

Abstract
Sphingolipid metabolites have emerged as critical players in a number of fundamental biological processes. Among them, sphingosine-1-phosphate (S1P) promotes cell survival and proliferation, in contrast to ceramide and sphingosine, which induce cell growth arrest and apoptosis. These sphingolipids with opposing functions are interconvertible inside cells, suggesting that a finely tuned balance between them can determine cell fate. Sphingosine kinases (SphKs), which catalyze the phosphorylation of sphingosine to S1P, are critical regulators of this balance. Of the two identified SphKs, sphingosine kinase type 1 (SphK1) has been shown to regulate various processes important for cancer progression and will be the focus of this review, since much less is known of biological functions of SphK2, especially in cancer. SphK1 is overexpressed in various types of cancers and upregulation of SphK1 has been associated with tumor angiogenesis and resistance to radiation and chemotherapy. Many growth factors, through their tyrosine kinase receptors (RTKs), stimulate SphK1 leading to a rapid increase in S1P. This S1P in turn can activate S1P receptors and their downstream signaling. Conversely, activation of S1P receptors can induce transactivation of various RTKs. Thus, SphK1 may play important roles in S1P receptor RTK amplification loops. Here we review the role of SphK1 in tumorigenesis, hormonal therapy, chemotherapy resistance, and as a prognostic marker. We will also review studies on the effects of SphK inhibitors in cells in vitro and in animals in vivo and in some clinical trials and highlight the potential of SphK1 as a new target for cancer therapeutics.
Keywords: Sphingosine kinase, cancer, transactivation, angiogenesis
1. INTRODUCTION: SPHINGOSINE KINASES AND S1P
Similar to phospholipids whose metabolites are well known potent mediators, metabolites of sphingolipids have emerged as signaling molecules with pleiotropic effects on important cellular processes including cancer progression [1]. The key players among the sphingolipid metabolites are ceramide (N-acyl sphingosine), sphingosine, and sphingosine-1-phosphate (S1P). Ceramide can be produced de novo or by breakdown of sphingomyelin and more complex sphingolipids. Sphingosine is only formed as a result of ceramidase catalyzed hydrolysis of ceramide and can either be acylated to ceramide or phosphorylated by sphingosine kinases (SphKs) to S1P.
Ceramide and sphingosine mediate cell cycle arrest and induce apoptosis, whereas S1P promotes cell growth, proliferation, and survival. Interestingly, because these oppositely acting sphingolipid metabolites are interconvertible within cells, the balance between ceramide/sphingosine and S1P levels forms a so-called “sphingolipid rheostat”, where a cell is primed for death pathways when the balance leans towards ceramide or sphingosine, or toward cell survival and proliferattion when S1P levels are increased. Indeed, it has been suggested that modulation of the normal setting of this rheostat by increasing levels of ceramide and/or sphingosine at the expense of S1P might serve to drive successful cancer therapies [2,3]. There are several potential regulators of this rheostat. S1P can reversibly dephosphorylated by S1P phophatases (SPP1 and SPP2) back to sphingosine or cleaved irreversibly at the C2-C3 bond into palmitaldehyde, and phosphoethanolamine by S1P lyase (SPL). However, SphKs are uniquely situated to be key regulators of this rheostat [4], as they generate S1P and decrease sphingosine and ceramide.
SphKs are responsible for the conversion of sphingosine to S1P. Two distinct SphK isoforms have been identified, SphK1 and SphK2, numbered in order of their discovery and characterization. SphK1 and SphK2 are members of a family of evolutionarily conserved lipid kinases. Sequences of putative SphKs from about 30 species spanning four kingdoms and four phyla of Metazoa are available at NCBI (http://www.ncbi.nlm.nih.gov). Notably, these SphKs do not possess any identifiable domains besides the diacylglycerol kinase catalytic domain, which is common between sphingosine, ceramide, and diacylglycerol kinases. Numerous reviews have recently appeared on this family of lipid kinases and the reader is referred to these for extensive information about the SphKs [5-8].
S1P regulates diverse physiological and pathological processes important for cancer and inflammation, such as proliferation, migration, invasion, and angiogenesis, as demonstrated both in cell cultures and animals. S1P exerts most of its action as a specific ligand for a family of five cognate G protein-coupled receptors (GPCRs), designated as S1P receptor 1 to 5 (S1P1-5). S1P also has some still not well characterized receptor-independent intracellular functions in mammalian cells important for calcium homeostasis, cell growth, and protection of apoptosis (reviewed in [4]). S1P receptor independent functions are consistent with findings in lower organisms, such as plants and yeast, which express SphKs and respond to S1P, but lack S1P receptors. S1P is especially abundant in serum where its concentration (300-500 nM) is higher than the known Kd values for S1P receptors. Because S1P is tightly associated with serum proteins, particularly lipoproteins (e.g., high density lipoprotein) and albumin, it has been suggested that the bio-available or free fraction of S1P is not sufficient to constitutively activate S1P receptors [9].
Recently, the role of SphKs in cancer (SphK1 in particular) has received considerable attention. This review focuses on current knowledge of the involvement of SphK1 in cancer, particularly in colon cancer, breast cancer, prostate cancer, and glioblastoma, and its roles in tumorigenesis, hormonal therapy, chemotherapy resistance, and as a prognostic marker. We also review the literature that implicates SphK1 as a potential therapeutic target for cancer.
2. SPHK1 - AN ONCOGENE
Sphk1 has been shown to have many of the characteristics of a bona fide oncogene. Non-transformed NIH 3T3 fibroblasts overexpressing SphK1 acquire a transformed phenotype, as determined by focus formation, colony growth in soft agar, and ability to form tumors in nude mice [10]. SphK1 expression is also required for oncogenic Ras-mediated transformation [10]. Further, translocation of SphK1 to the plasma membrane, a common mechanism of its activation by growth factors, enhances foci formation and growth in soft agar [11].
SphK1 also appears to act as an oncogene in erythroleukemia. Microarray transcriptome analysis of pro-erythroblasts from spi-1-transgenic mice, a model for multiple stages of erythroleukemia, revealed that transcriptional upregulation of SphK1 is repeatedly associated with the tumorigenic phenotype [12]. Moreover, overexpression of SphK1 in non-tumorigenic pro-erythroblasts increased their clonogenicity as well as resistance to apoptosis, and they acquired tumorigenicity when engrafted in vivo [12]. These results suggest that high expression of SphK1 may be an oncogenic event required for progression of erythroleukemia.
3. SPHK1: EXPRESSION, ACTIVATION, AND TRANSLOCATION
Elevated expression of SphK1 has been observed in multiple types of cancer. The levels of SphK1 mRNA were approximately 2-fold higher in tumors of the breast, colon, lung, ovary, stomach, uterus, kidney, and rectum compared with normal tissue from the same patient when measured by the Cancer Profiling Array (Clontech) that contains 241 paired human samples [13,14]. SphK1 is also overexpressed in acute leukemia patients [15]. Analyses of microarray data available online (http://www.oncomine.org/, http://www.ncbi.nlm.nih.gov/geo/) show statistically significant increases in SphK1 expression in: N-methyl-N-nitrosourea-induced rat breast cancer model [16]; recurrent breast cancer following tamoxifen therapy [17]; squamous cell carcinoma and it’s precursor actinic keratotic lesions in non-melanoma [18] and melanoma skin cancers [19]; advanced stages of cervical cancer [20]; invasive carcinoma of bladder [21]; oligodendrogliomas [22]; head and neck cancer [23,24]; leukemia, including B- and T-cell acute lymphoblastic leukemia and acute myeloid leukemia [25]; and in adult male germ cell tumors [26] (Fig. 1). Studies of the early onset of colorectal cancer showed increases in SphK1 levels which did not reach statistical significance, indicating that further classification of these tumor samples may be required [27] (Fig. 1).
Fig. (1). Expression of SphK1 in various cancers.

NCBI microarray repository data and data from Oncomine were analyzed for the expression of SphK1 in different types of cancer. Normalized values of SphK1 RNA levels were compared with control tissues and plotted as fold. Error bars indicate standard deviations and all p values were less than 0.05 for all except for colorectal cancers.
Immunohistochemical analyses of human breast cancer, colon cancer, and lung cancer tissues revealed that carcinoma cells themselves are the major source of SphK1 expression in the tumor [14,28,29]. This observation supports the notion that cancer takes advantage of the growth promoting properties of S1P by upregulating levels of the enzyme that produces it. Since SphK1 forms S1P at the expense of its pro-apoptotic precursors sphingosine and ceramide, its increased expression shifts the ‘sphingolipid rheostat’ toward pro-survival. Indeed, increased levels of S1P have been detected in the ascites fluid and plasma of ovarian cancer patients [30,31].
There are multiple pieces of evidence suggesting that it is not only activation of SphK1, but also its translocation to the plasma membrane that is critical for it to produce S1P that mediates its biological effects. Diverse external stimuli, particularly growth factors and chemoattractants, such as platelet derived growth factor (PDGF), epidermal growth factor (EGF), nerve growth factor (NGF), insulin growth factor (IGF), transforming growth factor beta (TGFβ), and phorbol 12-myristate 13-acetate (PMA), cause a rapid, transient stimulation of SphK1 (reviewed in [8]). The mechanism of SphK1 activation involves extracellular signal-regulated kinase (ERK) 1/2-mediated phosphorylation of SphK1 on Ser225 [32]. Notably, this single phosphorylation site not only controls the catalytic activity of SphK1, but is also necessary for agonist-induced translocation of SphK1. SphK1 is predominately a cytosolic enzyme, and translocation from the cytosol to the plasma membrane [32], where its substrate sphingosine resides allows precise temporal and localized formation of S1P that subsequently activates appropriate cell surface S1P receptors [33]. Phosphorylation of SphK1 has been shown to induce conformational changes that enhance its binding to membrane acidic phospholipids such as phosphatidylserine which can prolong its retention at the plasma membrane [34]. This phosphorylation-dependent localization of SphK1 to the plasma membrane appears essential for cell proliferation and survival [11]. This notion is supported by the observation that although expression of a non-phosphorylatable mutant SphK1 provided no survival or proliferative effects, artificial localization to the plasma membrane restored its pro-survival and pro-proliferative properties [11].
