

Abstract
Chemokine-mediated recruitment of regulatory cell subsets to the airway during inflammation and enhancement of their activities are potential strategies for therapeutic development in allergic asthma (AA). In this study, we aim to explore the role of XCL1, a chemokine associated with immune suppression and allergy, on CD4+CD25highCD127low/− regulatory T cell (Treg) function in AA. Flow cytometry and PCR analysis showed a reduction in XCL1 and XCR1 expression in AA Treg compared with healthy control and nonallergic asthmatic counterparts. This reduction in XCL1 expression was associated with the suboptimal regulatory function of Treg in AA. Interestingly, incubation with recombinant human XCL1 significantly increased Treg-mediated suppression and cytotoxicity by up-regulating expression of XCL1 and chief effector molecules of Treg function. Altogether, these results suggest an association between dysregulated XCL1 expression and reduced Treg activities in AA, as well as a potential role of XCL1 in reversing defective Treg function in the disease.
Allergic asthma is a chronic lung disease characterized by airway hyperreactivity, generally associated with Th2-biased immune responses. Such localized inflammatory responses have been attributed to the hyperresponsiveness of residential cell subsets and recruited populations of both innate immune cells and Th2-biased CD4+ T cells (1, 2). However, recent findings suggested that other regulatory cell types, such as naturally occurring regulatory T cells (Treg),3 also play a key role in mediating immune responses in allergic asthma (AA; Ref. 3). Treg are CD4+CD25highCD127low/− T cells that are chiefly involved in peripheral tolerance (4). In human AA, Treg do not suppress the proliferation and cytokine production of allergen-stimulated CD4+ T cells in AA subjects as well as in healthy individuals (5, 6). Furthermore, several murine studies have demonstrated that the induction of Treg function reverses airway hyperresponsiveness (7) and protects against experimentally induced asthma (8). Collectively, these findings suggest a role of Treg in controlling Th2 inflammation in AA.
Immune regulation by Treg has been suggested as a multifactorial phenomenon. Several molecules such as Foxp3, IL-10, TGF-β, CTLA-4, glucocorticoid-induced TNF receptor, and lymphocyte activation gene 3, have been associated with regulatory activities of Treg (9, 10). Treg also express multiple chemokine receptors controlling their migration to different tissues (11). In specific disease models, chemokines and their receptors have also been shown to be critical determinants for optimal Treg function. CCR2+ Treg are suggested to be more suppressive against pneumonitis progression than CCR2− counterparts (12). Chemotactic responses of Treg to CCL17 and CCL22 are beneficial in mounting effective immune responses in several forms of cancer (13, 14). CXCR3+ Treg are involved in immune tolerance in graft-vs-host diseases (15). Thus, modulation of Treg function by chemokines is a promising strategy for therapeutic development.
Although a large number of studies up-to-date have explored the role of chemokines in regulating inflammatory responses in AA, mechanisms of action of chemokines on Treg function in AA have not been well established. Thus, this study aims to investigate the role of chemokines in functional maintenance of Treg in AA. Our preliminary analysis of several T cell-associated chemokine/chemokine receptor mRNA transcripts showed distinct profiles between Treg and CD4+CD25− responder T cells (see Fig. 1B). More interestingly, our analysis showed significant differences in the expression of XCL1 and XCR1 between healthy control (HC) Treg and AA Treg, suggesting an involvement of this chemokine and its receptor in AA pathobiology (see Fig. 1B).
FIGURE 1.
