

Abstract
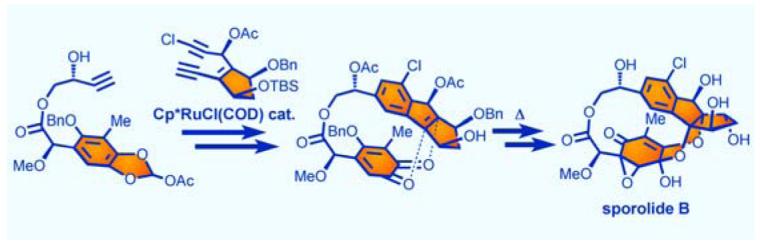
The first total synthesis of the highly oxygenated, marine derived, natural product sporolide B was achieved through a convergent strategy involving a ruthenium-catalyzed [2+2+2] cycloaddition to assemble the indene structural motif and a thermally-induced Diels-Alder-type reaction to forge the macrocycle.
Keywords: cycloaddition, Diels-Alder, natural product, o-quinone, sporolide
Sporolides A (1a) and B (1b) (Figure 1) are two highly unusual natural products reported by the Fenical group in 2005.[1] Isolated from the marine actinomycete Salinospora tropica, these molecules possess no obvious biological activity, yet their intriguing molecular architectures imply the existence of an as yet unidentified, secondary metabolite of the enediyne class,[2] whose fleeting nature may explain the incorporation of the chloro-substituted aryl rings within their structures[2, 3] through a Bergman cycloaromatization reaction.[4] In view of the importance of the enediyne family of natural products[5] and in order to better understand the biosynthetic origins of the sporolides A and B and their postulated enediyne precursor, we embarked on the total synthesis of these molecules. In this communication we report the first total synthesis of sporolide B (1b) in its naturally occurring enantiomeric form through a highly stereoselective and convergent strategy that involves two important cycloaddition reactions.
Figure 1.
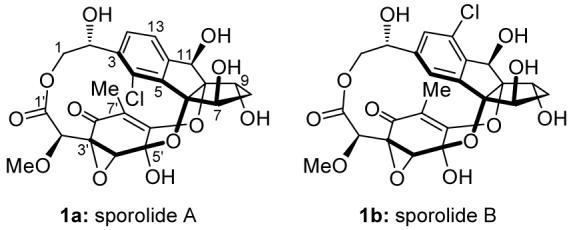
Molecular structures of sporolides A (1a) and B (1b).
The unprecedented C24 polycyclic structure of sporolide B (1b) includes 12 oxygen atoms, 10 stereogenic centers, a 13-membered macrolide ring, a chlorobenzene nucleus embedded within an indane structural motif, and two oxygen bridges that, together with the ester bond, connect the two domains of the molecule into its cage-like structure.
These special structural elements and unique connectivities amounted to a formidable synthetic challenge that was eventually met by adopting the devised synthetic strategy outlined retrosynthetically in Scheme 1. This strategy was based on two key retrosynthetic disconnections, a thermally-induced, intramolecular [4+2] cycloaddition reaction involving an o-quinone and a tetrasubstituted olefin to form the macrocyclic structure of the molecule (2 → 1b),[6] and a ruthenium-catalyzed, intermolecular [2+2+2] cycloaddition reaction between two acetylenic units,[7] building blocks 3 and 4, to forge its chlorobenzenoid indane structural motif. The complexity of the substrates involved in these planned reactions and the lack of any precedent for their application in complex natural product synthesis made them risky propositions with regards to both feasibility and topology (regio- and stereoselectivity). Nevertheless, model studies[6] and inspection of manual molecular models were encouraging.
Scheme 1.