In addition to this well-characterized rapid and transient SphK1 activation, more recent evidence has uncovered the existence of a pronounced delayed increase in SphK1 activity that is due to increased gene transcription. EGF [35], 17β-estradiol (E2) [36], PMA [37], histamine [38] extracellular nucleotides [39], 1,25-dihydroxyvitamin D3 [40], and prolactin [41], increase SphK1 by transcriptional upregulation, chronically increasing production of S1P perhaps to increase cells capability to grow and move [41]. Either ERK1/2 or PKC seems to be involved in upregulation of SphK1 expression, although involvement of intracellular signaling pathway slightly differ among each agonist [35-39]. These reported agents also induce rapid SphK1 activation, producing a biphasic activation of SphK1; the first peak occurs after minutes of stimulation and is followed by the second delayed activation after hours of stimulation (Fig. 2). This makes it hard to determine which phase is important because they cannot be distinguished by downregulation or inhibition of SphK1. In either event, SphK1 and its activation play a central and obligatory role.
Fig. (2). A model demonstrating biphasic increases of SphK1 by diverse stimuli.
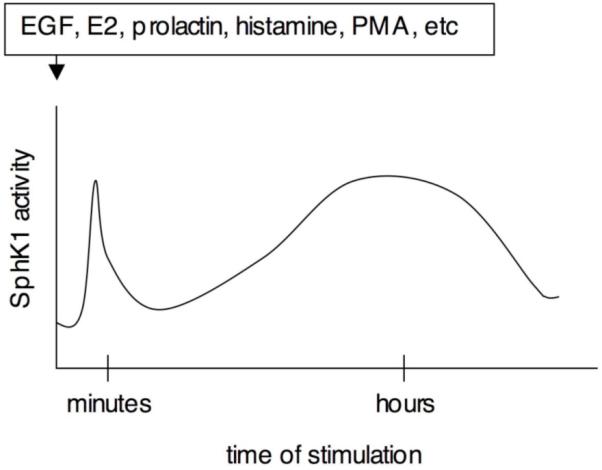
The first peak occurs due to phosphorylation of SphK1 after minutes of stimulation, followed by the second delayed activation due to transcriptional upregulation of SphK1 after hours of stimulation.
4. SPHK1: CELL MOTILITY
Several studies have implicated SphKs in cell migration, beginning with the demonstration that pharmacological SphK inhibitors, such as N,N-dimethylsphingosine (DMS) and D,L-threo-dihydrosphingosine (DHS), that inhibit both SphK1 and SphK2, suppress chemotaxis of many types of cells in response to many chemoattractants including growth factors (see [4] and references therein for the review of earlier work). Although in many studies, these SphK inhibitors did not seem to have a general inhibitory effect on cellular locomotion machinery, these results should be interpreted with caution since these inhibitors might also inhibit protein kinase C [42]. More recently, molecular approaches have been utilized to examine the functions of SphK1 and SphK2 primarily in human breast cancer cells. Downregulation of SphK1, but not SphK2 with specific siRNAs decreased EGF [43], prolactin- and E2-induced migration of MCF7 cells [41], while having no effect on migration towards fibronectin. Interestingly, downregulation of SphK1 appears to upregulate migration of MCF7 cells towards S1P, possibly due to reduced “inside out signaling” and desensitization of S1P2 receptors [43]. In more aggressive metastatic MDA-MB-453 breast cancer cells, downregulation of both SphK1 and SphK2 inhibits migration towards EGF, while overexpression of either SphK1 or SphK2 enhances migration to EGF [44]. Therefore, it is possible that SphK1 and SphK2 have distinct but sometimes overlapping functions, depending on the cellular localization of SphK2.
5. SPHK1: CROSSTALK, TRANSACTIVATION, AND SIGNAL AMPLIFICATION LOOPS
Crosstalk between members of different receptor families has emerged as an important concept in signal transduction. As described above, diverse growth factors and mitogens stimulate SphK1 resulting in production of S1P, which can act either intracellularly as a second messenger, or be exported from cells to activate cell-surface S1P receptors to initiate “inside-out” signaling cascades in an autocrine and/ or paracrine manner. The first demonstration of this paradigm that was later extended to many other growth factors was the observation that binding of PDGF to the PDGF tyrosine kinase receptor activates and translocates SphK1 to the plasma membrane. This was followed by spatially restricted formation of S1P and activation of cell surface S1P1 receptors, which are essential for PDGF-directed cell movement [33]. On the other hand, similar crosstalk between PDGF and S1P2 inhibits chemotaxis toward PDGF [45]. Several recent studies have shown that export of S1P from cells is mediated at least in part by the ATP-binding cassette (ABC) transporter family including ABCC1 and ABCA1 [46-48].
A well established reciprocal mechanism of receptor crosstalk has been shown to regulate cell motility and growth. Tyrosine phosphorylation of various receptor tyrosine kinases (RTKs) in response to activation of many GPCRs, such as S1PRs, named transactivation, is important for amplification of signaling. Among many RTKs, EGFR is the most well-known example [49,50]. EGFR have been linked to diverse biological processes in human cancer cells, including migration, invasion and proliferation, and thus considered to be closely related to cancer progression [50,51]. Indeed, as a molecular target therapy for cancer in clinical practice, cetuximab (Erbitux; ImClone Systems, NY), a chimeric monoclonal antibody against EGFR, was shown to significantly reduce cancer progression and improve the overall survival of patients with chemotherapy-resistant metastatic colorectal cancer in phase III large randomized clinical trials, indicating the significance of EGFR in cancer progression [52]. Like many other GPCR ligands, S1P can transactivate EGFR in certain cell types such as gastric cancer cells [53]. S1P-stimulated transactivation of EGFR requires matrix metalloproteinase activation, which induces proteolytic cleavage of transmembrane pro- heparin-binding EGF-like growth factor (HB-EGF) that subsequently activates EGFR [53].
Transactivation of RTKs have been demonstrated not only for EGFR, but also in other receptors. HER2/Neu (ErbB2) receptor belongs to the EGFR family of RTKs and functions as a homo- or heterodimer with other members of the EGFR family upon interaction with agonistic ligands, such as EGF. HER2 is currently attracting a great deal of attention because a new adjuvant therapy using an antibody against HER2, trastuzumab (Herceptin; Genentech, California), has proved effective in treating certain cancers [54]. S1P induced transactivation of HER2 in gastric cancer cells, which was dependent on EGFR tyrosine kinase activity and MMP function [55].
Hepatocyte growth factor (HGF) receptor, c-Met, is another RTK that is overexpressed in many solid tumors. Activation of c-Met results in cell proliferation, increased survival, altered motility, and enhanced invasion into the extracellular matrix. S1P also transactivates c-Met in gastric cancer cells [53]. Additionally, S1P transactivated PDGFbe-taR in ovarian cancer cells [56] and the VEGF receptors, Flk-1/KDR and VEGFR2 in human umbilical vein endothelial cells (HUVEC) [57,58]. Therefore, S1P can act upstream of various RTK signaling pathways, suggesting that it may be a potent stimulator of cancer progression.
In summary, diverse growth factor ligands of RTKs activate SphK1. S1P, which is produced by activated SphK1, not only exerts its biological effect through S1P receptors and subsequent signaling to control cellular functions, but also transactivates various RTKs. Therefore, it is tempting to speculate that positive feedback amplification loops of SphK1, S1P, and RTKs, can provide even more amplification of signal transduction pathways that contribute to cancer progression (Fig. 3).
Fig. (3). S1P receptor and RTK signal amplification loops.

SphK1 activation by various agonists via their receptor tyrosine kinases (RTKs) is followed by translocation of SphK1, which converts S1P from sphingosine (Sph). Exported S1P not only induces S1P receptor signaling, but also transactivation of various RTKs, that form signaling amplication loops. Solid arrows represent intracellular transductions, dotted arrows represent translocation, and open arrows represent transactivation of RTKs.
6. SPHK1 AND ANGIOGENESIS
Angiogenesis is a crucial component of the growth and metastasis of tumors. Multiple phases of this process, such as migration, proliferation, morphogenesis, and vascular stabilization, are needed for optimal tumor growth beyond a diffusion-limited size. The anti-angiogenic approach to cancer has been greatly advanced by the development of bevacizumab (Avastin; Genentech, CA), an anti-VEGF antibody, to treat colorectal cancer in combination regimen with conventional chemotherapeutic agents.
Recent growing evidence implicates S1P as one of the most potent pro-angiogenic factors. S1P induces various effects on endothelial cells that support its role as an angiogenic molecule. S1P promotes endothelial cell migration much more potently than either VEGF or HGF, thereby promoting blood vessel formation [59]. S1P strongly induces migration of bone marrow-derived endothelial cell precursors to neovascularisation sites [60]. S1P stimulates tube formation by HUVECs in extracellular matrix Matrigel. In vivo studies showed that S1P synergized with polypeptide angiogenic factors such as FGF-2 and VEGF to induce angiogenesis and vascular maturation in which Matrigel plugs were implanted subcutaneously in mice [61]. However, the most direct evidence that S1P contributes to tumor angiogenesis comes from use of a murine monoclonal antibody (mAb) against S1P as a molecular sponge to selectively absorb and neutralize S1P [62]. Treatment of mice bearing various human xenograft tumors with anti-S1P mAb blocked endothelial cell migration, capillary formation, inhibited blood vessel formation induced by VEGF and bFGF, and arrested tumor-associated angiogenesis [62]. The S1P mAb also prevented the release of proangiogenic cytokines (VEGF, IL-8 and IL-6) from tumor cells. Importantly, anti-S1P mAb substantially reduced tumor progression in mouse xenograft and allograft models [62].
