

Abstract
The activation of signal transducer and activator of transcription 3 (STAT3) has been identified as a key mediator that drives the fundamental components of melanoma malignancy, including immune suppression in melanoma patients. Increasing evidence also suggests that regulatory T cells (Tregs) are important in suppressing anti-tumor immunity and play a dominant role in negating efficacious immunotherapy approaches. We hypothesized that WP1066, a novel inhibitor of STAT3 signaling, reverses immune suppression through the inhibition of Tregs and that this contributes to the antitumor activity of this agent against melanoma brain metastases. We found that the mean percentage of peripheral blood mononuclear cells expressing phosphorylated STAT3 (p-STAT3) was significantly elevated in samples from patients with melanoma brain metastases compared to healthy donors, 16.13 ± 2.48% versus 4.17 ± 1.79%. The p-STAT3 inhibitor WP1066 enhanced CD3+ (which contained Tregs) but not CD8+ T cell cytotoxicity against human A375 melanoma cells, indicating that this p-STAT3 blockade agent did not directly activate CD8+ T cells. Furthermore, the p-STAT3 inhibitor did not enhance the cytotoxicity of CD3+CD25− T cells (from which Tregs were excluded), indicating that the enhanced cytotoxicity of WP1066 is secondary to its inhibition of Tregs. This was confirmed by demonstrating that WP1066 inhibited FoxP3+ Treg induction in a dose-dependent manner. Moreover, CD3+ T cells exhibited markedly enhanced levels of phosphorylated ZAP-70, a critical proximal signal in T cell activation, after exposure to WP1066. Similar effects were not observed in Treg-depleted CD3+CD25− T cell populations, confirming that the T cell activation by WP compounds is secondary to their inhibition of the Tregs. These results suggest that WP1066 enhances T cell cytotoxicity against melanoma through inhibition of Tregs.
Keywords: STAT3, STAT3 inhibitors, Melanoma, Cytotoxic T cells, Regulatory T cells, Central nervous system
Introduction
Upon metastasis to the central nervous system (CNS), melanoma is associated with a median survival time of less than 1 year. The development of new effective therapies for metastatic melanoma to the CNS is a major unmet clinical need. Signal transducer and activator of transcription 3 (STAT3) is a clinically significant latent transcription factor whose phosphorylation results in its activation, its nuclear translocation, and the upregulation of various oncogenic gene products [16]. Phosphorylated STAT3 (p-STAT3) serves as an intersection point for many upstream pathways including interleukin (IL)-6, which is expressed in the CNS under a wide variety of conditions [9, 19, 32], that activate Jak2 then STAT3 by phosphorylation of the tyrosine residue in the transactivation domain of STAT3 [20]. Aberrant or overactive STAT3 is correlated with tumor cell proliferation, abnormal cell cycling, vascular endothelial growth factor overproduction and angiogenesis, impaired apoptotic mechanisms [16], and, of critical importance, immune escape. This last process, by which tumor cells are able to avoid detection and destruction by a patient’s immune system, promotes tumor aggression, malignancy, and metastasis [12, 23, 30, 33].
STAT3-mediated immune escape is particularly relevant in cases of metastatic melanoma not only due to the strong correlation between p-STAT3 expression and melanoma malignancy, but also because of the potential susceptibility of melanoma to immunotherapy [26]. In human melanoma patients, constitutive STAT3 was observed in the majority of tested melanoma cell lines and primary tumors, but not in normal skin specimens matched from the same patients [22] or from compound or dysplastic nevus tissues [37]. A study by Xie et al. [35] confirmed the importance of STAT3 in melanoma metastasis by showing that only highly metastatic melanoma cell lines, not poorly metastatic ones, have elevated levels of p-STAT3. In addition, blockade of p-STAT3 by the expression of dominant-negative STAT3 significantly suppresses matrix metalloproteinase 2 (MMP-2) expression and the invasiveness of melanoma cells, inhibits tumor growth, and prevents metastasis in nude mouse model systems [35]. A subsequent study of brain metastasis in humans with melanoma have demonstrated high levels of activated STAT3 more commonly in brain metastasis specimens than in parenchymal tumors, indicating that p-STAT3 is a key enhancer of malignancy in melanoma [34].
p-STAT3 also becomes upregulated within the immune cells upon association with a variety of cancers [17]. Mice with ablation of Stat3 in their hematopoietic cells have marked antitumor clearance by the immune system and a reduction in the number of tumor-infiltrating regulatory T cells (Tregs) [17]. Further, STAT3 is required for both transforming growth factor (TGF)-β and IL-10 production by CD4+ T cells [14], factors necessary for the generation of tumor-associated Tregs. In addition, IL-2 has been shown to regulate FoxP3 expression in human CD4+CD25+ Tregs by STAT3 binding of the first intron of the FoxP3 gene [38]. Thus, STAT3 is a key signaling molecule for the generation of the immune suppressive Treg component in patients with cancer. Furthermore, CD3+CD25+FoxP3+ Tregs are present within CNS melanoma metastases and peritumoral tissue [15].
We previously showed that WP1066, a novel small molecule inhibitor of p-STAT3, induces phosphorylated ZAP-70 (p-ZAP-70; Tyr319), a critical signal in T cell activation, and increases T cell proliferation [13]. We hypothesized that STAT3 blockade agents could enhance T cells’ cytotoxic activity against melanoma through the inhibition of Tregs and that this would contribute to the antitumor activity of this agent against melanoma brain metastases. To investigate how p-STAT3 inhibitors act upon T cells from immune suppressed melanoma patients, T cells from melanoma brain metastasis patients were isolated and analyzed for their cytotoxicity against melanoma cells with and without the presence of Tregs. Delineating the mechanism of activity of STAT3 inhibitors is crucial since these compounds could be utilized as immune therapeutics, especially for cancers within the CNS.