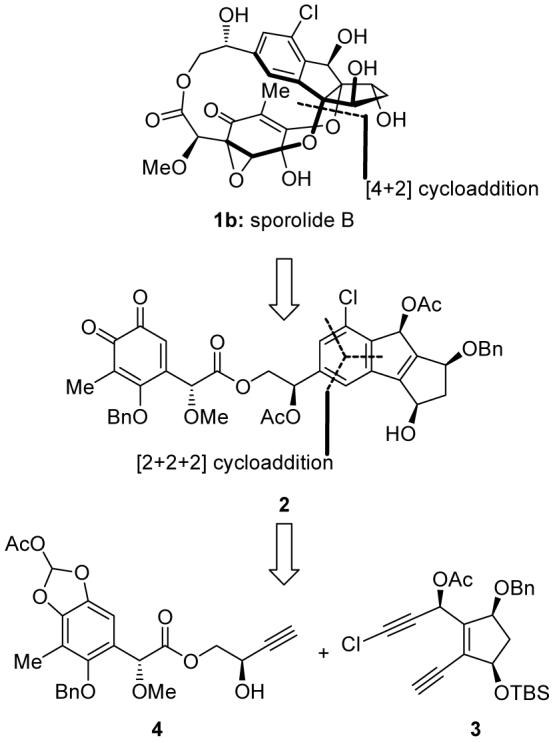
Retrosynthetic analysis of sporolide B (1b). Ac = acetyl, Bn = benzyl, TBS = tert-butyldimethylsilyl.
Scheme 2 summarizes the construction of chloroacetylene building block 3. Thus, enantiomerically pure iodoenone 5 (ee > 99 %)[8] was reduced under Luche conditions[9] (NaBH4, CeCl3·7H2O, -78 °C) to afford, upon benzylation (NaH, BnBr, THF), vinyl iodide 6 (ca. 10:1 diastereomeric ratio, 95 % combined yield).[10] Carboxymethylation of the latter compound under palladium-catalyzed conditions [Pd(PPh3)2Cl2 cat., CO, MeOH] led to methyl ester 7 (95 % yield), whose reduction (DIBAL-H, 95 % yield), protection as a THP ether (DHP, TsOH·H2O), and desilylation (TBAF) furnished allylic alcohol 8. Oxidation of 8 with Dess-Martin periodinane[11] (DMP) gave the corresponding enone (83 % overall yield for the last three steps), which was iodinated (I2, CH2Cl2/pyridine) to afford iodoenone 9 in 80 % yield. Luche reduction of the latter (NaBH4, CeCl3·7H2O, -78 °C) proceeded stereoselectively (ca. 5:1 diastereomeric ratio)[10] to afford, upon silylation (TBSCl, imidazole, 4-DMAP), vinyl iodide 10 in 94 % yield for the two steps. Sonogashira coupling of 10 with TMS-acetylene [Pd(PPh3)2Cl2 cat., CuI cat., Et2NH, 98 % yield] followed by removal of the THP protecting group (Et2AlCl, -25 → 25 °C, 99 % yield) and oxidation of the resulting primary alcohol (DMP, 79 % yield) furnished aldehyde 11. The required chloroacetylene structural motif was then installed within the growing molecule by reaction of aldehyde 11 with a preformed solution of lithiochloroacetylene (cis-1,2-dichloroethylene, MeLi, Et2O, 0 °C) to furnish the expected secondary alcohol, which, however, possessed the undesired stereochemistry at C11.[12] This stereochemistry was inverted through an oxidation-reduction protocol. Thus, oxidation of this alcohol with DMP led to ketone 12 (93 % overall yield for the last two steps), which was reduced stereoselectively (chelation controlled, ca. 7:1 diastereomeric ratio) with DIBAL-H (toluene, -78 °C)[12] to afford the desired alcohol 13 with 81 % yield after silica gel chromatographic separation from its undesired diastereomer. Finally, removal of the TMS group from its acetylene host in 13 (K2CO3, MeOH, 99 % yield), followed by acetylation (Ac2O, Et3N, 4-DMAP, 98 % yield), led to the targeted acetoxy chloroacetylene 3 via hydroxy precursor 14.
Scheme 2.