The S1P1 receptor plays a specific and crucial role in vasculogenesis and angiogenesis. During embryonic development, S1P1 is required for stabilization of nascent blood vessels, and S1P1 null mice die from extensive hemmorhaging in utero between embryonic days 12.5 and 14.5, suggesting a critical role of S1P1 for basal vascular integrity during development (vasculogenesis) [63]. Knockdown of S1P1 in endothelial cells significantly impaired angiogenic responses and abolished the induction of cell adhesion molecules [64]. Interestingly, VEGF induces S1P1 mRNA and protein expression in endothelial cells and potentiates the vascular effects mediated by S1P [65]. Moreover, expression of S1P1 is upregulated in the tumor vasculature during angiogenesis in vivo [66] and downregulating its expression was effective in inhibiting angiogenesis and tumor growth in vivo, suggesting that S1P1 receptor is also a critical component of the tumor angiogenic response [66].
There is also some evidence suggesting that SphK1 is involved in angiogenesis. Overexpression of SphK1 in endothelial cells enhanced survival to serum deprivation and detachment from the extracellular matrix, suggesting that SphK1 may play an important role in vascular phenomena that occur under stressed conditions [67]. Importantly, the SphK1-SphK2 double deletion in mice confirms that these kinases are essential for vascular development and angiogenesis [68]. Taken together, these results suggest that S1P has effects not only on tumor cells themselves but also is permissive or required for the actions of angiogenic factors and provide proof of concept that targeting of S1P or SphK are novel strategies for development of new types of cancer treatments.
7. SPHK2 AND CANCER
SphK2 as well as SphK1 have important roles in the immune system. The potent immunosuppressive drug FTY720 (2-amino-2-[2-(4-octylphenyl)ethyl]-1,3-propanediol hydrochloride), a sphingosine analogue, has great clinical potential for treatment of multiple sclerosis (reviewed in [69]). FTY720 is a pro-drug that is phosphorylated in vivo primarily by SphK2 to a mimetic of S1P, FTY720-P. A large body of evidence has established that FTY720-P prevents egress of T cells from secondary lymphoid organs to circulation by functionally desensitizing S1P1 (reviewed in [70]).
Although SphK1 and SphK2 are closely related and use the same substrate, sphingosine, and generate the same product, S1P, they appear to have contrasting roles in cells. In contrast to SphK1, overexpression of SphK2 suppresses growth and enhances apoptosis, preceded by cytochrome c release and activation of caspase-3 [71,72]. In NIH 3T3 cells, SphK2 localizes to the endoplasmic reticulum (ER) and functions to increase ceramide levels, which contribute to its apoptogenic effects [73]. Interestingly, artificial targeting of SphK1 to the ER converts the normally anti-apoptotic kinase into one with a pro-apoptotic function [73]. These results imply that the cellular location where S1P is produced determines how SphK1 and SphK2 have opposite effects on cell survival and opposing functions in regulation of sphingolipid metabolism and thus levels of ceramide.
On the other hand, downregulation of SphK2 in some cancer cells unexpectedly resulted in cell growth inhibition. In U1242 and U87 MG glioblastoma cells, downregulation of SphK2 inhibited cell proliferation more potently than downregulation of SphK1 [74]. Likewise, in MCF7 breast cancer cells as well as HCT116 colon cancer cells, downregulation of SphK2 sensitized them to apoptosis induced by doxorubicin and reduced decreased basal and doxorubicin-induced expression of p21 without affecting increased expression of p53. This study shows that endogenous SphK2 is important for p53-independent induction of p21 expression by doxorubicin and suggest that SphK2 may influence the balance between cytostasis and apoptosis of human cancer cells [75]. Surprisingly, CD4+ T cells from SphK2 knockout mice exhibit a hyperactivated phenotype with significantly enhanced proliferation and cytokine secretion in response to IL-2 and increased STAT5 phosphorylation as well as reduced sensitivity to regulatory T cell-mediated suppression in vitro. Yet treatment with S1P did not counteract these effects [76]. Thus, it was suggested that Sphk2 modulates intestinal autoimmunity and IL-2 pathways in T cells independent of S1P [76].
Some studies suggest that SphK2 has an overlapping function with SphK1. For example, EGF, which contributes to the invasiveness of human breast cancer cells stimulated SphK2 and SphK1 in MDA-MB-453 breast cancer cells. Downregulation of SphK2 in these cells completely eliminated migration towards EGF suggesting that similar to SphK1, SphK2 also plays an important role in migration of MDA-MB-453 cells towards EGF [44]. In vivo studies of SphK1 or SphK2 knockout mice also suggest that these kinases may have compensatory or overlapping roles, at least in mouse development [68,77] as SphK1/SphK2 double-knockout mice which completely lack S1P die in utero due to severe defects in neurogenesis and vasculogenesis [68]. These results suggest that the complete absence of S1P may not be compatible with life, suggesting that pan-SphK inhibitors may not be ideal therapeutics. Instead, specific targeting of SphK1 may be a novel and reasonable therapeutic option.
8. COLON CANCER AND SPHK1: TUMORIGENESIS
Colorectal cancers (cancers of colon or rectum) are often associated with local bleeding and thus, at the tumor site, platelets are activated and may secrete large amounts of S1P, leading us to speculate that S1P could also be important for colorectal cancer progression. Imunohistochemical analysis of SphK1 expression has been performed in human colorectal cancer tissue as well as normal mucosa adjacent to the tumor [28]. In normal colonic mucosa, stromal inflammatory cells express high levels of SphK1. Differentiated colonic epithelial cells near the lumen also express high levels of SphK1, suggesting that SphK1 is expressed during differentiation of epithelial cells. In colon carcinoma cells, a high level of expression was seen in the cancer cells themselves. Analysis of 27 colorectal cancer specimens indicated that 63% of tumors express high levels of SphK1 [28]. In agreement with this observation, overexpression of SphK1 was observed immunohistochemically in a well-established rat colon carcinogenesis model induced by azoxymethane (AOM) [78]. Of interest, in this AOM-induced colon cancer model, there was a correlation between expression of SphK1 and adenoma to carcinoma sequence, that is, there was little or no SphK1 in normal colon mucosa, low SphK1 in adenomas (benign tumor), and high SphK1 expression in carcinomas [78]. In addition, expression of S1P lyase (SPL), which irreversibly degrades S1P, was significantly downregulated in human colon cancer tissues in comparison with normal adjacent tissues, as determined by quantitative real-time PCR (qPCR) and immunohistochemical analysis [79]. Downregulation of S1P phosphatases was also observed, suggesting that colon cancer cells survive, at least in part, by reducing S1P catabolism [79].
The adenoma-carcinoma sequence of carcinogenesis of colorectal cancers in humans is well characterized. First, adenomas arise from normal colon mucosa, and then grow in size, and finally carcinomas appear to arise from adenomas and a series of genetic alterations leading to the progressive disordering of the normal mechanisms controlling cell growth are associated. Pioneering work by Vogelstein and colleagues over the last 20 years has identified a number of critically important genetic alterations that contribute, through their multiplicity over many years, to the eventual development of colorectal cancer [80]. The earliest event appears to involve the APC (adenomatous polyposis coli) gene, which is mutated in individuals affected by familial adenomatous polyposis (FAP). APC mutations are sufficient for the growth of early colorectal adenomas and are very common in sporadic colorectal cancer. Thus, MIN mice model (APCMIN/+ mice) are an ideal animal model to investigate colorectal tumorigenesis.
To investigate the role of SphK1 in intestinal tumors, the development of small intestinal adenomas in APCMIN/+ SphK-/- mice was examined. The most important finding was that the incidence of adenomas was unchanged but the size distribution was dramatically reduced [28]. This suggests that SphK1 expression is critical for the progression phase of tumor development, and also suggests that SphK1 does not regulate the initiation of intestinal tumors. Another interesting observation was that neither the deficiency of S1P1, S1P2, nor S1P3 receptors had an effect on adenoma incidence or size, suggesting that extracellular S1P signaling via its receptors was not involved in adenoma progression [28]. On the other hand, SPL expression and activity in Min mouse adenomas were downregulated compared to adjacent normal tissues [79], which also suggests that intracellular metabolism of S1P is crucial in adenoma progression. These recent studies have established a novel link between S1P metabolism and colorectal tumorigenesis [81] and suggest that SphK1 may also be a biomarker for colon malignancy. Further, SphK1 may be a good target for the mechanism-based chemoprevention and for therapeutic strategies against colon cancer development.
An important question is how does expression of SphK1 contribute to tumorigenesis? It is possible that SphK1 and production of S1P regulate induction of cyclooxygenase-2 (COX-2), a well-described important player in colon tumorigenesis. Several lines of evidence implicate SphK1/S1P in COX-2 induction and production of the inflammatory mediator, prostaglandin E2 (PGE2), in response to pro-inflammatory cytokines [82,83]. In rat colon cancers induced by AOM, mRNA and protein expression of COX-2 as well as SphK1 are upregulated [78] and both have a similar cellular distribution. Furthermore, SphK1 was also critical for both basal and cytokine-induced COX-2 expression in this cancer model [78].
9. BREAST CANCER AND SPHK1: HORMONAL THERAPY
Breast cancer is the most common cancer and the second leading cause of death from malignancy in women in the United States, and thus has been the object of intensive research that is now revealing the complexity of this disease. The levels of SphK1 mRNA in breast cancer and normal tissues from the same patient have been measured and compared [13]. SphK1 expression was generally higher in breast cancers compared to adjacent normal breast tissue and increases up to 4-fold were found in 80% of the cases, whereas low levels in tumors compared with the normal specimens were rare [13]. Recently, using microarray technology was used to correlate expression of several enzymes of sphingolipid metabolism in breast cancer [29]. Survival analysis revealed a worse outcome of patients with high SphK1 expression, suggesting its value as a prognostic marker.