Materials and methods
Cell line
The human melanoma cell line A375 was provided by Dr. Elizabeth Grimm (The University of Texas M. D. Anderson Cancer Center, Houston, TX) and was maintained in RPMI 1640 medium supplemented with 10% fetal bovine serum at 37°C in a humidified atmosphere of 5% CO2 and 95% air and was free of mycoplasma contamination.
STAT3 inhibitor
WP1066 [11, 24], which blocks p-STAT3 [21, 25], was synthesized and supplied by Dr. Waldemar Priebe (M. D. Anderson). WP1066 was dissolved in dimethylsulfoxide (DMSO, Sigma-Aldrich, St Louis, MO) as stock solution and serially diluted to the desired concentration with RPMI 1640 medium. The final concentration of DMSO in cell culture systems was <0.05%.
Human subjects
Patients with brain metastases from melanoma who were to undergo surgical resection were recruited for the study. Peripheral blood was drawn from the patients intraoperatively. Peripheral blood was also drawn from healthy volunteer donors. The melanoma metastasis, after resection and verification of pathology by a neuropathologist, was transported on ice from the operating room and was frozen immediately upon arriving at the laboratory for protein isolation and Western blot analysis. This study was approved by the institutional review board of The University of Texas M. D. Anderson Cancer Center (LAB03-0687), and written informed consent was obtained.
Isolation of peripheral blood mononuclear cells and staining for p-STAT3 expression
Blood samples were mixed with an equal volume of sterile phosphate-buffered saline (PBS) and subjected to density gradient centrifugation using Ficoll-Paque (Amersham Biosciences, Piscataway, NJ). Peripheral blood mononuclear cells (PBMCs) were isolated and washed twice with PBS. For fixation, 5 × 106 cells were resuspended in 0.5 ml of PBS, 37°C pre-warmed paraformaldehyde was added to achieve a final concentration of 1%, and the solution was incubated for 10 min at 37°C and then chilled on ice for 1 min. For permeabilization, the paraformaldehyde was removed by pelleting the cells at 1,500 rpm for 5 min and resuspending them in 90% methanol, and then the cells were incubated for 30 min on ice and pelleted at 1,500 rpm for 5 min; 1 × 106 cells were seeded in duplicate into wells of 96 v-bottom well plates, centrifuged for 5 min at 1,500 rpm, and washed once with fluorescence-activated cell sorter (FACS) buffer (PBS with 0.5% bovine serum albumin [BSA]) for 5 min at 1,500 rpm. The cells were resuspended in 45 μl of FACS buffer/well and 5 μl of mouse phycoerythrin (PE)-labeled anti-human p-STAT3 (Y705) antibody (BD Biosciences, San Jose, CA). Matched control wells included 5 μl of PE-labeled IgG2a-κ isotype control (eBioscience, San Diego, CA). The cells were incubated for 60 min at room temperature, washed with 200 μl of FACS buffer/well, centrifuged for 5 min at 1,500 rpm, resuspended in 250 μl of FACS buffer/well, and transferred to FACS tubes for flow analysis with FACSCalibur (BD Biosciences).
T cell enrichment
PBMCs from healthy donors were pelleted and resuspended in FACS buffer and 2 mM EDTA to achieve a final cell concentration of 1 × 107 cells/ml. The Pan T cell isolation kit II protocol (Miltenyi Biotec, Auburn, CA) was utilized as specified in the kit instructions. Briefly, 1 μl of T cell biotin-antibody cocktail (Miltenyi Biotec) was added to the PBMCs suspended in FACS buffer, and the solution was gently vortexed and incubated at 4°C for 10 min. Thereafter, 1 μl of anti-biotin microbeads (Miltenyi Biotec) per 106 cells was added. The cells were vortexed, incubated at 4°C for 15 min, and then washed and resuspended in FACS buffer to achieve a final concentration of 2 × 108 cells/ml. The T cells were fractionated using negative-selection LS columns (Miltenyi Biotec) loaded with 2 × 108 PBMCs per column. The T cell fractions were pooled and counted, spun down, and resuspended in RPMI 1640 medium to be used for purification of T cell subpopulations and cytotoxicity assays.
Purification of T cell subpopulations
T cells suspended in RPMI 1640 medium were labeled with 75 μl each of PE-conjugated anti-CD3, allophycocyanin (APC)-conjugated anti-CD8, and fluorescein isothiocyanate (FITC)-conjugated anti-CD25 antibodies (Miltenyi Biotec) per 1 × 108 cells. After incubation at 4°C for 40 min, the labeled cells were washed twice with FACS buffer and resuspended in FACS buffer with 1 mM EDTA to achieve a final concentration of 4 × 107 cells/ml. The cells were then passed through a 40-μm cell strainer. Single-color samples were made for calibration, and the CD3+, CD3+CD25−, and CD8+ T cells were sorted on a FACSAria Cell Sorter (BD Biosciences). Since the intracellular staining process for FoxP3 in Tregs would permeabilize the membrane and affect Treg viability and subsequent response to the STAT3 blockade agents, we elected to obtain Treg-depleted CD3 T cells (CD3+CD25−) by sorting Tregs (CD3+CD25+) out from the CD3+ population. Functional Tregs have the surface phenotype CD3+CD25+; thus, the presence of the Tregs should be minimized in this post-sorted cell population.
