaded with 500 pmol of GST-FKBP52 (lanes 1–3) or 500 pmol of GST-FKBP12 (lanes 4–6) were treated or not with 10 nmol of FK506 before incubation with 500 pmol of PAHX, as indicated in Materials and Methods. Bound proteins were subjected to Western blotting analysis by using a polyclonal antibody directed against PAHX. Lanes 1 and 4, no PAHX was added to the resin. Recombinant purified PAHX was loaded as control in lane 7. Migration of molecular size markers (New England Biolabs) is indicated on the left.
In Vitro Effect of FK506 on the Complex PAHX/FKBP52.
Because FK506 had been found to increase the interaction between FKBP52-I and ΔPAHX, the effect on the interaction between the complete proteins was investigated using the same in vitro system. The GST-FKBP52 resin first was incubated with increasing amounts of FK506 and then with PAHX. The capacity of the resin to retain different amounts of PAHX was monitored as before. As shown in Fig. 5B, the FK506 had no significant effect on FKBP52/PAHX complex formation in this in vitro system. There are several possible explanations for this apparent discrepancy between in vitro and in vivo studies: (i) there may be in the yeast cell—and absent from the purified protein preparations—a third (protein) component involved in complex stabilization by FK506; (ii) in the absence of FK506, FKBP52-I may be complexed with some other endogenous yeast component, but such binding may be prevented by FK506, liberating FKBP52-I to participate in high-affinity binding with PAHX; (iii) the modulation of the PAHX-FKBP52 interaction by FK506 may require a structural modification of PAHX or FKBP52 that does not occur in vitro.
In Vitro Investigation of PAHX Interaction with FKBP12.
The capacity of PAHX to associate with FKBP12 was investigated under the same experimental conditions as for FKBP52. As shown in Fig. 5A, the FKBP12 resin does not retain PAHX, corroborating results obtained with the yeast two-hybrid system, where no significant β-galactosidase activity could be detected in the L40 strain containing pLexA-FKBP12 and pGAD-PAHX (data not shown). Even in the presence of FK506, PAHX failed to stably interact with FKBP12 (Fig. 5B Lower). This indicates that PAHX is a specific target or substrate for the immunophilin FKBP52, unlike FAP48, which interacts with both FKBP52 and FKBP12.
The FK506-binding domains of FKBP52 (FKBP52-I) and FKBP12 share high structural homology (12–14) but have different affinities for FK506: a dissociation constant of 0.4 nM has been described for FKBP12 (34) whereas 40 and 66 nM have been reported for FKBP52 (18, 35). Likewise, a Ki of approximately 1 nM for FK506 has been found with FKBP12 (36), but it is 10 nM with FKBP52 (13). These observations suggest that these two immunophilins have different targets—as supported by FKBP12’s binding to calcineurin in the presence of FK506, whereas no interaction between calcineurin and FKBP52 has been detected.
Significance of the Interaction of PAHX or LN1 with FKBP52.
The mouse MRL/lpr strains, used for the isolation of LN1 (31), spontaneously develop a severe autoimmune disease similar to human systemic lupus erythematosus (SLE). Studies on MRL/lpr mice suggest that excessive numbers of self-reactive T lymphocytes released into peripheral organs could be responsible for the autoimmune disease in this strain (37). It has been pointed out that the gene encoding LN1 has a low expression in the early stage of lupus nephritis in the renal cortex of MRL/lpr mice, so the protein could be involved in a reaction against the progression of the disease (31). Correlated with this, the immunosuppressant FK506 has been found to have the potential for preventing the progression of nephropathy in MRL/lpr mice with spontaneous lupus nephritis (38–40), though the mechanism of action remains unknown. These findings are to be juxtaposed with our result that the homolog of LN1 interacts specifically with the FK506-binding protein FKBP52, and this interaction possibly may be potentiated by FK506 via a bridging molecule.
The human protein PAHX is a peroxisomal enzyme catalyzing the first step in phytanic acid oxidation: the α-oxidation of phytanoyl-CoA to α-hydroxyphytanoyl-CoA. This enzyme is targeted to peroxisomes by the protein PEX7, the receptor for the type 2 peroxisomal targeting signal (PTS2)—an amino-terminal motif with the consensus sequence RLX5H/QL, which is sufficient to direct proteins into the peroxisomes (32, 33, 41). PAHX is the defective enzyme in Refsum disease that is a peroxisomal disorder characterized by accumulation of the fatty acid phytanic acid, originating from food of direct or indirect vegetable origin, resulting in the classic triad of peripheral neuropathy, retinis pigmentosa, and cerebellar ataxia (42).