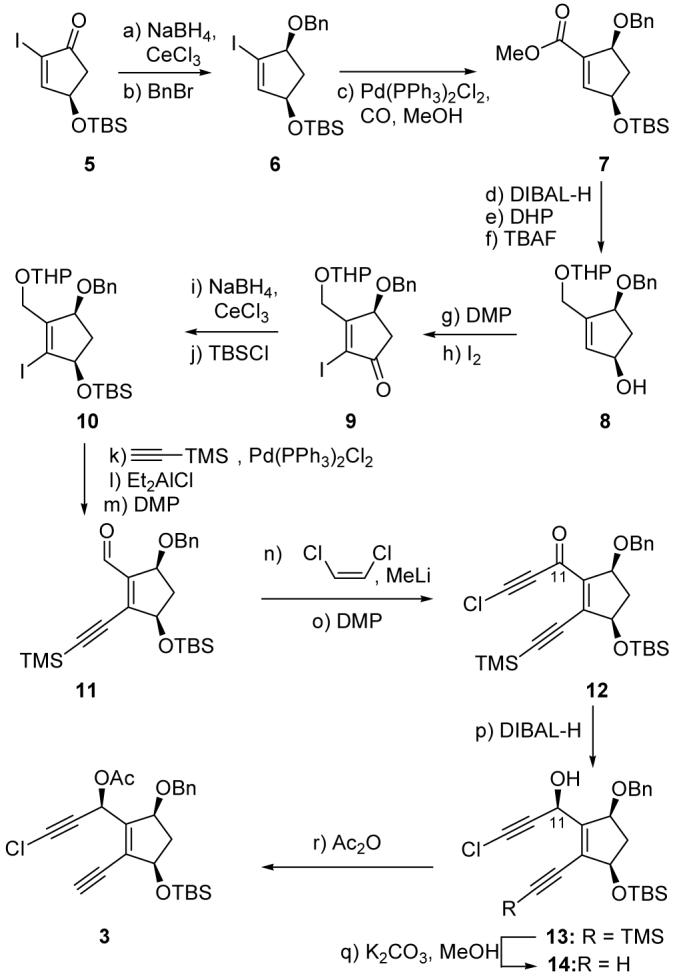
Construction of building block 3. Reagents and conditions: a) NaBH4 (1.2 equiv), CeCl3·7H2O (1.2 equiv), MeOH, -78 °C, 1 h; b) NaH (60 % in mineral oil, 1.5 equiv), THF, 0 °C, 30 min; then BnBr (1.5 equiv), TBAI (0.2 equiv), THF, 0 → 25 °C, 16 h, 95 % for two steps; c) Pd(PPh3)2Cl2 (0.05 equiv), Et3N (5.0 equiv), CO (balloon pressure), MeOH, 70 °C, 3 h, 95 %; d) DIBAL-H (1.0 M in toluene, 2.5 equiv), toluene, -78 → -10 °C, 1 h, 95 %; e) DHP (1.5 equiv), TsOH·H2O (0.1 equiv), CH2Cl2, 0 °C, 30 min; f) TBAF (1.0 M in THF, 1.5 equiv), THF, 25 °C, 3 h; g) DMP (1.2 equiv), NaHCO3 (5.0 equiv), CH2Cl2, 25 °C, 30 min, 83 % for three steps; h) I2 (3.0 equiv), CH2Cl2/pyridine (1:1), 25 °C, 15 h, 80 %; i) NaBH4 (1.2 equiv), CeCl3·7H2O (1.2 equiv), MeOH, -78 °C, 1 h; j) TBSCl (2.0 equiv), imidazole (2.0 equiv), 4-DMAP (0.1 equiv), CH2Cl2, 25 °C, 3 h, 94 % for two steps; k) TMS-acetylene (1.2 equiv), Pd(PPh3)2Cl2 (0.02 equiv), CuI (0.04 equiv), Et2NH, 25 °C, 16 h, 98 %; l) Et2AlCl (1.8 M in toluene, 2.0 equiv), CH2Cl2, -25 → 25 °C, 2 h, 99 %; m) DMP (1.2 equiv), NaHCO3 (5.0 equiv), CH2Cl2, 25 °C, 1 h, 79 %; n) cis-1,2-dichloroethylene (4.5 equiv), MeLi (1.6 M in Et2O, 3.0 equiv), Et2O, 0 °C, 30 min; then 11, Et2O, 0 °C, 10 min; o) DMP (1.5 equiv), NaHCO3 (5.0 equiv), CH2Cl2, 25 °C, 1 h, 93 % for two steps; p) DIBAL-H (1.0 M in toluene, 1.5 equiv), toluene, -78 °C, 30 min, 81 %; q) K2CO3 (1.5 equiv), MeOH, 25 °C, 1 h, 99 %; r) Ac2O (1.5 equiv), Et3N (1.5 equiv), 4-DMAP (0.1 equiv), CH2Cl2, 0 °C, 30 min, 98 %. DHP = 3,4-dihydro-2H-pyran, DIBAL-H = diisobutylaluminum hydride, 4-DMAP = 4-dimethylaminopyridine, DMP = Dess-Martin periodinane, TBAF = tetra-n-butylammonium fluoride, TBAI = tetra-n-butylammonium iodide, THF = tetrahydrofuran, TMS = trimethylsilyl, Ts = 4-toluenesulfonyl.