T cell cytotoxicity assay against melanoma cells
A375 cells in RPMI 1640 medium were cultured for 3 days, trypsinized, pelleted, and resuspended in FACS buffer at room temperature to achieve a concentration of 106 cells/ml. Carboxy–fluorescein diacetate succinimidyl ester (CFSE) stock solution (CellTrace CFSE Cell Proliferation Kit; Invitrogen, Eugene, OR) was added to achieve a final concentration of 4 μM, the mixture was incubated at 37°C for 10 min, and then the staining reaction was quenched by the addition of five volumes of ice-cold PBS for 5 min. The A375 cells were washed 3 times in RPMI 1640 medium and plated for the cytotoxicity assay. The ratio of CD3+ or CD3+CD25− T effector cells to A375 target cells was 3.5:1 when using cells from healthy donors and 10:1 when using cells from melanoma brain metastasis patients because of immune incompetence in cancer patients. Treatment groups consisted of A375 target cells alone; A375 cells in the presence of 2 μM WP1066; A375 cells with CD3+ or CD3+CD25− T cells; A375 cells with CD3+ or CD3+CD25− T cells in the presence of 2 μM WP1066. In another group of experiments, the CD8+ T effector cell to A375 target cell ratio was 5:1. Treatment groups consisted of A375 target cells alone; A375 cells in the presence of 2 μM WP1066; CD8+ T cells with A375 cells; and CD8+ T cells with A375 cells in the presence of 2 μM WP1066. After 48 h of incubation, the CFSE-labeled A375 melanoma cells were removed from the plates with trypsin and analyzed by FACS. The A375 cells were stained with propidium iodide (PI; BD Biosciences) to distinguish viable cells from nonviable cells. A375 cells that were stained with CFSE and PI were considered nonviable. Flow cytometry acquisition of the A375 target cells was done with a FACS Calibur flow cytometer (BD Biosciences), and data analysis was done using FlowJo software (TreeStar, Ashland, OR).
Immunoblotting analysis
Resected melanoma brain metastases were used for protein isolation and immunoblotting analysis as described below. PBMCs, CD3+ T cells, and CD3+CD25− T cells from healthy donors and melanoma brain metastasis patients were purified as described above. PBMCs, CD3+ T cells, or CD3+CD25− T cells were seeded at a density of 1 × 106 cells/well in 6-well culture plates and incubated at 37°C, in an atmosphere containing 5% CO2, with the RPMI 1640 medium in absence or presence of 5 μM WP1066. After 2 h, the PBMCs were further cultured in absence or presence of 10 ng/ml of IL-6 for 15 min. Afterwards, all above cells were pelleted and rinsed with ice-cold PBS at 1,500 rpm for 5 min. The cells were lysed for 30 min in ice-cold lysis buffer [50 mM Tris–HCl (pH 8.0), 150 mM NaCl, 1 mM EDTA] containing 1% Triton-X-100 and phosphatase and protease inhibitors (Sigma-Aldrich). The lysates were centrifuged at 14,000 rpm for 10 min at 4°C. The supernatants were collected and quantified for protein content. Equal amounts of proteins (20 μg) were electrophoretically fractionated in 8% sodium dodecyl sulfate (SDS)-polyacrylamide gels, transferred to nitrocellulose membranes, and subjected to immunoblot analysis with specific antibodies against p-STAT3 (Tyr705), STAT3, p-ZAP-70 (Tyr319), ZAP-70 (Cell Signaling Technology, Inc., Danvers, MA), and β-actin (Sigma-Aldrich). Autoradiography of the membranes was performed using Amersham ECL Western blotting detection reagents (Amersham Biosciences). The densities of the protein bands compared to the β-actin protein control were measured with Image J program provided by the NIH (http://rsb.info.nih.gov/ij/index.html).
Induction Treg assays
Because of limitations on blood sample volumes that can be obtained from patients undergoing surgery as well as the low percentage of Tregs in PBMCs, it was impractical to isolate sufficient Tregs for analysis of their response to STAT3 blockade in melanoma brain metastasis patients. To assess whether WP1066 is capable of inhibiting Tregs, especially since this immune population is a key factor mediating immune suppression in cancer patients [6], including melanoma patients, we used an in vitro Treg induction system in which FoxP3 expression was induced in naïve CD4+ T cells isolated from mice whose spleens can provide a far more enriched source of naïve CD4+ cells than human blood samples, which must be limited to volumes insufficient for CD4+CD25−CD62Lhi extraction. FoxP3+ Treg generation was directly measured by intracellular staining for FoxP3 protein expression. In this system, naïve CD4+ T cells underwent robust FoxP3+ Treg differentiation when they were activated by polyclonal stimulation in the presence of exogenous TGF-β.
Single cell suspensions were prepared from pooled lymph nodes and spleens of C57BL/6 J mice (MD Anderson Cancer Center, Houston, TX). The CD4+ cells were enriched using CD4 MACS beads (Miltenyi Biotec) and were stained with PerCP (peridinin–chlorophyll–protein complex)-labeled anti-CD4 (L3T4), FITC-labeled anti-CD62L (MEL-14), and APC-labeled anti-CD25 (PC61) antibodies (Miltenyi Biotec). The cells were further sorted on a FACSAria cell sorter (BD Biosciences) to obtain pure populations of naïve CD4+CD25−CD62Lhi T cells. Naïve CD4+CD25−CD62Lhi cells were then incubated on anti-CD3-coated plastic plates (2 μg/ml) in complete RPMI 1640 medium with soluble anti-CD28 (2 μg/ml) antibody and rTGF-β1 (1 ng/ml) to induce FoxP3+ Tregs. The cultures were supplemented with WP1066 at concentrations of 0, 1, 2, and 4 μM for 48 h. PerCP-conjugated anti-CD4 (L3T4) and APC-conjugated anti-CD25 (PC61) monoclonal antibodies were used for cell surface staining. To detect FoxP3 protein expression, the surface-stained cells were further subjected to intracellular staining with PE-conjugated monoclonal antibodies to FoxP3 (clone FJK-16s; eBioscience) using the staining buffers and conditions specified by the manufacturer.
Statistical analysis
Data are presented as mean ± standard errors (SEs) of three repeated experiments. Student’s t test was performed. A P value below 0.05 was considered statistically significant.