Many studies have utilized MCF7 breast cancer cells to investigate the role of SphK1 and its product S1P. Endogenous SphK1 activity in breast cancer cells has been shown to be decreased by cellular stresses such as actinomycin D, doxorubicin, etoposide, and gamma irradiation, which were likely caused by post-transcriptional modulations as SphK1 mRNA levels remained unchanged [84]. Downregulation of SphK1 also reduced the viability of MCF7 cells [43,84] by inducing cell cycle arrest and apoptosis via the mitochondrial pathway, accompanied by significant increases in ceramide levels [43,84]. Furthermore, overexpression of SphK1 in MCF7 cells significantly enhanced migratory responses towards EGF [43], whereas its downregulation completely blocked EGF-mediated migration and proliferation and enhanced sensitivity to doxorubicin, a potent chemotherapeutic agent [43]. Taken together, these results suggest that SphK1 may be a critical component of EGFR signaling in breast cancer progression. This has important implications as EGFR expression is associated with poor prognosis in breast cancer and correlates with lack of response to anti-estrogen therapy [85].
The sex steroid hormone estrogen, 17-beta estradiol (E2), acts as a mitogenic factor contributing to the development of human breast cancer. In fact, anti-estrogen therapy is the most successful treatment for patients with estrogen receptor alpha positive breast tumors, which itself is as effective as any other anticancer drugs in breast cancer treatment [86]. E2 stimulates mitogenesis of MCF7 cells in the absence of any other growth factors. E2 has dual actions on SphK1 in MCF7 cells - rapid and transient activation and a delayed but prolonged activation resulting in part from the nuclear transcriptional activity of estrogen receptors [36] (Fig. 2). Overexpression of SphK1 in MCF7 cells significantly increases cell growth in response to E2 stimulation, as determined by colony growth in soft agar and solid focus formation [36]. Moreover, MCF7-SphK1 cells produce larger tumors in nude mice with higher microvessel density in the periphery in an E2-dependent manner [87]. The growth promoting effects of E2 have also been suggested to result from increased extracellular EGF and transactivation of EGFR. As described above, this transactivation mechanism involves complicated crosstalk in which E2 stimulates SphK1 and production and release of S1P, leading to activation of the S1P3 receptor, whose downstream signaling stimulates matrix metalloproteinase cleavage of secreted HB-EGF to release EGF and transactivate the growth promoting EGFR [88]. Interestingly, prolactin (PRL), another mammogenic hormone, also upregulates SphK1 biphasically in MCF7 cells [41]. Importantly, downregulation of SphK1 abolished PRL-induced, as well as E2-induced, cell proliferation and migration [41].
Estrogen receptor (ER) status is a major determinant of breast cancer. Therefore, pharmacological inhibition of E2 synthesis or the ER are effective treatments for ER-positive tumors against risk of recurrence after surgical removal. When comparing ER positive and ER negative tumors, SphK1 is more highly expressed in ER negative tumors that are known for their higher proliferative activity [29]. In the ER positive group, higher expression of SphK1 was associated with worse prognosis. Moreover, immunohistochemical analysis identified the carcinoma cells in breast tumors as the major source of SphK1 expression [29]. The positive correlation of SphK1 expression with poor prognosis, as well as its high expression among ER negative tumors is consistent with the anti-apoptotic and proliferative properties of its product, S1P.
10. PROSTATE CANCER AND SPHK1: RESISTANCE TO THERAPY
Most pharmacological therapies against cancer utilize the apoptotic machinery of cells and the participation of ceramide generation in many of them has been suggested. For example, in prostate cancer cells, taxol (paclitaxel) has been shown to induce ceramide generation [89]. Conversely, inability to generate ceramide has been linked to anti-cancer therapy resistance [90]. The defect in radiation-resistant and chemo-resistant cell lines that do not generate ceramide after irradiation or chemotherapy can be bypassed by treatment with cell-permeable ceramide [91,92]. However, increased ceramide generation alone may not be sufficient in some cases to successfully push cells toward apoptosis without also inhibiting the SphKs that produce S1P. In this regard, a correlation was originally noted between SphK1 activity and resistance to irradiation in prostate cancer cells. SphK1 activity in radiation-resistant LNCaP cells was not affected by ionizing radiation, whereas SphK1 activity was markedly inhibited in radiation-sensitive TSU cells [93]. The relationship between SphK1 and resistance to chemotherapy has also been shown in prostate cancer cells. Prostate cancer PC-3 cells and LNCaP cells differ in hormonal requirements, as LNCaP growth is exclusively dependent on androgens, whereas PC-3 is not. Thus, these two prostate cancer cell lines exhibit contrasting sensitivities to hormonal therapy and chemotherapy, mimicking the progression from hormone-sensitive to hormone-refractory disease that is observed in clinical practice. Using these two cell lines, it was shown that docetaxel (Taxotere) and camptothecin induced strong inhibition of SphK1 and elevation of the ceramide/S1P ratio only in cell lines sensitive to these drugs [94]. SphK1 overexpression in both cell lines impaired the efficacy of chemotherapy by decreasing the ceramide/S1P ratio [94]. Alternatively, downregulation of SphK1 by siRNA or pharmacologic inhibition induced apoptosis coupled with ceramide elevation and loss of S1P [94]. In mouse xenografts, SphK1-overexpressing PC-3 cells developed remarkably larger tumors and resistance to docetaxel treatment, which provided the first in vivo demonstration of SphK1 as a sensor of chemotherapy effects [94]. Accordingly, ceramide elevation normally triggered by docetaxel treatment was attenuated in tumors derived from PC3-SphK1 cells. In agreement with in vitro results, the increase of the ceramide/S1P ratio was strongly diminished in PC3-SphK1 tumors compared with the PC3-vector tumors [94]. In addition, another study showed that PC3 cells are resistant to camptothecin because of their higher SphK1 expression and activity and elevated expression of S1P1 and S1P3 receptors [95]. If these conclusions can be applied to other types of cancer, they suggest that upregulated expression of SphK1 by cancer cells not only promotes tumorigenesis, it also protects them against chemo and radiation therapy.
Indeed, similar effects of altering SphK activity have been reported in other cancer cell types. In melanoma cells, overexpression of SphK1 reduced sensitivity to Fas- and ceramide-mediated apoptosis, and conversely, its downregulation decreased resistance to apoptosis [96]. The sensitivity of MCF7 breast cancer cells to the anticancer drug doxorubicin was potentiated by the pan SphK inhibitor, DMS [87]. Relevant to this, induction of apoptosis in in MCF7 cells by doxorubicin was accompanied by degradation of SphK1 [84]. In addition, chemosensitive HL-60 acute myeloid leukemia cells had low SphK1 expression and produced ceramide, whereas chemoresistant HL-60 variants had sustained high levels of SphK1 activity and did not produce ceramide upon drug treatment [97].
Development of multidrug resistance (MDR) in cancer represents a major obstacle to successful chemotherapy, and involves diverse mechanisms including up-regulation of the drug efflux pumps, MDR-associated protein (MRP-1) and P-glycoprotein (Pgp). In a recent study, overexpression of SphK1 in RBE-4 cerebral endothelial cells was shown to enhance the expression of Pgp at the mRNA and protein levels [98]. Furthermore, as demonstrated in these brain tumor-derived endothelial cells, S1P also stimulated the transport activity of Pgp via activation of S1P1 and S1P3 receptors [98]. Moreover, downregulation of SphK1 in Molt-4 and HL-60 leukemia cells responsive to DNA-damaging agents was reported, but not in MDR1-positive or MRP1-positive cells refractory to doxorubicin and etoposide [97,99]. These findings suggest that SphK1 may play an important role in the development and maintenance of MDR.
Collectively, this information indicates that (i) SphK1 activity is downregulated in cancer cells that are responsive to apoptosis-inducing anticancer treatments and (ii) that upregulation of SphK1 can protect against various chemotherapies and ionizing radiation, combination therapy of a SphK1 inhibitor with cytotoxic drugs or radiation could provide a new strategy for the treatment of therapeutic-resistant cancers.
11. GLIOBLASTOMA AND SPHK1: A PROGNOSTIC INDICATOR
Gliomas are primary neoplasms of the central nervous system that arise from glial cells. Astrocytoma is one major type of glioma that occurs in adults. The most aggressive type of astrocytoma, known as glioblastoma multiforme (GBM), is the most common and most devastating type of brain cancer with median survival of less than a year, and currently available therapies only minimally improve the prognosis. The malignant nature of these tumors is due to their rapid cellular proliferation and diffuse invasion into the surrounding brain, leading to inevitable recurrence of tumors even after radical resection.
Survival analysis of 48 patients with GBM indicated a strong correlation of SphK1 expression at the mRNA level with short patient survival [74]. Patients whose tumors were among the highest one-third with regard to SphK1 expression survived a median of 102 days, whereas those within the lower two-thirds survived a median of 357 days [74]. Consistent with the higher expression of SphK1, ceramide levels are lower in human gliomas compared to normal surrounding brain tissue, and are inversely related to malignant progression and poor prognosis [100]. In contrast to SphK1, SphK2 expression did not correlate with GBM patient survival [74]. Likewise, expression of S1P1, S1P2, and S1P3 receptors were highly variable among individual tumors, and no significant correlations with histological grade or patient survival were found [74]. SphK1 is important for proliferation of glioma cells, as pharmacological inhibition or down-reglation of its expression significantly decreased cell growth by preventing cells from exiting the G1 phase of the cell cycle [74]. In addition, among a panel of human glioma cell lines, exogenously added S1P stimulated proliferation of half of the cell lines tested by signaling through the ERK/MAP kinase and PI-3 kinase pathways [101].
Patients with gliomas expressing high levels of epidermal growth factor receptor (EGFR) and plasminogen activator inhibitor-1 (PAI-1) have a shorter overall survival prognosis. Moreover, EGF enhances PAI-1 expression in glioma cells [102]. Although multiple known signaling cascades are activated by EGF in glioma cells, recently it was shown that EGF enhances expression of PAI-1 via sequential activation of c-Src, protein kinase C delta (PKCdelta), and SphK1. EGF induced rapid phosphorylation of c-Src and PKCdelta and concomitant translocation of PKCdelta as well as SphK1 to the plasma membrane. Furthermore, SphK1 was indispensable for both EGF-induced c-Jun phosphorylation and PAI-1 expression [103], suggesting another role for SphK1 in EGF-dependent glioblastoma progression.