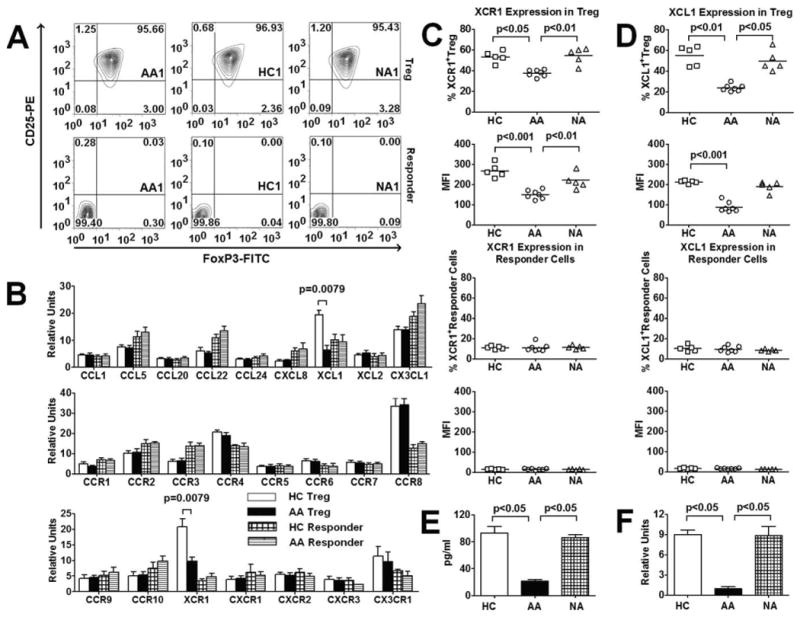
XCL1 and XCR1 expression by Treg and responder T cells among three subject groups. A, Purity of flow cytometry-sorted Treg (top row) and responder T cells (bottom row) in AA, HC, and NA representatives. FACS plots showed expression of CD25 and Foxp3 in sorted cells. B, Expression of mRNA transcripts of chemokines and chemokine receptors by Treg and responder T cells in HC and AA subjects (n = 5 for each group). Data were presented as fold expression of a candidate gene over the expression of the housekeeping gene β2-microglobulin. p values represented significant differences from unpaired t tests that had been corrected for multiple comparisons with Bonferroni method. C and D, Protein expression of XCR1 and XCL1 by Treg and responder T cells from AA (n = 7), HC (n = 5), and NA (n = 5) subjects. p values represented significant differences from ANOVA tests. E, Expression of XCL1 in BAL from AA, HC, and NA subjects by ELISA. F, Expression of XCL1 by BAL Treg from AA, HC, and NA subjects by QT-PCR. In E and F, p values represented significant differences from Mann-Whitney tests that had been corrected for multiple comparisons with Bonferroni method (n = 4 for each group). Bar heights with SD error bars represented the average value of a triplicate sample. Dot plot graphs with horizontal bars represented median values.
XCL1 (lymphotactin) is a chemokine with relatively similar structure to that of CC and CXC chemokines with its associated receptor XCR1 (16). XCL1 and XCR1 are expressed in many lymphocyte subsets such as T cells and NK cells (17). Besides its specific chemotactic properties on these cell populations, XCL1 can exert a potent stimulatory effect on their suppression and cytotoxicity against tumors and responder T cells (18–20). Moreover, XCL1 has been reported to play a role in recruitment of lymphocytes to areas of allergic inflammation (21, 22). Therefore, along with our preliminary mRNA analysis of XCL1/XCR1 expression by Treg, these evidences prompt us to examine possible interaction between XCL1 and Treg in AA. In this study we show that reduced Treg function in AA is associated with dysregulation of their XCL1 expression, and this chemokine is involved in the modulation of Treg-mediated suppression and cytotoxicity in AA.
Materials and Methods
Human subjects
The study was approved by the Stanford Administrative Panel on Human Subjects in Medical Research. All subjects signed informed consent forms before participating in the study. The diagnosis of asthma was assessed via National Heart, Lung, and Blood Institute guidelines (www.nhlbi.nih.gov/guidelines/asthma/asthgdln.pdf), using clinical symptoms, signs, and forced expiratory volume in 1 s (FEV1). Asthma was defined by airflow obstruction that is at least partially reversible and is measured by spirometry. Reversibility is determined by an increase in FEV1 of >200 ml and >12% from baseline measure after inhalation of short-acting β2-agonist. Comprehensive clinical data were collected at each patient visit, including history, disease severity, medication status, common allergens, IgE level, and FEV1. AA and nonallergic asthmatics (NA) were classified based on physical exam, history, and elevated blood IgE levels (>50 IU/ml for AA) and positive skin tests to allergens (for AA). Patients with FEV1 <60% were considered severe. Those in the range of 60–80% were considered moderate, and those with FEV1 >80% were considered mild. HC were defined as nonsmoking subjects >13 years of age with a total serum IgE of <25 IU/ml, negative skin testing as compared with positive histamine control, and no evidence of lung diseases. In addition, on spirometry testing, there was no evidence of obstructive or restrictive lung disease for HC. Bronchoalveolar lavage fluid (BAL) samples were collected with a standardized protocol for clinical research at Lucile Packard Children's Hospital (Palo Alto, CA).