Results
Expression of p-STAT3 is enhanced in PBMCs from melanoma brain metastasis patients and can be blocked with WP1066
Melanoma brain metastases commonly express p-STAT3 (Fig. 1a). Since p-STAT3 expressing tumors have been shown to induce p-STAT3 expression in tumor associated immune cells and since these immune cells can regain access back to the peripheral circulation, we next determined if the PBMCs had increased expression of p-STAT3. PBMCs were isolated from both healthy donors (n = 14) and melanoma brain metastasis patients (n = 10) and analyzed via FACS for p-STAT3 levels. The percentage of p-STAT3-expressing PBMCs from melanoma patients (16.13 ± 2.48%) was significantly elevated compared to the mean from healthy donors (4.17 ± 1.79%, P < 0.05) (Fig. 1b). Of note, the expression of p-STAT3 is lost within 24 h in PBMCs (data not shown). We next determined if the p-STAT3 inhibitor WP1066 could down modulate both constitutive and induced expression of p-STAT3 in PBMCs. In order to recapitulate the CNS biology of PBMCs within the CNS, IL-6 was added. By scanning densitometry, in comparison to respective non-treated controls, WP1066 significantly reduced the levels of both constitutive and IL-6-induced p-STAT3 by 52.4 and 61.5%, respectively. Regardless of the systemic or CNS microenvironment, WP1066 was capable of down modulating p-STAT3 signaling (Fig. 1c).
Fig. 1.

Expression of p-STAT3 is enhanced in PBMCs from melanoma brain metastasis patients and is blocked with WP1066. a Melanoma brain metastases express p-STAT3. The resected melanoma metastases were lysed, electrophoretically fractionated in 8% SDS-polyacrylamide gels, transferred to nitrocellulose membranes, and immunoblotted with antibodies to p-STAT3 (Tyr705), STAT3, or β-actin. b Expression of p-STAT3 is enhanced in PBMCs from melanoma brain metastasis patients. PBMCs were isolated from blood samples obtained from healthy donors (n = 14) and melanoma brain metastasis patients (n = 10), fixed in paraformaldehyde, permeabilized, stained with mouse PE-labeled anti-human p-STAT3 (Y705) antibody, and analyzed by FACS. The percentage of p-STAT3-positive PBMCs differed significantly (P < 0.05) between healthy donors and melanoma brain metastasis patients. c WP1066 inhibits p-STAT3 in PBMCs from melanoma brain metastasis patients. The PBMCs were incubated with either the RPMI 1640 medium alone or supplemented with WP1066 in the presence or absence of 10 ng/ml of IL-6. Subsequently, cells were lysed, electrophoretically fractionated in 8% SDS-polyacrylamide gels, transferred to nitrocellulose membranes, and immunoblotted with antibodies to p-STAT3 (Tyr705), STAT3, or β-actin
STAT3 inhibitor WP1066 enhances CD3+ T cell cytotoxicity against melanoma
The ratio of CD3+ or CD3+CD25−T effector cells to A375 target cells was 3.5:1 when using cells from healthy donors and 10:1 when using cells from melanoma brain metastasis patients because of immune incompetence in cancer patients. WP1066 markedly enhanced the cytotoxicity of CD3+ T cells from healthy donors, 26.3 ± 3.1% versus 13.4 ± 0.6% (P < 0.05) (Fig. 2a), and melanoma brain metastasis patients, 24.1 ± 2.1% versus 15.2 ± 1.0% (P < 0.05) (Fig. 2b), against human melanoma A375 cells, indicating that this compound can enhance T cell cytotoxicity. Minimal apoptosis of A375 cells, 2.7 ± 0.2%, was seen when 2 μM of WP1066 (below the IC50) was used alone (Fig. 2).
Fig. 2.

WP1066 enhances the cytotoxicity of CD3+ T cells against human melanoma A375 cells. A375 cells were labeled with CFSE and exposed to multiple experimental conditions in the presence of CD3+ cells from healthy donors (Fig. 2a, effector:target cell ratio = 3.5:1) and melanoma brain metastasis patients (Fig. 2b, effector:target cell ratio = 10:1). A375 cells were grown in triplicate cultures in RPMI 1640 medium alone or supplemented with 2 μM of WP1066, CD3+ T cells, or CD3+ T cells plus 2 μM of WP1066. Flow cytometry was used to determine the percentage of PI-positive cells for each experimental condition. *P < 0.05 in comparison with the CD3+ PBMC group. These experiments were reproduced four times with similar results
WP1066 does not primarily activate CD8+ T cells
To determine whether the STAT3 blockade agent WP1066 directly activates CD8+ T cells, A375 cytotoxicity assays were performed in various combinations with CD8+ T cells obtained from healthy donors as well as from melanoma brain metastasis patients. WP1066 did not enhance the cytotoxicity against human melanoma A375 cells by CD8+ cells obtained from either healthy donors or melanoma brain metastasis patients (Fig. 3).
Fig. 3.

The effect of WP1066 on the cytotoxicity of CD8+ T cells against human melanoma A375 cells. A375 cells labeled with CFSE were exposed to multiple experimental conditions in the presence or absence of CD8+ T cells from healthy donors (effector:target cell ratio = 5:1). Cells were grown in triplicate cultures in RPMI 1640 medium alone or supplemented with 2 μM of WP1066, CD8+ T cells, or CD8+ T cells plus 2 μM of WP1066. Flow cytometry was used to determine the percentage of PI-positive cells for each experimental condition. There were no statistically significant differences between the CD8+ T cell group and CD8+ T cell plus WP1066 group. These experiments were reproduced four times with similar results
Depletion of Tregs abolishes WP1066-enhanced CD3+ T cell cytotoxicity against melanoma
Since there was no direct enhancement of CD8+ T cell cytotoxicity by WP1066, we suspected that the mechanism by which STAT3 blockade enhanced the cytotoxicity of CD3+ populations could involve the inhibition of immunosuppressive agents, such as Tregs, within that population. The CD3+CD25− population (Treg depleted) was assayed for cytotoxicity against human melanoma A375 cells with and without the WP1066 (Fig. 4). As before, 2 μM of WP1066 by itself did not increase apoptosis in human melanoma A375 target cells. Furthermore, WP1066 did not enhance the cytotoxicity of CD3+CD25− T cells from healthy donors, 17.1 ± 0.5% versus 18.7 ± 0.2% (P > 0.05) (Fig. 4a), or melanoma brain metastasis patients, 12.1 ± 0.9% versus 13.8 ± 0.6% (P > 0.05) (Fig. 4b), indicating that the enhanced cytotoxicity of WP1066 observed with the CD3+ T cells (Fig. 2) likely derives from the ability of WP1066 to inhibit Tregs.