12. SPHK1 AS A CANCER CHEMOTHERAPEUTIC TARGET
As described above, ample evidence supports SphK1 as a promising target to treat cancer; first, SphK1 itself is an oncogene; second, SphK1 is overexpressed in various types of tumors; third, many growth factors and mitogens cause biphasic activation of SphK1 and increases in S1P production; fourth, S1P has potent stimulaltory effects on tumor angiogenesis; fifth, SphK1 overexpression as well as S1P itself strongly stimulate cell motility, which is important for invasion and metastasis; sixth, resistance to chemotherapy or radiation therapy is associated with SphK1 upregulation, suggesting that inhibition of SphK1 may result in regaining sensitivity to those therapies; seventh, S1P, SphK, S1P receptors, and RTKs provide a complicated crosstalk network that suggests SphK1 may also be a key regulator of several signal amplification loops that contribute to tumorigenesis; finally, because SphK1 is a key controller of the “sphingolipid rheostat”, inhibitors of SphK1 are expected not only to inhibit S1P production, but also to increase sphingosine and ceramide, pushing cancer cells more toward cell death.
Examination of the effects of specifically downregulating SphK1 expression support this notion. For example, down-regulation of SphK1 with siRNA triggers apoptosis in many types of cancer cells including breast [84], glioblastoma [74], prostate, and leukemia [97,99]. Apoptosis induced by knocking down SphK1 was demonstrated by activation of effector caspases and cytochrome c release [84,97], which were associated with significant increases in ceramide and sphingosine levels, precursors of S1P that are both known to induce apoptosis by actions upstream of the mitochondrial pathway [2].
13. SPHK INHIBITORS IN VITRO
Despite the high level of interest in sphingolipid-derived signaling, there are very few established inhibitors of the enzymes of sphingolipid metabolism. In particular, the field suffers from a lack of potent and selective inhibitors of SphK. The most commonly used pharmacological inhibitor to date are sphingosine analogues, especially N,N-dimethylsphingosine (DMS) and DL-threo-dihydrosphingosine (DHS). DMS and DHS are potent competitive inhibitors of SphK1 and SphK2, with Ki values of around 5 and 10 μM, respectively [104,105]. Both elicit growth inhibition and cause apoptosis in a variety of human cancer cells including colon [106,107], breast [87,108], prostate [93], gastric [106,107], and lung [107]. More interestingly, these sphingosine analogues can also potentiate anti-cancer regimen-induced cell death and overcome radiation and chemo resistance in various solid tumor cell lines [109].
Even though sphingosine analogues were found to be equipotent for many types of cancer cells in vitro and capable of inhibiting tumor growth in vivo, they caused severe hemolysis in mice. Moreover, like sphingosine, DMS and DHS can also inhibit protein kinase C [110]. Clearly, potent inhibitors of SphK1 that can be readily synthesized (or purified from natural sources), and that are not toxic to animals are needed to evaluate the potential clinical utility of SphK1 inhibitors.
By screening a library of synthetic compounds, a series of inhibitors were found that were selective towards SphK and did not inhibit other lipid (phosphatidylinostiol 3-phosphate kinase) and protein kinases (PKCα and extracellular kinase 2), and demonstrated activity at sub- to micromolar concentrations, making them more potent than other known SphK inhibitors [13]. These SphK inhibitors were anti-proliferative toward panel of tumor cell lines, including those with multi-drug resistance phenotype [13].
14. SPHK INHIBITORS IN VIVO
A synthetic variant of one of the inhibitors discussed above, called compound V (IC50 for inhibition of SphK and cell viability was approximately 2 μmol/l), was tested for its effect on tumors produced from JC mammary adenocarcinoma cells subcutaneously implanted in immunocompetent BALB/c mice [13]. Interestingly, tumor growth in mice treated with compound V was 50-85% inhibited compared to growth in untreated mice. Importantly, there were no significant differences in body weights of the two groups, suggesting compound V was not toxic. Another inhibitor from this series, 4-(4-(4-chloro-phenyl) -thiazol-2-ylamino)-phenol, designated as SKI-II, was orally bioavailable, detectable in the blood for at least 8 hours, and showed significant inhibition of xenograft tumor growth [111]. The correlation with pharmacokinetic and pharmacodynamic properties, in addition to oral bioavailability and anti-tumor activity of SKI-II, provides strong support for moving this approach forward for cancer therapy.
Recently, these orally bioavailable SphK inhibitors also suppressed inflammation in the dextran sulfate sodium (DSS) model of ulcerative colitis in mice [112]. Since it is well known that some ulcerative colitis patients develop colon and rectal cancer after years of severe inflammation, SphK inhibitors may be valuable from the standpoint of prevention of carcinogenesis.
15. SPHK INHIBITORS AND CLINICAL TRIALS
L-threo-Dihydrosphingosine (Safingol), a protein kinase C inhibitor with SphK-inhibiting properties, has reached the phase I clinical evaluation level. Administration of Safingol alone proved to be nontoxic and in combination with doxorubicin did not alter the pharmacokinetics of the anticancer drug [113]. Although the initial phase I trial with Safingol ended before achieving a maximally tolerated dose and had entered too few patients to assess activity, a modest positive effect was noted [113].
The analogue of sphingosine, FTY720, which is known to be phosphorylated in vivo by SphK2, is an immunomodulator in Phase III trials for patients with multiple sclerosis. Recently, a new action of FTY720 as an activator of protein phosphatase 2A [114], led to a study assessing its therapeutic potential in blast crisis chronic myelogenous leukemia (CML-BC) and Philadelphia chromosome-positive (Ph1-positive) acute lymphocytic leukemia (Ph1 ALL) patient cells and in in vitro and in vivo models of these BCR/ABL+ leukemias. It was found that FTY720 is a potent inhibitor of BCR/ABL leukemogenesis, as it induces marked apoptosis of CML-BCCD34+ and Ph1 ALLCD34+/CD19+ patient cells by impairing p210/p190-BCR/ABL activity and expression [115]. Furthermore, in vivo long-term administration of pharmacologic FTY720 doses does not induce adverse effects and significantly inhibits wild-type and T315I p210 and p190 BCR/ABL leukemogenesis in mice [115]. Because, CML-BC and Ph1 ALL are fatal BCR/ABL-driven leukemias against which Abl kinase inhibitors fail to induce a long-term response and FTY720 has already received FDA approval, this could lead rapidly to new clinical trials.
Phenoxodiol, an anticancer genistein derivative, directly induces mitotic arrest and apoptosis in most cancer cells and is currently undergoing clinical trials as a chemotherapeutic in ovarian and prostate cancers. Recently, SphK was identified as a new target for phenoxodiol [116]. It was shown that phenoxodiol’s major action is to block the activation of SphK [116]. Although phenoxodiol is not a SphK1 specific inhibitor, clinical trials using this compound may provide some important messages for clinical usage of SphK1 inhibitors.
16. CONCLUSIONS
Because of the important roles of sphingolipids in cancer progression as summarized above, SphK1 targeted therapy is expected to join the next generation of cancer treatments. The results of many studies described in this review emphasize the role of SphK1 in cancer progression, suggesting that SphK1 could be a novel molecular target for cancer therapy. In addition to investigating functional activity of SphKs, much of current research in this area is directed toward the continuing search for effective SphK1 inhibitors that are effective in vivo.
ACKNOWLEDGEMENTS
This work was supported by NRSA Training Grant 5T32GM008695-08 (K.T.), R01AI50094 (S.S.), R01CA6-1774 (S.S.) and R37GM043880 (S.S.). S.M. was supported by the Intramural Research Program of the National Institute of Mental Health.