Scheme 3 depicts the construction of propargyl alcohol building block 4 starting from aldehyde 15.[13] Thus, 15 was subjected to Wittig olefination (Ph3P=CH2, -78 → 0 °C, 98 % yield) to afford styrene derivative 16, which entered a highly enantioselective Sharpless asymmetric dihydroxylation reaction[14] (AD-mix β, 96 % yield, 98 % ee) to afford, after sequential silylation (TBSCl, Et3N, 4-DMAP, 99% yield) and methylation (tBuOK, MeI, 95 % yield), fully protected pentasubstituted aryl system 18. The latter compound was converted to carboxylic acid 19 through a three-step sequence involving desilylation (TBAF, 99 % yield) and oxidation of the resulting primary alcohol, first with DMP (78 % yield), and then with NaClO2 (96 % yield). Coupling of 19 with acetylenic alcohol 20,[15] as facilitated by EDCI and 4-DMAP, led to the selective formation of hydroxy ester 21 (73 % yield). Reaction of the latter intermediate with Pb(OAc)4 in benzene at 75 °C led smoothly to the required hydroxy acetylenic building block 4 in 89 % yield (ca. 6:1 diastereoisomeric mixture).
Scheme 3.
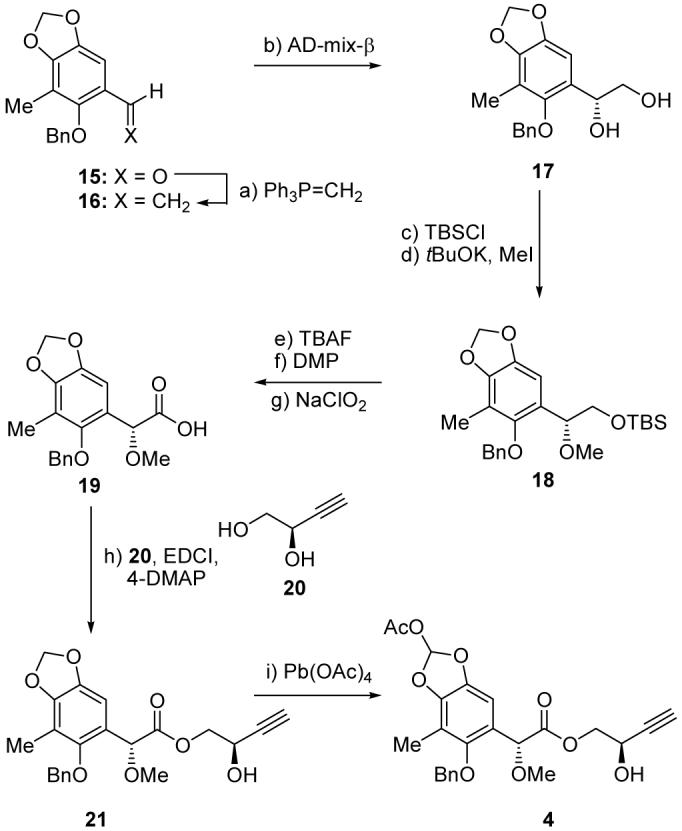
Construction of building block 4. Reagents and conditions: a) MePPh3Br (1.5 equiv), KHMDS (1.0 M in toluene, 1.2 equiv), THF, 0 °C, 30 min; then 15, THF, -78 → 0 °C, 30 min, 98 %; b) AD-mix β (1.4 g per mmol 16), tBuOH/H2O (1:1), 25 °C, 8 h, 96 %; c) TBSCl (1.5 equiv), Et3N (1.5 equiv), 4-DMAP (0.1 equiv), CH2Cl2, 25 °C, 8 h, 99 %; d) tBuOK (3.0 equiv), MeI (4.0 equiv), MeCN, 0 → 25 °C, 16 h, 95 %; e) TBAF (1.0 M in THF, 1.5 equiv), THF, 25 °C, 16 h, 99 %; f) DMP (1.5 equiv), NaHCO3 (5.0 equiv), CH2Cl2, 25 °C, 1 h, 78 %; g) NaClO2 (4.5 equiv), NaH2PO4·2H2O (3.0 equiv), 2-methyl-2-butene (2.5 equiv), tBuOH/H2O (1:1), 25 °C, 30 min, 96 %; h) 20 (1.3 equiv), EDCI (1.2 equiv), 4-DMAP (0.2 equiv), CH2Cl2, 25 °C, 3 h, 73 %; i) Pb(OAc)4 (1.5 equiv), benzene, 75 °C, 1 h, 89 %. EDCI = 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride, KHMDS = potassium bis(trimethylsilyl)amide.