Cell isolation
CD4+ T cells were first isolated from buffy coats derived from up to 400 ml of whole blood with CD4+ Rosette Kit (StemCell Technologies). CD4+ T cells were incubated with anti-CD127-allophycocyanin and anti-CD25-PE and anti CD4-FITC Abs (Miltenyi Biotech) before undergoing flow cytometry sorting for CD4+CD25highCD127low/− Treg and CD4+CD25− responder T cells.
Suppression assays
Autologous Treg and responder T cells were cultured at 75,000 cells per ml of RPMI 1640 plus 10% FBS plus 1% l-glutamine (C medium) while allogeneic irradiated APCs (CD3+ depleted cells) were cultured at 750,000 per ml of C medium. Anti-CD3 Abs (clone HIT3; BD Biosciences) were used at 2 μg/ml. Cells and Ab solution were plated at 50 μl per well in 96-well U-bottom plates. Additional medium was added to wells so that the final volume in each well reached 200 μl. Cells were pulsed with 1 μCi of thymidine (25 μl) per well on day 6 and harvested on day 7 with a cell harvester. Thymidine incorporation was determined using a 1450 Microbeta Wallac Trilux liquid scintillation counter. To evaluate effects XCL1 and other chemokines on Treg-mediated suppression, recombinant human XCL1 (gifts from the Krensky Laboratory, Stanford University, Stanford, CA), CCL1, CCL5, CCL17, and CCL22 (PeproTech) were introduced into the assays at various concentrations at day 0. For analysis of ex vivo effects of steroids and IL-4 on Treg function, prednisone (Novartis Pharmaceuticals) and IL-4 (PeproTech) were added at day 0 to cell cultures at 1000 U/ml. Proliferation assays with Treg alone and responder T cells alone were also performed in the presence of XCL1 and other chemokines. All assays were performed in triplicates.
Flow-based killing assay
Treg were left inactivated in C medium with supplemented IL-2 at 10U/ml for 4 days after purification. Alternatively, they were stimulated with plate-bound anti-CD3 (clone HIT3; BD Biosciences)/CD46 (clone E4.3; BD Biosciences) Abs, each at 5 μg/ml, for 4 days in C medium with supplemented IL-2 as previously described (23, 24). Responder T cells (CD4+CD25− targets) were stimulated with plate-bound anti-CD3/CD28 Abs (BD Biosciences), each at 5 μg/ml, in C medium with supplemented IL-2 for 4 days. Responder T cells were then labeled with CFSE (Invitrogen) at the final concentration of 2 μM at 37°C in the dark for 10 min and washed with PBS. Treg were cocultured with CFSE-labeled targets for 4 h at ratio of 5 × 105 or 5 × 104 Treg to 5 × 104 responder T cells (10:1 and 1:1) at 37°C. 7-aminoactinomycin D (7-AAD; Sigma-Aldrich) was added after 4 h at 1 μg/ml in PBS with 1% paraformaldehyde before flow cytometry analysis.
Cell staining
For granzyme A and granzyme B detection, Treg were stimulated with plate-bound CD3/CD46 Abs, each at 5 μg/ml, in C medium with supplemented IL-2 at 10U/ml for 4 days. For Foxp3, XCL1, and XCR1 detection, cells were analyzed right after purification, except for XCL1 incubation experiments, where cells were incubated with XCL1 in C medium and supplemented IL-2 for 4 days. For IL-10 and TGF-β detection, cells were stimulated with PMA (10 ng/ml; Sigma-Aldrich) and ionomycin (1 μg/ml; Sigma-Aldrich) for 5 h in the presence of brefeldin A (diluted 1× solution was added in the last 2.5 h; BioLegend). Cells were fixed and permeabilized with Cytofix/Cytoperm solution (BD Biosciences), then stained with Abs for Foxp3-FITC (clone 206D), IL-10-PE (BioLegend), granzyme A-PE (BD Biosciences), and granzyme B-PE (Caltag Laboratories). XCL1/XCR1 and TGF-β detection were performed with purified anti-human XCL1/XCR1 and TGF-β Abs (R&D Systems) and conjugated with FITC and PE secondary Abs per manufacturers' standard protocols. Data were collected on a FACSCalibur (BD Biosciences) and then analyzed with FlowJo software (Tree Star).