ABBREVIATIONS
- COX-2
Cyclooxygenase-2
- E2
17-Beta estradiol
- EGF
Epidermal growth factor
- EGFR
Epidermal growth factor receptor
- ER
Estrogen receptor
- GPCR
G protein-coupled receptor
- MDR
Multidrug resistance
- MMP
Matrix metalloproteinase
- RTK
Receptor tyrosine kinase
- PCR
Polymerase chain reaction
- PGE2
Prostaglandin E2
- Pgp
P-glycoprotein
- S1P
Sphingosine-1-phosphate
- SphK
Sphingosine kinase
- SPL
S1P lyase
- SPP
S1P phosphohydrolase
REFERENCES
- [1].Ogretmen B, Hannun YA. Nature Rev. Cancer. 2004;4:604–616. doi: 10.1038/nrc1411. [DOI] [PubMed] [Google Scholar]
- [2].Cuvillier O, Nava VE, Murthy SK, Edsall LC, Levade T, Milstien S, Spiegel S. Cell Death Differ. 2001;8:162–171. doi: 10.1038/sj.cdd.4400793. [DOI] [PubMed] [Google Scholar]
- [3].Herr DR, Chun J. Curr. Drug Targets. 2007;8:155–167. doi: 10.2174/138945007779315669. [DOI] [PubMed] [Google Scholar]
- [4].Spiegel S, Milstien S. Nature Rev. Mol. Cell Biol. 2003;4:397–407. doi: 10.1038/nrm1103. [DOI] [PubMed] [Google Scholar]
- [5].Spiegel S, Milstien S. J. Biol. Chem. 2007;282:2125–2129. doi: 10.1074/jbc.R600028200. [DOI] [PubMed] [Google Scholar]
- [6].Taha TA, Hannun YA, Obeid LM. J. Biochem. Mol. Biol. 2006;39:113–131. doi: 10.5483/bmbrep.2006.39.2.113. [DOI] [PubMed] [Google Scholar]
- [7].Leclercq TM, Pitson SM. IUBMB Life. 2006;58:467–472. doi: 10.1080/15216540600871126. [DOI] [PubMed] [Google Scholar]
- [8].Alvarez SE, Milstien S, Spiegel S. Trends Endocrinol. Metab. 2007;18:300–307. doi: 10.1016/j.tem.2007.07.005. [DOI] [PubMed] [Google Scholar]
- [9].Murata N, Sato K, Kon J, Tomura H, Yanagita M, Kuwabara A, Ui M, Okajima F. Biochem. J. 2000;352:809–815. [PMC free article] [PubMed] [Google Scholar]
- [10].Xia P, Gamble JR, Wang L, Pitson SM, Moretti PA, Wattenberg BW, D’Andrea RJ, Vadas MA. Curr. Biol. 2000;10:1527–1530. doi: 10.1016/s0960-9822(00)00834-4. [DOI] [PubMed] [Google Scholar]
- [11].Pitson SM, Xia P, Leclercq TM, Moretti PA, Zebol JR, Lynn HE, Wattenberg BW, Vadas MA. J. Exp. Med. 2005;201:49–54. doi: 10.1084/jem.20040559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Le Scolan E, Pchejetski D, Banno Y, Denis N, Mayeux P, Vainchenker W, Levade T, Moreau-Gachelin F. Blood. 2005;106:1808–1816. doi: 10.1182/blood-2004-12-4832. [DOI] [PubMed] [Google Scholar]
- [13].French KJ, Schrecengost RS, Lee BD, Zhuang Y, Smith SN, Eberly JL, Yun JK, Smith CD. Cancer Res. 2003;63:5962–5969. [PubMed] [Google Scholar]
- [14].Johnson KR, Johnson KY, Crellin HG, Ogretmen B, Boylan AM, Harley RA, Obeid LM. J. Histochem. Cytochem. 2005;53:1159–1166. doi: 10.1369/jhc.4A6606.2005. [DOI] [PubMed] [Google Scholar]
- [15].Sobue S, Iwasaki T, Sugisaki C, Nagata K, Kikuchi R, Murakami M, Takagi A, Kojima T, Banno Y, Akao Y, Nozawa Y, Kannagi R, Suzuki M, Abe A, Naoe T, Murate T. Leukemia. 2006;20:2042–2046. doi: 10.1038/sj.leu.2404386. [DOI] [PubMed] [Google Scholar]
- [16].Chan MM, Lu X, Merchant FM, Iglehart JD, Miron PL. Carcinogenesis. 2005;26:1343–1353. doi: 10.1093/carcin/bgi100. [DOI] [PubMed] [Google Scholar]
- [17].Ma XJ, Wang Z, Ryan PD, Isakoff SJ, Barmettler A, Fuller A, Muir B, Mohapatra G, Salunga R, Tuggle JT, Tran Y, Tran D, Tassin A, Amon P, Wang W, Enright E, Stecker K, Estepa-Sabal E, Smith B, Younger J, Balis U, Michaelson J, Bhan A, Habin K, Baer TM, Brugge J, Haber DA, Erlander MG, Sgroi DC. Cancer Cell. 2004;5:607–616. doi: 10.1016/j.ccr.2004.05.015. [DOI] [PubMed] [Google Scholar]
- [18].Nindl I, Dang C, Forschner T, Kuban RJ, Meyer T, Sterry W, Stockfleth E. Mol. Cancer. 2006;5:30. doi: 10.1186/1476-4598-5-30. doi:10.1186/1476-4598-5-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Talantov D, Mazumder A, Yu JX, Briggs T, Jiang Y, Backus J, Atkins D, Wang Y. Clin. Cancer Res. 2005;11:7234–7242. doi: 10.1158/1078-0432.CCR-05-0683. [DOI] [PubMed] [Google Scholar]
- [20].Wong YF, Selvanayagam ZE, Wei N, Porter J, Vittal R, Hu R, Lin Y, Liao J, Shih JW, Cheung TH, Lo KW, Yim SF, Yip SK, Ngong DT, Siu N, Chan LK, Chan CS, Kong T, Kutlina E, McKinnon RD, Denhardt DT, Chin KV, Chung TK. Clin. Cancer Res. 2003;9:5486–5492. [PubMed] [Google Scholar]
- [21].Dyrskjot L, Kruhoffer M, Thykjaer T, Marcussen N, Jensen JL, Moller K, Orntoft TF. Cancer Res. 2004;64:4040–4048. doi: 10.1158/0008-5472.CAN-03-3620. [DOI] [PubMed] [Google Scholar]
- [22].French PJ, Swagemakers SM, Nagel JH, Kouwenhoven MC, Brouwer E, van der Spek P, Luider TM, Kros JM, van den Bent MJ, Sillevis Smitt PA. Cancer Res. 2005;65:11335–11344. doi: 10.1158/0008-5472.CAN-05-1886. [DOI] [PubMed] [Google Scholar]
- [23].Pyeon D, Newton MA, Lambert PF, den Boon JA, Sengupta S, Marsit CJ, Woodworth CD, Connor JP, Haugen TH, Smith EM, Kelsey KT, Turek LP, Ahlquist P. Cancer Res. 2007;67:4605–4619. doi: 10.1158/0008-5472.CAN-06-3619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Ginos MA, Page GP, Michalowicz BS, Patel KJ, Volker SE, Pambuccian SE, Ondrey FG, Adams GL, Gaffney PM. Cancer Res. 2004;64:55–63. doi: 10.1158/0008-5472.can-03-2144. [DOI] [PubMed] [Google Scholar]
- [25].Andersson A, Ritz C, Lindgren D, Eden P, Lassen C, Heldrup J, Olofsson T, Rade J, Fontes M, Porwit-Macdonald A, Behrendtz M, Hoglund M, Johansson B, Fioretos T. Leukemia. 2007;21:1198–1203. doi: 10.1038/sj.leu.2404688. [DOI] [PubMed] [Google Scholar]
- [26].Korkola JE, Houldsworth J, Chadalavada RS, Olshen AB, Dobrzynski D, Reuter VE, Bosl GJ, Chaganti RS. Cancer Res. 2006;66:820–827. doi: 10.1158/0008-5472.CAN-05-2445. [DOI] [PubMed] [Google Scholar]
- [27].Hong Y, Ho KS, Eu KW, Cheah PY. Clin. Cancer Res. 2007;13:1107–1114. doi: 10.1158/1078-0432.CCR-06-1633. [DOI] [PubMed] [Google Scholar]
- [28].Kohno M, Momoi M, Oo ML, Paik JH, Lee YM, Venkataraman K, Ai Y, Ristimaki AP, Fyrst H, Sano H, Rosenberg D, Saba JD, Proia RL, Hla T. Mol. Cell Biol. 2006;26:7211–7223. doi: 10.1128/MCB.02341-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Ruckhaberle E, Rody A, Engels K, Gaetje R, von Minckwitz G, Schiffmann S, Grosch S, Geisslinger G, Holtrich U, Karn T, Kaufmann M. Breast Cancer Res. Treat. 2008 doi: 10.1007/s10549-007-9836-9. in press. [DOI] [PubMed] [Google Scholar]
- [30].Sutphen R, Xu Y, Wilbanks GD, Fiorica J, Grendys EC, Jr., LaPolla JP, Arango H, Hoffman MS, Martino M, Wakeley K, Griffin D, Blanco RW, Cantor AB, Xiao YJ, Krischer JP. Cancer Epidemiol. Biomarkers Prev. 2004;13:1185–1191. [PubMed] [Google Scholar]
- [31].Hong G, Baudhuin LM, Xu Y. FEBS Lett. 1999;460:513–518. doi: 10.1016/s0014-5793(99)01400-3. [DOI] [PubMed] [Google Scholar]
- [32].Pitson SM, Moretti PA, Zebol JR, Lynn HE, Xia P, Vadas MA, Wattenberg BW. EMBO J. 2003;22:5491–5500. doi: 10.1093/emboj/cdg540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Hobson JP, Rosenfeldt HM, Barak LS, Olivera A, Poulton S, Caron MG, Milstien S, Spiegel S. Science. 2001;291:1800–1803. doi: 10.1126/science.1057559. [DOI] [PubMed] [Google Scholar]
- [34].Stahelin RV, Hwang JH, Kim JH, Park ZY, Johnson KR, Obeid LM, Cho W. J. Biol. Chem. 2005;280:43030–43038. doi: 10.1074/jbc.M507574200. [DOI] [PubMed] [Google Scholar]
- [35].Doll F, Pfeilschifter J, Huwiler A. Biochim. Biophys. Acta. 2005;1738:72–81. doi: 10.1016/j.bbalip.2005.12.001. [DOI] [PubMed] [Google Scholar]
- [36].Sukocheva OA, Wang L, Albanese N, Pitson SM, Vadas MA, Xia P. Mol. Endocrinol. 2003;17:2002–2012. doi: 10.1210/me.2003-0119. [DOI] [PubMed] [Google Scholar]