Fig. 4.

Depletion of Tregs eliminates WP1066-enhanced CD3+ T cell cytotoxicity against human melanoma A375 cells. The experiment in Fig. 2 was repeated with CD3+CD25−T cells after depleting Tregs (CD3+CD25+) with a FACSAria cell sorter. A375 cells were labeled with CFSE and exposed to multiple experimental conditions in the presence or absence of CD3+CD25− cells from healthy donors (Fig. 3a, effector:target cell ratio = 3.5:1) and melanoma brain metastasis patients (Fig. 3a, effector:target cell ratio = 10:1). Cells were grown in triplicate cultures in RPMI 1640 medium alone or supplemented with 2 μM of WP1066, CD3+CD25− T cells, or CD3+CD25− T cells plus 2 μM of WP1066. Flow cytometry was used to determine the percentage of PI-positive cells for each experimental condition. *P < 0.05 in comparison with the CD3+CD25− PBMC group. These experiments were reproduced four times with similar results
Depletion of Tregs abolishes WP1066-enhanced phosphorylation of ZAP-70 in CD3+ T cells
Since the phosphorylation of ZAP-70 is immediately downstream of the T cell receptor but proximal to other markers of T cell activation including STAT3, we examined the effects of WP1066 on p-ZAP-70 in CD3+ T cells with and without the presence of Tregs from healthy donors as well as melanoma brain metastasis patients. After exposure to WP1066, the CD3+ T cells (that would contain Tregs) from the healthy donors and the melanoma brain metastasis patients, demonstrated a 78.8 and 56% enhancement of p-ZAP-70, respectively (Fig. 5). However, WP1066 did not enhance p-ZAP-70 in the CD3+CD25−T cells (that would lack Tregs), indicating that WP1066 enhancement of p-ZAP-70 is secondary to the inhibition of Tregs (Fig. 5).
Fig. 5.

Depletion of Tregs abolishes WP1066-enhanced phosphorylation of ZAP-70 (p-ZAP-70) in CD3+ T cells. CD3+ and CD3+CD25− T cells were isolated from the PBMCs of healthy donors, and melanoma brain metastasis patients. The cells were incubated with either the RPMI 1640 medium alone or supplemented with WP1066. Subsequently, cells were lysed, electrophoretically fractionated in 8% SDS-polyacrylamide gels, transferred to nitrocellulose membranes, and immunoblotted with antibodies to p-ZAP-70 (Tyr319), ZAP-70, phosphorylated AKT (p-AKT: Ser473), AKT, or β-actin. These experiments were reproduced four times with similar results
WP1066 markedly inhibits inducible Tregs
To determine the effects of WP1066 on the de novo synthesis of Tregs, we tested WP1066 in an in vitro Treg induction assay. WP1066 inhibited FoxP3+ Treg induction in a dose-dependent manner, with 70.6% of inhibition at 2.0 μM concentration (Fig. 6), which is achievable in vivo [13]. Thus, inhibition of p-STAT3 can diminish the induction of Tregs, which may contribute to the antitumor responses observed with the use of STAT3 blockade agents in vivo.
Fig. 6.
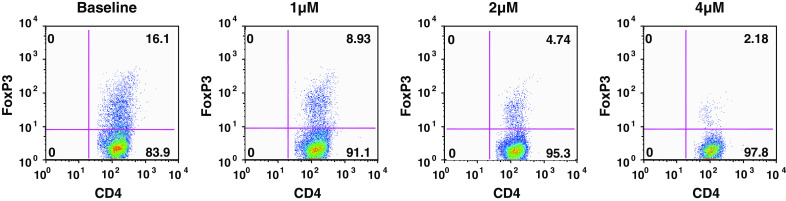
WP1066 inhibits inducible Tregs. CD4+ cells enriched from pooled lymph nodes and spleens of C57BL/6 J mice were stained with PerCP-labeled anti-CD4 (L3T4), FITC-labeled anti-CD62L (MEL-14), and APC-labeled anti-CD25 (PC61) antibodies. A FACSAria cell sorter was used to obtain naïve CD4+CD25− CD62Lhi T cells, which were stimulated with anti-CD3 antibody (2 μg/ml), anti-CD28 antibody (2 μg/ml), and rTGF-β1 (1 ng/ml) to induce FoxP3+ Tregs. The cultures were supplemented with WP1066 at the concentrations shown for 48 h, then stained with PerCP-conjugated anti-CD4 and APC-conjugated anti-CD25 antibodies, permeabilized, and stained with PE-conjugated mAbs to FoxP3. Tregs (high CD4, high FoxP3) are plotted as the percentage of overall T cells for each experimental condition
Discussion
Clarification of the mechanisms underlying p-STAT3-mediated immune suppression will assist in the development of adjuvant immunotherapies for metastatic melanoma but is complicated by the fact that p-STAT3 interacts with the immune system in multiple ways. Mechanisms of immune evasion mediated by p-STAT3 include down regulation of the functional activity of antigen-presenting cells such as dendritic cells, including decreased expression of MHC class I molecules [17]; inhibition of apoptotic signal receptors; down modulation of co-stimulation; production of immunosuppressive cytokines [1, 29]; induction of TGF-β, and IL-10 [5, 36]; and an increase of Tregs, which possess immunosuppressive activity towards antigens from the self, infectious agents, and tumors [4, 16, 30]. Tregs are abundantly present within melanoma metastasis to the CNS [15], and this likely contributes to the immunosuppression and tumor immune evasion that counteract immunological clearance of the tumor. Our data demonstrate that the p-STAT3 inhibitor WP1066 inhibits Tregs and enhances the cytotoxicity of T cells from melanoma brain metastasis patients against melanoma and that the antitumor activity of WP1066 derives from its ability to inhibit Tregs.