Quantitative PCR (QT-PCR) analysis
RNA was isolated using RNeasy kits (Qiagen) according to the manufacturer's protocol. Similar cell number (200,000 for peripheral blood cells and 100,000 for BAL cells) were used for each subject. For cDNA synthesis, 500 ng of total RNA was transcribed with cDNA transcription reagents (Applied Biosystems) using random hexamers, according to the manufacturer's protocol. Gene expression was measured in real time using primers and other reagents purchased from Applied Biosystems and SuperArray. All PCR assays were performed in triplicates. Data were presented as relative fold expression of the candidate gene to the expression of the housekeeping gene β2-microglobulin.
Statistical analysis
Data were tested for normality and variance equality before being subjected to appropriate statistical tests. All statistical procedures were performed with Prism software (GraphPad). Differences with p < 0.05 were considered statistically significant. Correction for multiple comparisons was performed via Bonferroni method.
Results
Decreased XCL1 and XCR1 expression by allergic asthmatic Treg
Subjects included 11 HC, 11 mild-to-severe AA, and 7 mild-to-moderate NA (Table I). CD4+CD25highCD127low/− Treg and CD4+CD25− cells (responder T cells) were isolated by flow cytometry sorting. Purity of Treg and responder T cells was confirmed with intracellular staining for Foxp3 to be more than 95 and 99%, respectively (Fig. 1A). To confirm our preliminary QT-PCR data (Fig. 1B), we performed flow cytometry analysis of XCL1/XCR1 protein expression in Treg among three subject groups. Our results showed a reduced expression of XCL1/XCR1 in Treg from AA subjects compared with HC and NA in both percentage of XCL1-positive cells and the mean fluorescent intensity (MFI) (Fig. 1, C and D). We also examined responder T cells that showed significantly lower expression of XCL1/XCR1 compared with Treg, but no significant differences among three subject groups (Fig. 1, C and D), suggesting that the reduction in XCL1 expression in AA was specific to Treg.
Table I. Subject demographicsa.
| ID | Age (years) | Duration (years) | Severity | Medications | IgE (IU/ml) | Allergen Status | FEV1 (%) |
|---|---|---|---|---|---|---|---|
| AA1 | 35 | 3 | Severe | ICS, OCS, Salmeterol | 164 | Tree, Grass, Mold, Weed | 54 |
| AA2 | 16 | 14 | Severe | OCS, Xolair | 3470 | Tree, Weed, Cat | 52 |
| AA3 | 23 | 22 | Moderate | ICS, Salmeterol | 2361 | Weed, Dust Mite | 71 |
| AA4 | 20 | 16 | Moderate | ICS, Salmeterol | 1493 | Weed, Dust Mite | 73 |
| AA5 | 13 | 10 | Moderate | Albuterol | 186 | Weed, Grass | 73 |
| AA6 | 15 | 5 | Mild | ICS, Salmeterol | 275 | Grass | 82 |
| AA7 | 29 | 25 | Mild | Albuterol | 205 | Grass, Mold | 90 |
| AA8 | 19 | 5 | Mild | Albuterol | 508 | Weed | 92 |
| AA9 | 38 | 36 | Mild | Albuterol | 292 | Tree, Grass | 93 |
| AA10 | 18 | 2 | Mild | Albuterol | 455 | Dust Mite | 90 |
| AA11 | 17 | 11 | Mild | Albuterol | 141 | Weed, Dog | 95 |
| 22.1 ± 2.5 | 13.6 ± 3.2 | 868.2 ± 334.4 | 87.5 ± 3.0 | ||||
| NA1 | 42 | 21 | Moderate | ICS, Albuterol | less than 50 | N/A | 69 |
| NA2 | 44 | N/A | Mild | Albuterol | less than 50 | N/A | 85 |
| NA3 | 55 | N/A | Mild | Albuterol | less than 50 | N/A | 88 |
| NA4 | 25 | 5 | Mild | Albuterol | less than 50 | N/A | 95 |
| NA5 | 38 | N/A | Mild | Albuterol | less than 50 | N/A | 92 |
| NA6 | 19 | N/A | Mild | Albuterol | less than 50 | N/A | 91 |
| NA7 | 45 | 25 | Mild | Albuterol | less than 50 | N/A | 85 |
| 38.3 ± 4.7 | 17.0 ± 6.1 | less than 50 | 86.4 ± 3.2 |
ICS, inhaled corticosteroids; N/A, not available.