- [37].Nakade Y, Banno Y, Hagiwara K, Sobue S, Koda M, Suzuki M, Kojima T, Takagi A, Asano H, Nozawa Y, Murate T. Biochim. Biophys. Acta. 2003;1635:104–116. doi: 10.1016/j.bbalip.2003.11.001. K. T.K. [DOI] [PubMed] [Google Scholar]
- [38].Huwiler A, Doll F, Ren S, Klawitter S, Greening A, Romer I, Bubnova S, Reinsberg L, Pfeilschifter J. Biochim. Biophys. Acta. 2006;1761:367–376. doi: 10.1016/j.bbalip.2006.02.007. [DOI] [PubMed] [Google Scholar]
- [39].Klawitter S, Hofmann LP, Pfeilschifter J, Huwiler A. Br. J. Pharmacol. 2007;150:271–280. doi: 10.1038/sj.bjp.0706983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Manggau M, Kim DS, Ruwisch L, Vogler R, Korting HC, Schafer-Korting M, Kleuser B. J. Invest. Dermatol. 2001;117:1241–1249. doi: 10.1046/j.0022-202x.2001.01496.x. [DOI] [PubMed] [Google Scholar]
- [41].Doll F, Pfeilschifter J, Huwiler A. Endocr. Relat. Cancer. 2007;14:325–335. doi: 10.1677/ERC-06-0050. [DOI] [PubMed] [Google Scholar]
- [42].Khan W, Dobrowsky R, Touny S, Hannun Y. Biochem. Biophys. Res. Commun. 1990;172:683–691. doi: 10.1016/0006-291x(90)90728-6. [DOI] [PubMed] [Google Scholar]
- [43].Sarkar S, Maceyka M, Hait NC, Paugh SW, Sankala H, Milstien S, Spiegel S. FEBS Lett. 2005;579:5313–5317. doi: 10.1016/j.febslet.2005.08.055. [DOI] [PubMed] [Google Scholar]
- [44].Hait NC, Sarkar S, Le Stunff H, Mikami A, Maceyka M, Milstien S, Spiegel S. J. Biol. Chem. 2005;280:29462–29469. doi: 10.1074/jbc.M502922200. [DOI] [PubMed] [Google Scholar]
- [45].Goparaju SK, Jolly PS, Watterson KR, Bektas M, Alvarez S, Sarkar S, Mel L, Ishii I, Chun J, Milstien S, Spiegel S. Mol. Cell Biol. 2005;25:4237–4249. doi: 10.1128/MCB.25.10.4237-4249.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Mitra P, Oskeritzian CA, Payne SG, Beaven MA, Milstien S, Spiegel S. Proc. Natl. Acad. Sci. U.S.A. 2006;103:16394–16399. doi: 10.1073/pnas.0603734103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Kobayashi N, Nishi T, Hirata T, Kihara A, Sano T, Igarashi Y, Yamaguchi A. J. Lipid Res. 2006;47:614–621. doi: 10.1194/jlr.M500468-JLR200. [DOI] [PubMed] [Google Scholar]
- [48].Sato K, Malchinkhuu E, Horiuchi Y, Mogi C, Tomura H, Tosaka M, Yoshimoto Y, Kuwabara A, Okajima F. J. Neurochem. 2008 in press. [Google Scholar]
- [49].Daub H, Weiss FU, Wallasch C, Ullrich A. Nature. 1996;379:557–560. doi: 10.1038/379557a0. [DOI] [PubMed] [Google Scholar]
- [50].Schafer B, Gschwind A, Ullrich A. Oncogene. 2004;23:991–999. doi: 10.1038/sj.onc.1207278. [DOI] [PubMed] [Google Scholar]
- [51].Gschwind A, Prenzel N, Ullrich A. Cancer. Res. 2002;62:6329–6336. [PubMed] [Google Scholar]
- [52].Jonker DJ, O’Callaghan CJ, Karapetis CS, Zalcberg JR, Tu D, Au HJ, Berry SR, Krahn M, Price T, Simes RJ, Tebbutt NC, van Hazel G, Wierzbicki R, Langer C, Moore MJ. N. Engl. J. Med. 2007;357:2040–2048. doi: 10.1056/NEJMoa071834. [DOI] [PubMed] [Google Scholar]
- [53].Shida D, Kitayama J, Yamaguchi H, Yamashita H, Mori K, Watanabe T, Yatomi Y, Nagawa H. FEBS Lett. 2004;577:333–338. doi: 10.1016/j.febslet.2004.10.024. [DOI] [PubMed] [Google Scholar]
- [54].Tripathy D, Slamon DJ, Cobleigh M, Arnold A, Saleh M, Mortimer JE, Murphy M, Stewart SJ. J. Clin. Oncol. 2004;22:1063–1070. doi: 10.1200/JCO.2004.06.557. [DOI] [PubMed] [Google Scholar]
- [55].Shida D, Kitayama J, Yamaguchi H, Yamashita H, Mori K, Watanabe T, Nagawa H. Biochem. Biophys. Res. Commun. 2005;327:907–914. doi: 10.1016/j.bbrc.2004.12.088. [DOI] [PubMed] [Google Scholar]
- [56].Baudhuin LM, Jiang Y, Zaslavsky A, Ishii I, Chun J, Xu Y. FASEB J. 2004;18:341–343. doi: 10.1096/fj.03-0302fje. [DOI] [PubMed] [Google Scholar]
- [57].Endo A, Nagashima KI, Kurose H, Mochizuki S, Matsuda M, Mochizuki N. J. Biol. Chem. 2002;277:23747–23754. doi: 10.1074/jbc.M111794200. [DOI] [PubMed] [Google Scholar]
- [58].Tanimoto T, Jin ZG, Berk BC. J. Biol. Chem. 2002;277:42997–43001. doi: 10.1074/jbc.M204764200. [DOI] [PubMed] [Google Scholar]
- [59].Liu F, Verin AD, Wang P, Day R, Wersto RP, Chrest FJ, English DK, Garcia JG. Am. J. Respir. Cell Mol. Biol. 2001;24:711–719. doi: 10.1165/ajrcmb.24.6.4323. [DOI] [PubMed] [Google Scholar]
- [60].Annabi B, Thibeault S, Lee YT, Bousquet-Gagnon N, Eliopoulos N, Barrette S, Galipeau J, Beliveau R. Exp. Hematol. 2003;31:640–649. doi: 10.1016/s0301-472x(03)00090-0. [DOI] [PubMed] [Google Scholar]
- [61].Lee MJ, Thangada S, Claffey KP, Ancellin N, Liu CH, Kluk M, Volpi M, Sha’afi RI, Hla T. Cell. 1999;99:301–312. doi: 10.1016/s0092-8674(00)81661-x. [DOI] [PubMed] [Google Scholar]
- [62].Visentin B, Vekich JA, Sibbald BJ, Cavalli AL, Moreno KM, Matteo RG, Garland WA, Lu Y, Yu S, Hall HS, Kundra V, Mills GB, Sabbadini RA. Cancer Cell. 2006;9:225–238. doi: 10.1016/j.ccr.2006.02.023. [DOI] [PubMed] [Google Scholar]
- [63].Liu Y, Wada R, Yamashita T, Mi Y, Deng CX, Hobson JP, Rosenfeldt HM, Nava VE, Chae SS, Lee MJ, Liu CH, Hla T, Spiegel S, Proia RL. J. Clin. Invest. 2000;106:951–961. doi: 10.1172/JCI10905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].KrumpKonvalinkova V, Yasuda S, Rubic T, Makarova N, Mages J, Erl W, Vosseler C, Kirkpatrick CJ, Tigyi G, Siess W. Arterioscler. Thromb. Vasc Biol. 2005;25:546–552. doi: 10.1161/01.ATV.0000154360.36106.d9. [DOI] [PubMed] [Google Scholar]
- [65].Igarashi J, Erwin PA, Dantas AP, Chen H, Michel T. Proc. Natl. Acad. Sci. U.S.A. 2003;100:10664–10669. doi: 10.1073/pnas.1934494100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Chae SS, Paik JH, Furneaux H, Hla T. J. Clin. Invest. 2004;114:1082–1089. doi: 10.1172/JCI22716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Limaye VS, Li X, Hahn C, Xia P, Berndt MC, Vadas MA, Gamble JR. Blood. 2005;105:3169–3177. doi: 10.1182/blood-2004-02-0452. [DOI] [PubMed] [Google Scholar]
- [68].Mizugishi K, Yamashita T, Olivera A, Miller GF, Spiegel S, Proia RL. Mol. Cell Biol. 2005;25:11113–11121. doi: 10.1128/MCB.25.24.11113-11121.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Takabe K, Paugh SW, Milstien S, Spiegel S. Pharmacol. Rev. 2008 doi: 10.1124/pr.107.07113. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Rosen H, Goetzl EJ. Nature Rev. Immunol. 2005;5:560–570. doi: 10.1038/nri1650. [DOI] [PubMed] [Google Scholar]
- [71].Igarashi N, Okada T, Hayashi S, Fujita T, Jahangeer S, Nakamura SI. J. Biol. Chem. 2003;278:46832–46839. doi: 10.1074/jbc.M306577200. [DOI] [PubMed] [Google Scholar]
- [72].Liu H, Toman RE, Goparaju S, Maceyka M, Nava VE, Sankala H, Payne SG, Bektas M, Ishii I, Chun J, Milstien S, Spiegel S. J. Biol. Chem. 2003;278:40330–40336. doi: 10.1074/jbc.M304455200. [DOI] [PubMed] [Google Scholar]
- [73].Maceyka M, Sankala H, Hait NC, Le Stunff H, Liu H, Toman R, Collier C, Zhang M, Satin L, Merrill AH, Jr., Milstien S, Spiegel S. J. Biol. Chem. 2005;280:37118–37129. doi: 10.1074/jbc.M502207200. [DOI] [PubMed] [Google Scholar]
- [74].Van Brocklyn JR, Jackson CA, Pearl DK, Kotur MS, Snyder PJ, Prior TW. J. Neuropathol. Exp. Neurol. 2005;64:695–705. doi: 10.1097/01.jnen.0000175329.59092.2c. [DOI] [PubMed] [Google Scholar]
- [75].Sankala HM, Hait NC, Paugh SW, Shida D, Lepine S, Elmore LW, Dent P, Milstien S, Spiegel S. Cancer Res. 2007;67:10466–10474. doi: 10.1158/0008-5472.CAN-07-2090. [DOI] [PubMed] [Google Scholar]
- [76].Samy ET, Meyer CA, Caplazi P, Langrish CL, Lora JM, Bluethmann H, Peng SL. J. Immunol. 2007;179:5644–5648. doi: 10.4049/jimmunol.179.9.5644. [DOI] [PubMed] [Google Scholar]