With the two fragments 3 and 4 in hand, we set out to investigate their fusion into the desired polycyclic precursor for the final casting of the sporolide B macrocyclic structure. Although known to proceed well, the intermolecular, ruthenium-catalyzed [2+2+2] cycloaddition reaction of diynes with alkyl substrates to form benzenoid systems[7] was complicated in this instance by the presence of the chlorine atom and the complexity of the substrates involved. However, we were counting on the steric bulk of the chlorine residue on 3 (as compared to a hydrogen atom) and the coordinating ability of the free hydroxyl group in 4 to provide favorable conditions for the desired outcome of the reaction, including its regiochemistry with regards to the position of the chlorine atom on the aryl ring. In the event, combining 3 and 4 in 1,2-dichloroethane in the presence of Cp*RuCl(COD) catalyst[16] resulted, within 30 min, in the formation of compound 22 in 87 % yield and as a single regioisomer (Scheme 4). That this highly productive process led rapidly and exclusively to the desired meta-chloro isomer may be explained by inspection of transition states 3-TSa, 3TSb and 3-TSc, through which this reaction is presumed to proceed.
Scheme 4.

Synthesis of o-quinone 2. Reagents and conditions: a) 3 (1.0 equiv), 4 (1.1 equiv), Cp*RuCl(COD) (0.07 equiv), DCE, 25 °C, 30 min, 87 %; b) Ac2O (2.0 equiv), Et3N (2.0 equiv), 4-DMAP (0.1 equiv), CH2Cl2, 0 °C, 30 min, 92 %; c) HF (48 % aqueous solution, excess), MeCN, 25 °C, 30 min; then MeOH (excess), 25 °C, 3 h, 74 %; d) Ag2O (2.5 equiv), CH2Cl2, 25 °C, 30 min, 94 %. COD = 1,5-cyclooctadiene, Cp* = pentamethylcyclopentadienyl, DCE = 1,2-dichloroethane.
In preparation for the next challenging task, the forging of the cyclic framework of the sporolide molecule through the proposed [4+2] cycloaddition reaction,[6] compound 22 was converted to o-quinone 2 via catechol derivative 23 through sequential treatment with Ac2O-Et3N-4-DMAP (92 % yield) and aq.HF-MeOH (74 % yield), followed by Ag2O oxidation (94 % yield) as shown in Scheme 4.