The physiological significance of the interaction of PAHX with FKBP52 remains to be elucidated. Interestingly, the cleavable PTS2 signal is localized in the N-terminal part of PAHX, which is not required for interaction with FKBP52, thus reducing the probability that FKBP52 binding would be involved in recognition of the signal by the PTS2 receptor (PEX7). However, like mitochondrial proteins, peroxisomal proteins are synthesized on cytoplasmic ribosomes and posttranslationally imported into the organelle by a process that requires Hsp70-class cytoplasmic chaperones; so it would be of interest to investigate whether FKBP52 is involved in the folding and trafficking of this enzyme and whether binding of the FKBP52–FK506 complex with PAHX affects the latter’s enzymatic activity.
It is noteworthy that PAHX mRNA has been found in specific tissues such as liver and kidney but not in spleen, brain, heart, lung, and skeletal muscle (31), but here we show it also to be present in T cells. The results concerning the levels of expression and localization of PAHX mRNA raise an intriguing question with respect to the known function of PAHX: is phytanoyl-CoA the unique substrate of PAHX, or does PAHX have (an)other function(s)? This is also suggested by the finding that the mouse homolog protein LN1 could play a role in the development of an autoimmune disease.
In conclusion, the major finding of this work is that PAHX is another specific target for FKBP52. The observation that PAHX is also bound by an FKBP52–FK506 affinity matrix leads us to think that it is a good candidate for involvement in cellular signal transmission by FKBP52 in the presence of FK506, in a manner that may be compared with that of the calcineurin–FKBP12 complex. It is, however, too early to conclude that this physical interaction is important for the physiological effect of PAHX in vivo. Additional studies on this new FKBP-associated protein are required for a better understanding of its physiological role by itself and in conjunction with FKBP52.
Acknowledgments
We thank Dr. K. Murato (Fujisawa Pharmaceutical, Osaka, Japan) for the gift of FK506. We thank Dr. B. Standaert (Department of Chemistry, Harvard University, Cambridge, MA) for providing the FKBP12 cDNA. We are grateful to Dr. A. Chadli for helpful discussion throughout the course of this work. We are indebted to Jean-Claude Lambert for help with the figures. This study was supported, in part, by the Association pour la Recherche sur le Cancer Contract 9708 and the Ligue Nationale contre le Cancer Comité du Val de Marne.
ABBREVIATIONS
- FKBP
FK506-binding protein
- FAP
FKBP-associated protein
- GST
glutathione S-transferase
- GSH
glutathione
- LN1
lupus nephritis protein 1
- PAHX
phytanoyl-CoA α-hydroxylase
Footnotes
Data deposition: The sequence reported in this paper has been deposited in the GenBank database (accession no. AF112977).