- [77].Allende ML, Sasaki T, Kawai H, Olivera A, Mi Y, van Echten-Deckert G, Hajdu R, Rosenbach M, Keohane CA, Mandala S, Spiegel S, Proia RL. J. Biol. Chem. 2004;279:52487–52492. doi: 10.1074/jbc.M406512200. [DOI] [PubMed] [Google Scholar]
- [78].Kawamori T, Osta W, Johnson KR, Pettus BJ, Bielawski J, Tanaka T, Wargovich MJ, Reddy BS, Hannun YA, Obeid LM, Zhou D. FASEB J. 2006;20:386–388. doi: 10.1096/fj.05-4331fje. [DOI] [PubMed] [Google Scholar]
- [79].Oskouian B, Sooriyakumaran P, Borowsky AD, Crans A, Dillard-Telm L, Tam YY, Bandhuvula P, Saba JD. Proc. Natl. Acad. Sci. U.S.A. 2006;103:17384–17389. doi: 10.1073/pnas.0600050103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Vogelstein B, Fearon ER, Hamilton SR, Kern SE, Preisinger AC, Leppert M, Nakamura Y, White R, Smits AM, Bos JL. N. Engl. J. Med. 1988;319:525–532. doi: 10.1056/NEJM198809013190901. [DOI] [PubMed] [Google Scholar]
- [81].Oskouian B, Saba J. Cell Cycle. 2007;6:522–527. doi: 10.4161/cc.6.5.3903. [DOI] [PubMed] [Google Scholar]
- [82].Pettus BJ, Bielawska A, Spiegel S, Roddy P, Hannun YA, Chalfant CE. J. Biol. Chem. 2003;278:38206–38213. doi: 10.1074/jbc.M304816200. [DOI] [PubMed] [Google Scholar]
- [83].Billich A, Bornancin F, Mechtcheriakova D, Natt F, Huesken D, Baumruker T. Cell Signal. 2005;17:1203–1217. doi: 10.1016/j.cellsig.2004.12.005. [DOI] [PubMed] [Google Scholar]
- [84].Taha TA, Kitatani K, El-Alwani M, Bielawski J, Hannun YA, Obeid LM. FASEB J. 2006;20:482–484. doi: 10.1096/fj.05-4412fje. [DOI] [PubMed] [Google Scholar]
- [85].Lichtner RB. Biomed. Pharmacother. 2003;57:447–451. doi: 10.1016/j.biopha.2003.09.006. [DOI] [PubMed] [Google Scholar]
- [86].Thurlimann B, Keshaviah A, Coates AS, Mouridsen H, Mauriac L, Forbes JF, Paridaens R, Castiglione-Gertsch M, Gelber RD, Rabaglio M, Smith I, Wardley A, Price KN, Goldhirsch A. N. Engl. J. Med. 2005;353:2747–2757. doi: 10.1056/NEJMoa052258. [DOI] [PubMed] [Google Scholar]
- [87].Nava VE, Hobson JP, Murthy S, Milstien S, Spiegel S. Exp. Cell Res. 2002;281:115–127. doi: 10.1006/excr.2002.5658. [DOI] [PubMed] [Google Scholar]
- [88].Sukocheva O, Wadham C, Holmes A, Albanese N, Verrier E, Feng F, Bernal A, Derian CK, Ullrich A, Vadas MA, Xia P. J. Cell Biol. 2006;173:301–310. doi: 10.1083/jcb.200506033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Sumitomo M, Ohba M, Asakuma J, Asano T, Kuroki T, Hayakawa M. J. Clin. Invest. 2002;109:827–836. doi: 10.1172/JCI14146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Ogretmen B, Hannun YA. Drug Resist. Update. 2001;4:368–377. doi: 10.1054/drup.2001.0225. [DOI] [PubMed] [Google Scholar]
- [91].Chmura SJ, Nodzenski E, Beckett MA, Kufe DW, Quintans J, Weichselbaum RR. Cancer Res. 1997;57:1270–1275. [PubMed] [Google Scholar]
- [92].Itoh M, Kitano T, Watanabe M, Kondo T, Yabu T, Taguchi Y, Iwai K, Tashima M, Uchiyama T, Okazaki T. Clin. Cancer Res. 2003;9:415–423. [PubMed] [Google Scholar]
- [93].Nava VE, Cuvillier O, Edsall LC, Kimura K, Milstien S, Gelmann EP, Spiegel S. Cancer Res. 2000;60:4468–4474. [PubMed] [Google Scholar]
- [94].Pchejetski D, Golzio M, Bonhoure E, Calvet C, Doumerc N, Garcia V, Mazerolles C, Rischmann P, Teissie J, Malavaud B, Cuvillier O. Cancer Res. 2005;65:11667–11675. doi: 10.1158/0008-5472.CAN-05-2702. [DOI] [PubMed] [Google Scholar]
- [95].Akao Y, Banno Y, Nakagawa Y, Hasegawa N, Kim TJ, Murate T, Igarashi Y, Nozawa Y. Biochem. Biophys. Res. Commun. 2006;342:1284–1290. doi: 10.1016/j.bbrc.2006.02.070. [DOI] [PubMed] [Google Scholar]
- [96].Bektas M, Jolly PS, Muller C, Eberle J, Spiegel S, Geilen CC. Oncogene. 2005;24:178–187. doi: 10.1038/sj.onc.1208019. [DOI] [PubMed] [Google Scholar]
- [97].Bonhoure E, Pchejetski D, Aouali N, Morjani H, Levade T, Kohama T, Cuvillier O. Leukemia. 2006;20:95–102. doi: 10.1038/sj.leu.2404023. [DOI] [PubMed] [Google Scholar]
- [98].Pilorget A, Demeule M, Barakat S, Marvaldi J, Luis J, Beliveau R. J. Neurochem. 2007;100:1203–1210. doi: 10.1111/j.1471-4159.2006.04295.x. [DOI] [PubMed] [Google Scholar]
- [99].Taha TA, Osta W, Kozhaya L, Bielawski J, Johnson KR, Gillanders WE, Dbaibo GS, Hannun YA, Obeid LM. J. Biol. Chem. 2004;279:20546–20554. doi: 10.1074/jbc.M401259200. [DOI] [PubMed] [Google Scholar]
- [100].Riboni L, Campanella R, Bassi R, Villani R, Gaini SM, Martinelli-Boneschi F, Viani P, Tettamanti G. Glia. 2002;39:105–113. doi: 10.1002/glia.10087. [DOI] [PubMed] [Google Scholar]
- [101].Van Brocklyn J, Letterle C, Snyder P, Prior T. Cancer Lett. 2002;181:195–204. doi: 10.1016/s0304-3835(02)00050-2. [DOI] [PubMed] [Google Scholar]
- [102].Kasza A, Kowanetz M, Poslednik K, Witek B, Kordula T, Koj A. Cytokine. 2001;16:187–190. doi: 10.1006/cyto.2001.0957. [DOI] [PubMed] [Google Scholar]
- [103].Paugh BS, Paugh SW, Bryan L, Kapitonov D, Wilczynska KM, Gopalan SM, Rokita H, Milstien S, Spiegel S, Kordula T. FASEB J. 2008 doi: 10.1096/fj.07-8276com. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Olivera A, Kohama T, Tu Z, Milstien S, Spiegel S. J. Biol. Chem. 1998;273:12576–12583. doi: 10.1074/jbc.273.20.12576. [DOI] [PubMed] [Google Scholar]
- [105].Liu H, Sugiura M, Nava VE, Edsall LC, Kono K, Poulton S, Milstien S, Kohama T, Spiegel S. J. Biol. Chem. 2000;275:19513–19520. doi: 10.1074/jbc.M002759200. [DOI] [PubMed] [Google Scholar]
- [106].Sweeney EA, Sakakura C, Shirahama T, Masamune A, Ohta H, Hakomori S, Igarashi Y. Int. J. Cancer. 1996;66:358–366. doi: 10.1002/(SICI)1097-0215(19960503)66:3<358::AID-IJC16>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- [107].Endo K, Igarashi Y, Nisar M, Zhou QH, Hakomori S. Cancer Res. 1991;51:1613–1618. [PubMed] [Google Scholar]
- [108].Sachs CW, Safa AR, Harrison SD, Fine RL. J. Biol. Chem. 1995;270:26639–26648. doi: 10.1074/jbc.270.44.26639. [DOI] [PubMed] [Google Scholar]
- [109].Maurer BJ, Melton L, Billups C, Cabot MC, Reynolds CP. J. Natl. Cancer Inst. 2000;92:1897–1909. doi: 10.1093/jnci/92.23.1897. [DOI] [PubMed] [Google Scholar]
- [110].Igarashi Y, Hakomori S. Biochem. Biophys. Res. Commun. 1989;164:1411–1416. doi: 10.1016/0006-291x(89)91827-5. [DOI] [PubMed] [Google Scholar]
- [111].French KJ, Upson JJ, Keller SN, Zhuang Y, Yun JK, Smith CD. J. Pharmacol. Exp. Ther. 2006;318:596–603. doi: 10.1124/jpet.106.101345. [DOI] [PubMed] [Google Scholar]
- [112].Maines LW, Fitzpatrick LR, French KJ, Zhuang Y, Xia Z, Keller SN, Upson JJ, Smith CD. Dig. Dis. Sci. 2008 doi: 10.1007/s10620-007-0133-6. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].Schwartz GK, Ward D, Saltz L, Casper ES, Spiess T, Mullen E, Woodworth J, Venuti R, Zervos P, Storniolo AM, Kelsen DP. Clin. Cancer Res. 1997;3:537–543. [PubMed] [Google Scholar]
- [114].Matsuoka Y, Nagahara Y, Ikekita M, Shinomiya T. Br. J. Pharmacol. 2003;138:1303–1312. doi: 10.1038/sj.bjp.0705182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].Neviani P, Santhanam R, Oaks JJ, Eiring AM, Notari M, Blaser BW, Liu S, Trotta R, Muthusamy N, Gambacorti-Passerini C, Druker BJ, Cortes J, Marcucci G, Chen CS, Verrills NM, Roy DC, Caligiuri MA, Bloomfield CD, Byrd JC, Perrotti D. J. Clin. Invest. 2007;117:2408–2421. doi: 10.1172/JCI31095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [116].Gamble JR, Xia P, Hahn CN, Drew JJ, Drogemuller CJ, Brown D, Vadas MA. Int. J. Cancer. 2006;118:2412–2420. doi: 10.1002/ijc.21682. [DOI] [PubMed] [Google Scholar]