Furthermore, we collected BAL from allergen-challenged AA, NA, and HC subjects to investigate the possible involvement of XCL1 in airway inflammation in AA. We first examined XCL1 levels in BAL via ELISA. We found that XCL1 levels from AA BAL were significantly lower than those from HC and NA subjects (Fig. 1E). We subsequently purified Treg from these BAL samples via flow cytometry sorting to analyze their XCL1 expression. Due to the small numbers of Treg obtained from BAL, we performed QT-PCR analysis of XCL1 mRNA expression in these cells instead of examining XCL1/XCR1 protein expression. Similar numbers of Treg were used for each subject in PCR analysis. Our results showed that Treg from AA subjects expressed significantly lower levels of XCL1 mRNA transcripts compared with HC and NA subjects (Fig. 1F). Altogether, these data suggested that the reduced XCL1 expression in Treg in AA was associated with airway inflammation in the disease.
Allergic asthmatic Treg exhibited suboptimal suppressive and cytotoxic activities
Standard 3H incorporation assays were used to evaluate suppressive activities of Treg against autologous responder T cells. Responder T cells were exposed to plate-bound anti-CD3 Abs as TCR stimuli in the presence of irradiated CD3-depleted APCs from allogeneic HC and autologous Treg. To avoid potential individual variations in proliferative responses of responder T cells in the interpretation of suppressive function of Treg, the degree of suppression of responder T cell proliferation by Treg was quantitated by percentage reduction in 3H uptake in cocultures of Treg and responder T cells compared with cultures of responder T cells alone for each subject. Treg from both AA subjects and HC exhibited suppressive activities against responder T cell proliferation (Fig. 2A). However, AA Treg showed markedly reduced suppressive activities against autologous responder T cell proliferation in comparison to HC Treg. This phenomenon was observed at both 1:1 and 1:4 ratios of Treg to responder T cells (Fig. 2A). Interestingly, Treg from NA subjects showed no significant changes in suppressive activities against autologous responder T cell proliferation compared with HC Treg (Fig. 2A), suggesting that reduced Treg-mediated suppression is specific to AA.
FIGURE 2.
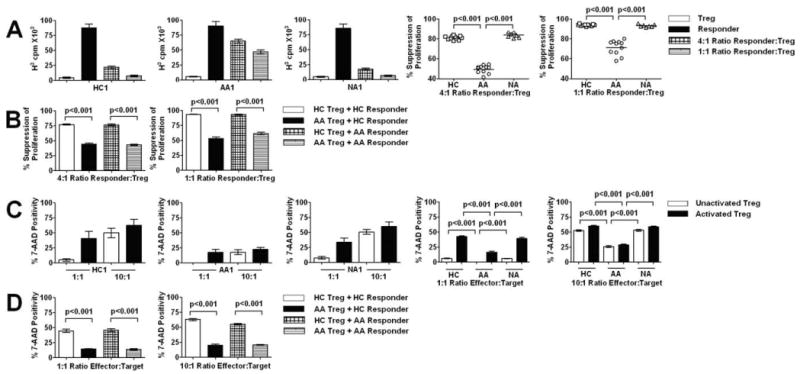
Quantitation of in vitro suppressive and cytotoxic activities of Treg among three subject groups. A, Bar graphs showed representatives of suppression assays in HC, AA, and NA subjects. Dot plot graphs showed summary of degree of suppression at 1:4 and 1:1 ratio of Treg to responder T cells. B, Allogeneic suppression assays at 1:4 and 1:1 ratio of Treg to responder T cells. C, Bar graphs showed representatives of killing assays in HC, AA, and NA subjects. Treg (inactivated or activated with anti-CD3/CD46 Abs) and responder T cells (activated with anti-CD3/CD28 Abs in all experiments) were cocultured at 1:1 and 10:1 ratios. Killing was quantitated by levels of 7-AAD incorporation by CFSE+ responder T cells in each cell culture. Dot plot graphs showed summary of degree of killing at 1:1 and 10:1 ratio of Treg to responder T cells. D, Allogeneic killing assays at 1:1 and 1:10 ratio of Treg to activated responder T cells. Horizontal bars in dot plot graphs represented median values. In A and C, bar heights with SD error bars represented the average value of a triplicate sample. p values represented significant differences from ANOVA tests of AA (n = 11), HC (n = 11), and NA (n = 7) subjects. In B and D, bar heights with SD error bars represented the average value of five subjects. p values represented significant differences from ANOVA tests (n = 5 for each group).