Scheme 5 presents the final stages and the completion of the total synthesis of sporolide B (1b). Thus, upon heating in toluene at 110 °C, o-quinone 2 underwent the much anticipated Diels-Alder reaction to afford desired product 24 (40 % yield based on ca. 50 % conversion),[17] apparently through transition state 2-TS. Although the remarkable diastereoselectivity (facial orientation of o-quinone) of this reaction can be rationalized by the steric bias of the dienophile (top face blocked by the substituents on the 5-membered rings), an explanation for its regioselectivity (o-quinone rotation around C2′-C3′ bond) remains elusive. It should be noted that the C-9 oxygen substituent within 24 and all its precursors resides (by design) in the opposite configuration from that required for sporolide B (1b), and therefore requires inversion. This task was to be achieved at some point downstream through an oxidation-reduction sequence, with the C-7 hydroxy group playing a directing role in the reduction. Therefore, subsequent steps had to accommodate, in addition to the obligatory dearomatization of the trioxygenated benzenoid ring, the two steps required for this inversion. To this end, a TES group was placed on the well positioned C-7 oxygen (TESOTf, Et3N, 95 % yield) before the two benzyl groups were removed [H2, Pd(OH)2 cat., 92 % yield] to afford hydroxy phenol 26 via intermediate 25. Exposure of 26 to PhI(OCOCF3)2 in the presence of PMBOH in acetonitrile furnished p-ketal quinone 27 in 75 % yield, providing the opening for the oxidation-reduction protocol to invert the C-9 stereochemistry. Indeed, oxidation of 27 (DMP, 90 % yield) followed by removal of the TES protecting group (aq.HF, MeCN, 85 % yield) gave, through the corresponding carbonyl compound, β-hydroxy ketone 28, whose reduction with Me4NBH(OAc)3 [MeCN:AcOH (10:1)] led to 1,3-diol 29 as a single stereoisomer, and in 85 % yield. This compound was treated sequentially with DDQ [CH2Cl2:H2O (10:1), 70 % yield] and DBU [CH2Cl2:MeOH (3:1), 78 % yield] to remove the PMB and acetate protecting groups, furnishing deoxysporolide B 30. The final step of the total synthesis of sporolide B (1b) involved regio- and stereoselective introduction of the missing oxygen atom from precursor 30 in the required epoxide form by reaction with tBuOOH in the presence of DBU in CH2Cl2 at 40 °C (3 h, 63 % yield). Synthetic sporolide B (1b) exhibited identical chromatographic data, spectroscopic data, and optical rotation sign to those of the authentic sample.[18]