We previously showed [13] that p-STAT3 blockade agents such as WP1066 can induce a cytotoxic T cell response and the phosphorylation of ZAP-70, a proximal signal directly associated with the activation of the T cell receptor/CD3 complex. Because Western blot analysis of key T cell receptor signaling pathways (MEKK/JNK, Raf/ERK, and NIK/NFKB) did not identify a mechanism by which the STAT3 blockade directly activated ZAP-70, we looked for an indirect means of activation. We suspected that because WP1066 can inhibit Tregs, activation of effector T cells may not necessarily result from direct interaction of STAT3 inhibitors and effector T cells but rather from inhibition of the immune inhibitory break (i.e., Tregs). In this report, we demonstrate that the increase in cytotoxicity against melanoma cells seen with the p-STAT3 inhibitors is attributable to the inhibition of Tregs rather than a direct effect on the cytotoxicity of T cells. Ideally, one would investigate the effects of STAT3 inhibition on human Treg activity, but this approach is problematic for several reasons. Given the low percentage of Tregs in human PBMCs, it is not possible to collect the volume of blood needed, especially during surgery, to isolate sufficient Tregs for both Western blot analysis and cytotoxicity assays under a multitude of experimental conditions from the same donor. Furthermore, cancer patients in general can have an overall decrease in the CD4 compartment [6]. Finally, because the consensus marker of Tregs, FoxP3, is intracellular that would require permeabilization for staining that would alter functional activity, we had to approach our analysis of STAT3 inhibitor interactions with Tregs using an indirect approach: looking at the effects of Treg depletion on whole T cell preparations. Western blotting experiments in which we depleted the CD25+ Treg-inclusive population indicated that the mechanism of T cell activation is secondary to inhibition of the Tregs. This is further supported by the inhibition of Tregs by STAT3 blockade in an in vitro induction assay. These findings are also consistent with the findings of others who have shown that Stat3 ablation in hematopoietic cells using the Mx1–Cre–loxP system is accompanied by a reduction in the number of tumor-infiltrating Tregs [17] and that IL-2 regulates FoxP3 expression in human CD4+CD25+ Tregs by STAT3 binding of the first intron of the FoxP3 gene [38]. Thus, the STAT3 blockade agents differ from other Treg modulatory agents like anti-CD25, which binds to the surface of Tregs and disrupts functional activity [8], and anti-CTLA4, which confers resistance to Treg-mediated immunosuppression but not through a direct effect on Tregs [7].
Our data provide a rationale to consider p-STAT3 targeting in the future in combination with other immunotherapies such as IFN-α, IL-2, granulocyte-macrophage colony-stimulating factor, monoclonal antibodies against tumor antigens, and anti-cancer vaccines [10, 26]. Additionally, because we have shown that STAT3 blockade has negligible inhibitory effects on effector T cell cytotoxicity against metastatic CNS melanoma, the unintended loss of CD8+ activity, which could potentially compromise immunological tumor clearance, can be minimized. IL-2, which induces favorable cytotoxic T cell responses, is approved by the US Food and Drug Administration to treat stage IV melanoma but has only a 15% objective response rate [2, 3, 28]. One factor underlying this poor response rate could be that a potent side effect of IL-2 therapy is the expansion of Tregs [38]. The synergistic activity of IL-2 and STAT3 blockade agents may overcome this limitation of IL-2 and yield a higher response rate, preserving the expansion of CD8+ cells while inhibiting the induction of Tregs. This is just one potential strategy of many that would rely upon inhibiting the immunosuppressive effects of Tregs via inhibition of p-STAT3. Because Tregs are present within CNS melanoma [15], the inhibitory effect of WP1066 on Tregs has clinical implications, especially considering that immunotherapeutic approaches for melanoma have so far been disappointing [27]. Because of their ability to cross the blood-brain barrier and establish physiologically relevant concentrations in the tumor microenvironment [13], the STAT3 blockade agents have great potential for the treatment of melanoma metastases to the CNS.
Acknowledgments
We are indebted to the patients and their families for participating in this study. We thank Adelina Fuentes and Melissa Burkett for their editorial assistance and Lamonne Crutcher for tissue acquisition. This work was supported by the Anthony Bullock III Foundation, an institutional research grant from The University of Texas M.D. Anderson Cancer Center, and National Institutes of Health grants to ABH (CA120813-01 and A177225-01) and to WP (SPORE in Melanoma, P50 CA093459).
Footnotes
L.-Y. Kong and J. Wei are the co-leader authors.