References
- 1.Fung J J, Starzl T E. Transplant Proc. 1994;26:3017–3020. [PubMed] [Google Scholar]
- 2.Thomson A W, Starzl T E. Autoimmunity. 1992;12:303–313. doi: 10.3109/08916939209148473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Galat A, Metcalfe S M. Prog Biophys Mol Biol. 1995;63:67–118. doi: 10.1016/0079-6107(94)00009-x. [DOI] [PubMed] [Google Scholar]
- 4.Fruman D A, Burakoff S J, Bierer B E. FASEB J. 1994;8:391–400. doi: 10.1096/fasebj.8.6.7513288. [DOI] [PubMed] [Google Scholar]
- 5.Schreiber S L. Science. 1991;251:283–287. doi: 10.1126/science.1702904. [DOI] [PubMed] [Google Scholar]
- 6.Bierer B E, Somers P K, Wandless T J, Burakoff S J, Schreiber S L. Science. 1990;250:556–559. doi: 10.1126/science.1700475. [DOI] [PubMed] [Google Scholar]
- 7.Kay J E. Biochem J. 1996;314:361–385. [PMC free article] [PubMed] [Google Scholar]
- 8.Liu J, Albers M W, Wandless T J, Luan S, Alberg D G, Belshaw P J, Cohen P, MacKintosh C, Klee C B, Schreiber S L. Biochemistry. 1992;31:3896–3901. doi: 10.1021/bi00131a002. [DOI] [PubMed] [Google Scholar]
- 9.Schreiber S L. Cell. 1992;70:365–368. doi: 10.1016/0092-8674(92)90158-9. [DOI] [PubMed] [Google Scholar]
- 10.Tai P K K, Maeda Y, Nakao K, Wakim N G, Duhring J L, Faber L E. Biochemistry. 1986;25:5269–5275. doi: 10.1021/bi00366a043. [DOI] [PubMed] [Google Scholar]
- 11.Renoir J M, Radanyi C, Faber L E, Baulieu E E. J Biol Chem. 1990;265:10740–10745. [PubMed] [Google Scholar]
- 12.Lebeau M C, Massol N, Herrick J, Faber L E, Renoir J M, Radanyi C, Baulieu E E. J Biol Chem. 1992;267:4281–4284. [PubMed] [Google Scholar]
- 13.Peattie D A, Harding M W, Fleming M A, DeCenzo M T, Lippke J A, Livingston D J, Benasutti M. Proc Natl Acad Sci USA. 1992;89:10974–10978. doi: 10.1073/pnas.89.22.10974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Callebaut I, Renoir J M, Lebeau M C, Massol N, Burny A, Baulieu E E, Mornon J P. Proc Natl Acad Sci USA. 1992;89:6270–6274. doi: 10.1073/pnas.89.14.6270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chambraud B, Rouvière-Fourmy N, Radanyi C, Hsiao K, Peattie D A, Livingston D J, Baulieu E E. Biochem Biophys Res Commun. 1993;196:160–166. doi: 10.1006/bbrc.1993.2229. [DOI] [PubMed] [Google Scholar]
- 16.Yem A W, Tomasselli A G, Heinrikson R L, Zurcher-Neely H, Ruff V A, Johnson R A, Deibel M R., Jr J Biol Chem. 1992;267:2868–2871. [PubMed] [Google Scholar]
- 17.Tai P K K, Chang H, Albers M W, Schreiber S L, Toft D O, Faber L E. Biochemistry. 1993;32:8842–8847. doi: 10.1021/bi00085a015. [DOI] [PubMed] [Google Scholar]
- 18.Renoir J M, Le Bihan S, Mercier-Bodard C, Gold A, Arjomandi M, Radanyi C, Baulieu E E. J Steroid Biochem Mol Biol. 1994;48:101–110. doi: 10.1016/0960-0760(94)90256-9. [DOI] [PubMed] [Google Scholar]
- 19.Le Bihan S, Renoir J M, Radanyi C, Chambraud B, Joulin V, Catelli M G, Baulieu E E. Biochem Biophys Res Commun. 1993;195:600–607. doi: 10.1006/bbrc.1993.2088. [DOI] [PubMed] [Google Scholar]
- 20.Radanyi C, Chambraud B, Baulieu E E. Proc Natl Acad Sci USA. 1994;91:11197–11201. doi: 10.1073/pnas.91.23.11197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Massol N, Lebeau M C, Renoir J M, Faber L E, Baulieu E E. Biochem Biophys Res Commun. 1992;187:1330–1335. doi: 10.1016/0006-291x(92)90448-t. [DOI] [PubMed] [Google Scholar]