Scheme 5.
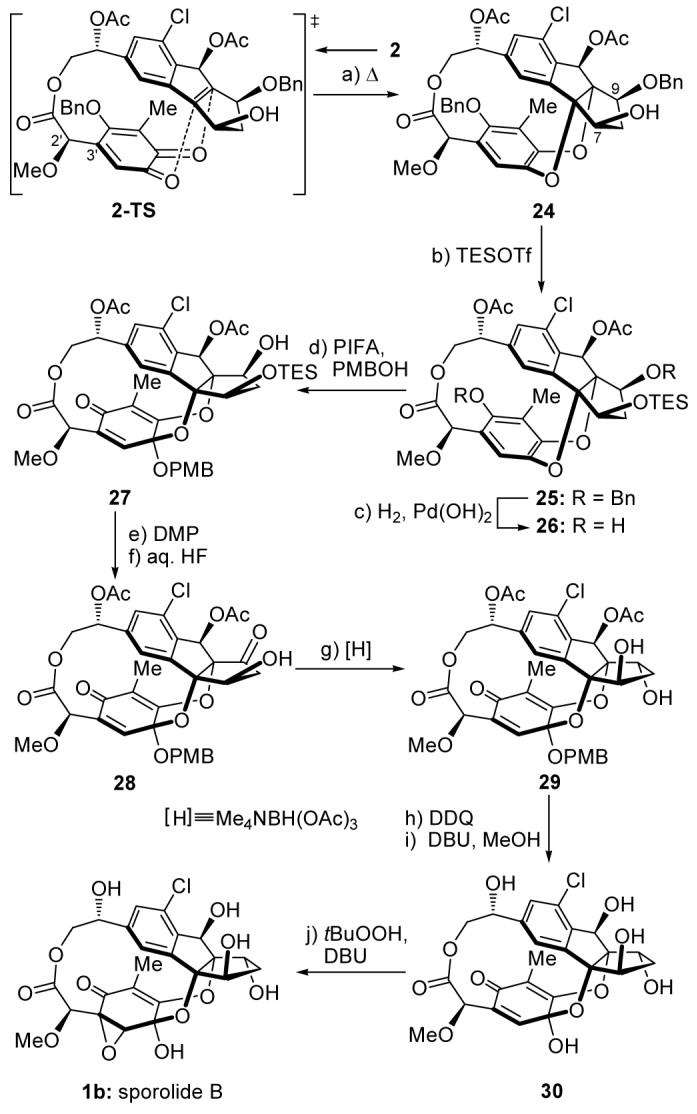
Completion of the total synthesis of sporolide B (1b). Reagents and conditions: a) toluene, 110 °C, 1.5 h, 40 % (based on 50 % recovered starting material); b) TESOTf (1.5 equiv), Et3N (2.0 equiv), CH2Cl2, 0 °C, 30 min, 95 %; c) H2 (balloon pressure), Pd(OH)2 (10 % on carbon, 2.0 equiv), EtOAc, 25 °C, 4 h, 92 %; d) PIFA (1.5 equiv), PMBOH (10 equiv), K2CO3 (5.0 equiv), MeCN, 0 °C, 30 min, 75 %; e) DMP (2.0 equiv), CH2Cl2, 25 °C, 1 h, 90 %; f) HF (48 % aqueous solution, excess), MeCN, 25 °C, 2 h, 85 %; g) Me4NBH(OAc)3 (10 equiv), MeCN/AcOH (10:1), 25 °C, 2 h, 85 %; h) DDQ (5.0 equiv), CH2Cl2/H2O (10:1), 25 °C, 5 h, 70 %; i) DBU (10 equiv), CH2Cl2/MeOH (3:1), 40 °C, 4 h, 78 %; j) tBuOOH (10 equiv), DBU (5.0 equiv), CH2Cl2, 40 °C, 3 h, 63 %. DBU = 1,8-diazabicyclo[5.4.0]undec-7-ene, DDQ = 2,3-dichloro-5,6-dicyano-1,4-benzoquinone, PIFA = phenyliodine(III) bis(trifluoroacetate), PMB = 4-methoxybenzyl, Tf = trifluoromethanesulfonyl.
The described regio- and stereocontrolled total synthesis of sporolide B (1b) demonstrates the power of the ruthenium-catalyzed intermolecular [2+2+2] cycloaddition reaction of acetylenic substrates and provides further insight into possible biosynthetic pathways to this novel secondary metabolite and its regioisomeric sibling sporolide A (1a).
Supplementary Material
Acknowledgments
We thank Prof. William Fenical for a sample of natural sporolide B.
Footnotes
We thank Drs. D. H. Huang and G. Siuzdak for NMR spectroscopic and mass spectroscopic assistance, respectively. We thank Dr. Randy Hungate and Randy Jensen of Amgen, Thousand Oaks, California, for the 13C NMR spectrum of synthetic sporolide B. Financial support for this work was provided by the National Institutes of Health (USA), the Skaggs Institute for Research and an Andrea Elizabeth Vogt Memorial Fellowship (to J.W.).