In addition to their ability to suppress cell proliferation, Treg have recently been shown to possess potent cytotoxicity against a wide range of target cells upon TCR stimulation by either anti-CD3/CD28 Abs or anti-CD3/CD46 Abs (23, 24). Therefore, this functional property of Treg was also examined in AA subjects by a flow-based killing assay previously described by Grossman et al. (24). Treg (effectors) were cultured for 4 days with IL-2 or with additional stimuli and IL-2 while responder T cells (targets) were activated with IL-2 and plate-bound anti-CD3/CD28 Abs for 4 days (please see Materials and Methods for detailed stimulation protocols). At the beginning of the assays, responder T cells were labeled with CFSE and incubated with Treg. Responder T cell death was detected by 7-AAD staining of CFSE-labeled cells before flow cytometry analysis. As previously demonstrated, anti-CD3/CD46 stimulation of Treg and anti-CD3/CD28 stimulation of responder T cells showed optimal killing of responder T cells by Treg (24). Thus, this method of activation was chosen to examine Treg function. We have performed killing assays with anti-CD3/CD28 stimulation of Treg and killings assays with unactivated responder T cells and found suboptimal cytotoxicity, similar to studies described by Grossman et al. (Ref. 23; data not shown). The 4-hour killing assays at 1:1 ratio of Treg to autologous responder T cells showed evidence of responder T cell death, which increased with IL-2 and anti-CD3/CD46 stimulation of Treg compared with Treg from cultures with IL-2 alone, in both HC and NA subjects (Fig. 2C). Surprisingly, killing was undetectable in AA subjects when Treg were not activated with anti-CD3/CD46 Abs (Fig. 2C). By either adding anti-CD3/CD46 Abs or increasing cell ratio to 10:1 of Treg to autologous responder T cells, responder T cell death became apparent in AA subjects but failed to reach the level from both HC and NA subjects under the same experimental conditions (Fig. 2C). Along with findings in suppression assays, these results suggested a reduced regulatory function of Treg from AA subjects.
To rule out the possibility that the suboptimal suppression and cytotoxicity observed above resulted from decreased susceptibility of responder T cells to Treg activities, we have performed suppression assays and killing assays with mixed allogeneic T cells from HC and AA subjects. Treg or responder T cells from each AA subject were paired with their HC counterparts in suppression and killing assays. AA Treg showed reduced killing and suppression of allogeneic responder T cells from HC subjects, whereas HC Treg exerted optimal cytotoxicity and suppression against allogeneic AA responder T cells (Fig. 2, B and D). These data suggested that the reduced suppression and killing observed in this study resulted from defective Treg function in AA.
Reduced Treg function in allergic asthmatics correlated with decreased XCL1 expression
Expression of molecules implicated in Treg-mediated suppression and cytotoxicity was examined next by intracellular staining on purified Treg. Our analysis focused on the expression of Foxp3, IL-10, TGF-β, granzyme A, and granzyme B. Treg from all subject groups express similar numbers of Foxp3+ cells (>95%); however, Treg from AA subjects expressed significantly lower levels of Foxp3 than HC and NA subjects via MFI analysis (Fig. 3A). IL-10 and TGF-β expression by Treg were similar among HC, AA, and NA subjects (Fig. 3, B and C). However, upon receiving anti-CD3/CD46 stimuli, AA Treg expressed markedly lower levels of granzyme A, in terms of granzyme A+ cells and MFI, and lower levels of granzyme B, in terms of MFI, than did HC and NA subjects (Fig. 3, D and E). Correlation analysis showed that Foxp3 expression positively correlated with Treg-mediated suppression of responder T cell proliferation, whereas granzyme A expression positively correlated with Treg-mediated cytotoxicity against responder T cells in AA subjects (Table II). Furthermore, XCL1 expression positively correlated with expression of Foxp3 and granzyme A and granzyme B (Table II). We have performed correlation analysis with XCR1 expression and found no significant correlations with Treg function (data not shown). Altogether, these data suggested that lower level of XCL1 was associated with dysfunctional Treg activity in AA.