- 22.Miyata Y, Chambraud B, Radanyi C, Leclerc J, Lebeau M C, Renoir J M, Shirai R, Catelli M G, Yahara I, Baulieu E E. Proc Natl Acad Sci USA. 1997;94:14500–14505. doi: 10.1073/pnas.94.26.14500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bose S, Weikl T, Bugl H, Buchner J. Science. 1996;274:1715–1717. doi: 10.1126/science.274.5293.1715. [DOI] [PubMed] [Google Scholar]
- 24.Lebeau M C, Myagkikh I, Rouvière-Fourmy N, Baulieu E E, Klee C B. Biochem Biophys Res Commun. 1994;203:750–755. doi: 10.1006/bbrc.1994.2246. [DOI] [PubMed] [Google Scholar]
- 25.Wiederrecht G, Hung S, Chan H K, Marcy A, Martin M, Calaycay J, Boulton D, Sigal N, Kincaid R L, Siekierka J J. J Biol Chem. 1992;267:21753–21760. [PubMed] [Google Scholar]
- 26.Yem A W, Reardon I M, Leone J W, Heinrikson R L, Deibel M R., Jr Biochemistry. 1993;32:12571–12576. doi: 10.1021/bi00210a004. [DOI] [PubMed] [Google Scholar]
- 27.Fields S, Song O. Nature (London) 1989;340:245–246. doi: 10.1038/340245a0. [DOI] [PubMed] [Google Scholar]
- 28.Chambraud B, Radanyi C, Camonis J H, Shazand K, Rajkowski K, Baulieu E E. J Biol Chem. 1996;271:32923–32929. doi: 10.1074/jbc.271.51.32923. [DOI] [PubMed] [Google Scholar]
- 29.Benichou S, Bomsel M, Bodéus M, Durand H, Douté M, Letourneur F, Camonis J, Benarous R. J Biol Chem. 1994;269:30073–30076. [PubMed] [Google Scholar]
- 30.Smith D B, Johnson K S. Gene. 1988;67:31–40. doi: 10.1016/0378-1119(88)90005-4. [DOI] [PubMed] [Google Scholar]
- 31.Iwano M, Ueno S, Miyazaki M, Harada T, Nagai Y, Hirano M, Dohi Y, Akai Y, Kurioka H, Dohi K. Biochem Biophys Res Commun. 1996;229:355–360. doi: 10.1006/bbrc.1996.1805. [DOI] [PubMed] [Google Scholar]
- 32.Mihalik S J, Morrell J C, Kim D, Sacksteder K A, Watkins P A, Gould S J. Nat Genet. 1997;17:185–189. doi: 10.1038/ng1097-185. [DOI] [PubMed] [Google Scholar]
- 33.Jansen G A, Ofman R, Ferdinandusse S, Ijlst L, Muijsers A O, Skjeldal O H, Stokke O, Jakobs C, Besley G, Wraith J E, Wanders R J A. Nat Genet. 1997;17:190–193. doi: 10.1038/ng1097-190. [DOI] [PubMed] [Google Scholar]
- 34.Siekierka J J, Hung S H Y, Poe M, Lin C S, Sigal N H. Nature (London) 1989;341:755–757. doi: 10.1038/341755a0. [DOI] [PubMed] [Google Scholar]
- 35.Tai P K K, Albers M W, Chang H, Faber L E, Schreiber S L. Science. 1992;256:1315–1318. doi: 10.1126/science.1376003. [DOI] [PubMed] [Google Scholar]
- 36.Harrison R K, Stein R L. Biochemistry. 1990;29:3813–3816. doi: 10.1021/bi00468a001. [DOI] [PubMed] [Google Scholar]
- 37.Cohen P L, Eisenberg R A. Annu Rev Immunol. 1991;9:243–269. doi: 10.1146/annurev.iy.09.040191.001331. [DOI] [PubMed] [Google Scholar]
- 38.Entani C, Izumino K, Iida H, Fujita M, Asaka M, Takata M, Sasayama S. Nephron. 1993;64:471–475. doi: 10.1159/000187375. [DOI] [PubMed] [Google Scholar]
- 39.Takabayashi K, Koike T, Kurasawa K, Matsumura R, Sato T, Tomioka H, Ito I, Yoshiki T, Yoshida S. Clin Immunol Immunopathol. 1989;51:110–117. doi: 10.1016/0090-1229(89)90211-0. [DOI] [PubMed] [Google Scholar]
- 40.Yamamoto K, Mori A, Nakahama T, Ito M, Okudaira H, Miyamoto T. Immunology. 1990;69:222–227. [PMC free article] [PubMed] [Google Scholar]
- 41.Waterham H R, Cregg J M. BioEssays. 1997;19:57–66. doi: 10.1002/bies.950190110. [DOI] [PubMed] [Google Scholar]
- 42.Steinberg D. In: The Metabolic and Molecular Basis of Inherited Disease. 7th Ed. Scriver C R, Beaudet A L, Sly W S, Valle D, editors. New York: McGraw–Hill; 1995. pp. 2351–2369. [Google Scholar]