References
- [1].Buchanan GO, Williams PG, Feling RH, Kauffman CA, Jensen PR, Fenical W. Org. Lett. 2005;7:2731–2734. doi: 10.1021/ol050901i. [DOI] [PubMed] [Google Scholar]
- [2].a) Fenical W, Jensen PR. Nature Chem. Biol. 2006;2:666–673. doi: 10.1038/nchembio841. [DOI] [PubMed] [Google Scholar]; b) Perrin CL, Rodgers BL, O’Connor JM. J. Am. Chem. Soc. 2007;129:4795–4799. doi: 10.1021/ja070023e. [DOI] [PubMed] [Google Scholar]
- [3].a) Udwary DW, Zeigler L, Asolkar RN, Singan V, Lapidus A, Fenical W, Jensen PR, Moore BS. Proc. Natl. Acad. Sci. U. S. A. 2007;104:10376–10381. doi: 10.1073/pnas.0700962104. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) McGlinchey RP, Nett M, Moore BS. J. Am. Chem. Soc. 2008;130:2406–2407. doi: 10.1021/ja710488m. [DOI] [PubMed] [Google Scholar]
- [4].Jones RR, Bergman RG. J. Am. Chem. Soc. 1972;94:660–661. [Google Scholar]
- [5].a) Nicolaou KC, Dai W-M. Angew. Chem. 1991;103:1453–1481. [Google Scholar]; Angew. Chem. Int. Ed. Engl. 1991;30:1387–1416. [Google Scholar]; b) Nicolaou KC, Smith AL, Yue EW. Proc. Natl. Acad. Sci. U. S. A. 1993;90:5881–5888. doi: 10.1073/pnas.90.13.5881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Nicolaou KC, Wang J, Tang Y. Angew. Chem. 2008;120:1454–1457. [Google Scholar]; Angew. Chem. Int. Ed. 2008;47:1432–1435. doi: 10.1002/anie.200705334. [DOI] [PubMed] [Google Scholar]
- [7].a) Yamamoto Y, Ogawa R, Itoh K. Chem. Commun. 2000:549–550. [Google Scholar]; b) Yamamoto Y, Arakawa T, Ogawa R, Itoh K. J. Am. Chem. Soc. 2003;125:12143–12160. doi: 10.1021/ja0358697. [DOI] [PubMed] [Google Scholar]
- [8].a) Johnson CR, Braun MP. J. Am. Chem. Soc. 1993;115:11014–11015. [Google Scholar]; b) Curran TT, Hay DA, Koegel CP. Tetrahedron. 1997;53:1983–2004. [Google Scholar]
- [9].Luche JL. J. Am. Chem. Soc. 1978;100:2226–2227. [Google Scholar]
- [10].Diastereomeric mixtures were taken through the sequences as such until conveniently separated at certain stages as noted in the text; yields after the [4+2] cycloaddition reaction refer to diastereomerically pure compound.
- [11].Dess DB, Martin JC. J. Org. Chem. 1983;48:4155–4156. [Google Scholar]
- [12].The C-11 stereochemistries of this alcohol and its epimer were determined by NMR spectroscopic analysis of tricyclic indene derivatives prepared by a sequence involving a ruthenium-catalyzed [2+2+2] cycloaddition reaction with a simple terminal acetylene [(S)-PMBOCH2CH(OTBS)C≡CH].
- [13].Saito N, Tashiro K, Maru Y, Yamaguchi K, Kubo A. J. Chem. Soc., Perkin Trans. 1. 1997;1:53–69. [Google Scholar]
- [14].Sharpless KB, Amberg W, Bennani YL, Crispino GA, Hartung J, Jeong KS, Kwong HL, Morikawa K, Wang ZM. J. Org. Chem. 1992;57:2768–2771. [Google Scholar]
- [15].Yadav JS, Chander MC, Joshi BV. Tetrahedron Lett. 1988;29:2737–2740. [Google Scholar]
- [16].a) Oshima N, Suzuki H, Moro-oka Y. Chem. Lett. 1984;13:1161–1164. [Google Scholar]; b) Yamamoto Y, Hattori K. Tetrahedron. 2008;64:847–855. [Google Scholar]
- [17].Heating for longer time or at lower temperatures resulted in significant decomposition or lower yields. Lewis acids failed to improve the outcome of this reaction.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.