References
- 1.Ahmad M, Rees RC, Ali SA. Escape from immunotherapy: possible mechanisms that influence tumor regression/progression. Cancer Immunol Immunother. 2004;53:844–854. doi: 10.1007/s00262-004-0540-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Atkins MB, Kunkel L, Sznol M, Rosenberg SA. High-dose recombinant interleukin-2 therapy in patients with metastatic melanoma: long-term survival update. Cancer J Sci Am. 2000;6(Suppl 1):S11–S14. [PubMed] [Google Scholar]
- 3.Atkins MB, Lotze MT, Dutcher JP, Fisher RI, Weiss G, Margolin K, Abrams J, Sznol M, Parkinson D, Hawkins M, Paradise C, Kunkel L, Rosenberg SA. High-dose recombinant interleukin 2 therapy for patients with metastatic melanoma: analysis of 270 patients treated between 1985 and 1993. J Clin Oncol. 1999;17:2105–2116. doi: 10.1200/JCO.1999.17.7.2105. [DOI] [PubMed] [Google Scholar]
- 4.Chattopadhyay S, Chakraborty NG, Mukherji B. Regulatory T cells and tumor immunity. Cancer Immunol Immunother. 2005;54:1153–1161. doi: 10.1007/s00262-005-0699-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Darnell JE., Jr Transcription factors as targets for cancer therapy. Nat Rev Cancer. 2002;2:740–749. doi: 10.1038/nrc906. [DOI] [PubMed] [Google Scholar]
- 6.Fecci PE, Mitchell DA, Whitesides JF, Xie W, Friedman AH, Archer GE, Herndon JE, 2nd, Bigner DD, Dranoff G, Sampson JH. Increased regulatory T-cell fraction amidst a diminished CD4 compartment explains cellular immune defects in patients with malignant glioma. Cancer Res. 2006;66:3294–3302. doi: 10.1158/0008-5472.CAN-05-3773. [DOI] [PubMed] [Google Scholar]
- 7.Fecci PE, Ochiai H, Mitchell DA, Grossi PM, Sweeney AE, Archer GE, Cummings T, Allison JP, Bigner DD, Sampson JH. Systemic CTLA-4 blockade ameliorates glioma-induced changes to the CD4+ T cell compartment without affecting regulatory T-cell function. Clin Cancer Res. 2007;13:2158–2167. doi: 10.1158/1078-0432.CCR-06-2070. [DOI] [PubMed] [Google Scholar]
- 8.Fecci PE, Sweeney AE, Grossi PM, Nair SK, Learn CA, Mitchell DA, Cui X, Cummings TJ, Bigner DD, Gilboa E, Sampson JH. Systemic anti-CD25 monoclonal antibody administration safely enhances immunity in murine glioma without eliminating regulatory T cells. Clin Cancer Res. 2006;12:4294–4305. doi: 10.1158/1078-0432.CCR-06-0053. [DOI] [PubMed] [Google Scholar]
- 9.Gijbels K, Van Damme J, Proost P, Put W, Carton H, Billiau A. Interleukin 6 production in the central nervous system during experimental autoimmune encephalomyelitis. Eur J Immunol. 1990;20:233–235. doi: 10.1002/eji.1830200134. [DOI] [PubMed] [Google Scholar]
- 10.Heimberger AB, Crotty LE, Archer GE, Hess KR, Wikstrand CJ, Friedman AH, Friedman HS, Bigner DD, Sampson JH. Epidermal growth factor receptor VIII peptide vaccination is efficacious against established intracerebral tumors. Clin Cancer Res. 2003;9:4247–4254. [PubMed] [Google Scholar]
- 11.Heimberger AB, Priebe W, Fokt I, Szymanski S, Hussain SF, Kong L-Y (2007) Small molecule inhibitors for immune modulation. US 0/908, 559
- 12.Heinrich PC, Behrmann I, Haan S, Hermanns HM, Muller-Newen G, Schaper F. Principles of interleukin (IL)-6-type cytokine signalling and its regulation. Biochem J. 2003;374:1–20. doi: 10.1042/BJ20030407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hussain SF, Kong L-Y, Jordan J, Conrad C, Madden T, Fokt I, Priebe W, Heimberger AB. A novel small molecule inhibitor of signal transducers and activators of transcription 3 reverses immune tolerance in malignant glioma patients. Cancer Res. 2007;67:9630–9636. doi: 10.1158/0008-5472.CAN-07-1243. [DOI] [PubMed] [Google Scholar]
- 14.Kinjyo I, Inoue H, Hamano S, Fukuyama S, Yoshimura T, Koga K, Takaki H, Himeno K, Takaesu G, Kobayashi T, Yoshimura A. Loss of SOCS3 in T helper cells resulted in reduced immune responses and hyperproduction of interleukin 10 and transforming growth factor-beta 1. J Exp Med. 2006;203:1021–1031. doi: 10.1084/jem.20052333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kong LY, Abou-Ghazal MK, Wei J, Chakraborty A, Sun W, Qiao W, Fuller GN, Fokt I, Grimm EA, Schmittling RJ, Archer GE, Jr, Sampson JH, Priebe W, Heimberger AB. A novel inhibitor of STAT3 activation is efficacious against established central nervous system melanoma and inhibits regulatory T cells. Clin Cancer Res. 2008;14:5759–5768. doi: 10.1158/1078-0432.CCR-08-0377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kortylewski M, Jove R, Yu H. Targeting STAT3 affects melanoma on multiple fronts. Cancer Metastasis Rev. 2005;24:315–327. doi: 10.1007/s10555-005-1580-1. [DOI] [PubMed] [Google Scholar]
- 17.Kortylewski M, Kujawski M, Wang T, Wei S, Zhang S, Pilon-Thomas S, Niu G, Kay H, Mule J, Kerr WG, Jove R, Pardoll D, Yu H. Inhibiting Stat3 signaling in the hematopoietic system elicits multicomponent antitumor immunity. Nat Med. 2005;11:1314–1321. doi: 10.1038/nm1325. [DOI] [PubMed] [Google Scholar]
- 18.Kurdi M, Booz GW. Can the protective actions of JAK-STAT in the heart be exploited therapeutically? Parsing the regulation of interleukin-6-type cytokine signaling. J Cardiovasc Pharmacol. 2007;50:126–141. doi: 10.1097/FJC.0b013e318068dd49. [DOI] [PubMed] [Google Scholar]
- 19.Lau LT, Yu AC. Astrocytes produce and release interleukin-1, interleukin-6, tumor necrosis factor alpha and interferon-gamma following traumatic and metabolic injury. J Neurotrauma. 2001;18:351–359. doi: 10.1089/08977150151071035. [DOI] [PubMed] [Google Scholar]
- 20.Li B, Chang CM, Yuan M, McKenna WG, Shu HK. Resistance to small molecule inhibitors of epidermal growth factor receptor in malignant gliomas. Cancer Res. 2003;63:7443–7450. [PubMed] [Google Scholar]
- 21.Madden T, Kazerooni R, Myer J, Culotta K, Donato N, Johansen M, Kondo Y, Mack D, Priebe W (2006) The preclinical pharmacology of WP1066, a potent small molecule inhibitor of the JAK2/STAT3 pathway. In: Proceedings of the 97th American Association for Cancer Research Annual Meeting. Washington, DC
- 22.Niu G, Bowman T, Huang M, Shivers S, Reintgen D, Daud A, Chang A, Kraker A, Jove R, Yu H. Roles of activated Src and Stat3 signaling in melanoma tumor cell growth. Oncogene. 2002;21:7001–7010. doi: 10.1038/sj.onc.1205859. [DOI] [PubMed] [Google Scholar]
- 23.Prendergast GC. Immune escape as a fundamental trait of cancer: focus on IDO. Oncogene. 2008;27:3889–3900. doi: 10.1038/onc.2008.35. [DOI] [PubMed] [Google Scholar]
- 24.Priebe W, Donato N, Talpaz M, Fokt I, Szymanski S (2004) Novel compounds for treatment of cell proliferative diseases. 832 WO/104013214
- 25.Priebe W, Fokt I, Szymanski S, Madden T, Bao JI, Lesyng B, Conrad C, Kupferman M, Abbruzzese J, Myer J (2006) Design, synthesis and structure-activity relationships of novel Jak2/STAT3 signaling inhibitors. In: Proceedings of the 97th American Association for Cancer Research Annual Meeting. Washington, DC
- 26.Rietschel P, Chapman PB. Immunotherapy of melanoma. Hematol Oncol Clin North Am. 2006;20:751–766. doi: 10.1016/j.hoc.2006.02.005. [DOI] [PubMed] [Google Scholar]
- 27.Rosenberg SA, Yang JC, Restifo NP. Cancer immunotherapy: moving beyond current vaccines. Nat Med. 2004;10:909–915. doi: 10.1038/nm1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rosenberg SA, Yang JC, Topalian SL, Schwartzentruber DJ, Weber JS, Parkinson DR, Seipp CA, Einhorn JH, White DE. Treatment of 283 consecutive patients with metastatic melanoma or renal cell cancer using high-dose bolus interleukin 2. JAMA. 1994;271:907–913. doi: 10.1001/jama.271.12.907. [DOI] [PubMed] [Google Scholar]
- 29.Ross JA, Nagy ZS, Cheng H, Stepkowski SM, Kirken RA. Regulation of T cell homeostasis by JAKs and STATs. Arch Immunol Ther Exp (Warsz) 2007;55:231–245. doi: 10.1007/s00005-007-0030-x. [DOI] [PubMed] [Google Scholar]
- 30.Sonabend AM, Rolle CE, Lesniak MS. The role of regulatory T cells in malignant glioma. Anticancer Res. 2008;28:1143–1150. [PubMed] [Google Scholar]
- 31.Sumimoto H, Imabayashi F, Iwata T, Kawakami Y. The BRAF-MAPK signaling pathway is essential for cancer-immune evasion in human melanoma cells. J Exp Med. 2006;203:1651–1656. doi: 10.1084/jem.20051848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tarkowski E, Rosengren L, Blomstrand C, Wikkelso C, Jensen C, Ekholm S, Tarkowski A. Early intrathecal production of interleukin-6 predicts the size of brain lesion in stroke. Stroke. 1995;26:1393–1398. doi: 10.1161/01.str.26.8.1393. [DOI] [PubMed] [Google Scholar]
- 33.Wang T, Niu G, Kortylewski M, Burdelya L, Shain K, Zhang S, Bhattacharya R, Gabrilovich D, Heller R, Coppola D, Dalton W, Jove R, Pardoll D, Yu H. Regulation of the innate and adaptive immune responses by Stat-3 signaling in tumor cells. Nat Med. 2004;10:48–54. doi: 10.1038/nm976. [DOI] [PubMed] [Google Scholar]
- 34.Xie TX, Huang FJ, Aldape KD, Kang SH, Liu M, Gershenwald JE, Xie K, Sawaya R, Huang S. Activation of stat3 in human melanoma promotes brain metastasis. Cancer Res. 2006;66:3188–3196. doi: 10.1158/0008-5472.CAN-05-2674. [DOI] [PubMed] [Google Scholar]
- 35.Xie TX, Wei D, Liu M, Gao AC, Ali-Osman F, Sawaya R, Huang S. Stat3 activation regulates the expression of matrix metalloproteinase-2 and tumor invasion and metastasis. Oncogene. 2004;23:3550–3560. doi: 10.1038/sj.onc.1207383. [DOI] [PubMed] [Google Scholar]
- 36.Yu H, Kortylewski M, Pardoll D. Crosstalk between cancer and immune cells: role of STAT3 in the tumour microenvironment. Nat Rev Immunol. 2007;7:41–51. doi: 10.1038/nri1995. [DOI] [PubMed] [Google Scholar]
- 37.Zhuang L, Lee C, Scolyer RA, McCarthy SW, Zhang X, Thompson JF, Hersey P. Mcl-1, Bcl-XL and STAT3 expression are associated with progression of melanoma whereas Bcl-2, AP-2 and MITF levels decrease during progression of melanoma. Mod Pathol. 2007;20:416–426. doi: 10.1038/modpathol.3800750. [DOI] [PubMed] [Google Scholar]
- 38.Zorn E, Nelson EA, Mohseni M, Porcheray F, Kim H, Litsa D, Bellucci R, Raderschall E, Canning C, Soiffer RJ, Frank DA, Ritz J. IL-2 regulates FOXP3 expression in human CD4+CD25+ regulatory T cells through a STAT-dependent mechanism and induces the expansion of these cells in vivo. Blood. 2006;108:1571–1579. doi: 10.1182/blood-2006-02-004747. [DOI] [PMC free article] [PubMed] [Google Scholar]