FIGURE 3.
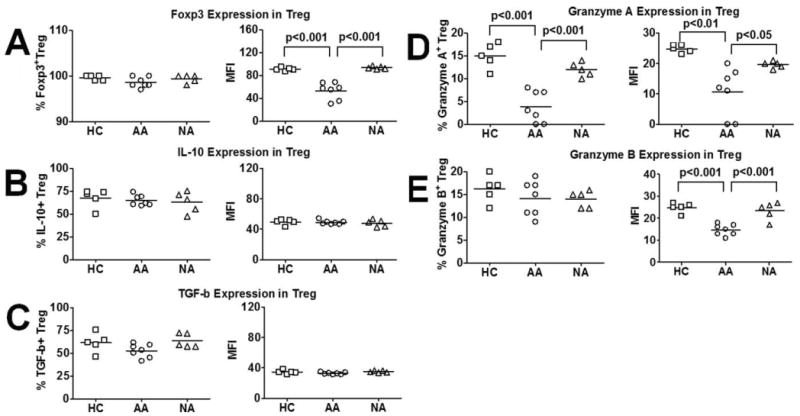
Expression of effector molecules of Treg function among three subject groups. A, Foxp3 expression by Treg in AA, HC, and NA subjects. B, IL-10 expression by Treg in AA, HC, and NA subjects. C, TGF-β expression by Treg in AA, HC, and NA subjects. D, Granzyme A expression by Treg in AA, HC, and NA subjects. E, Granzyme B expression by Treg in AA, HC, and NA subjects. Data were presented by means of percentages as well as MFI of cells that expressed candidate molecules. Horizontal bars in dot plot graphs represented median values. p values represented significant differences from ANOVA tests of AA (n = 7), HC (n = 5), and NA (n = 5) subjects.
Table II. Correlation analysis of selected parameters of Treg function, clinical assessment of allergic asthma, and XCL1 expression by Treg.
| FoxP3 MFI versus | Degree of Suppression 4:1 Ratio | Degree of Suppression 1:1 Ratio |
|---|---|---|
| N | 7 | 7 |
| RValue | 0.8929 | 0.7857 |
| p Value | 0.01239 | 0.048 |
| Granzyme A MFI versus | Degree of Killing 10:1 Ratio | Degree of Killing 10:1 Ratio with CD3/CD46 |
|
| ||
| N | 7 | 7 |
| RValue | 0.955 | 0.8829 |
| p Value | 0.0028 | 0.0123 |
| XCL1 MFI versus | Foxp3 MFI | |
|
| ||
| N | 7 | |
| RValue | 0.937 | |
| p Value | 0.0067 | |
| XCL1 MFI versus | Granzyme A MFI | Granzyme B MFI |
|
| ||
| N | 7 | 7 |
| RValue | 0.8469 | 0.8455 |
| p Value | 0.0238 | 0.0238 |
| FEV1 (%) versus | Degree of Suppression 4:1 Ratio | Degree of Suppression 1:1 Ratio |
|
| ||
| N | 11 | 11 |
| RValue | 0.621 | 0.7443 |
| p Value | 0.0414 | 0.0086 |
| FEV1 (%) versus | Degree of Killing 10:1 Ratio | Degree of Killing 10:1 Ratio with CD3/CD46 |
|
| ||
| N | 11 | 11 |
| RValue | 0.6995 | 0.8238 |
| p Value | 0.0166 | 0.0018 |
XCL1 enhanced Treg-mediated suppression and cytotoxicity in allergic asthmatics
XCL1 has been shown to improve immune suppression (18 – 20). Along with this finding, the association between reduced Treg function and decreased XCL1 expression in AA Treg prompted us to examine the hypothesis that exogenous XCL1 could possibly improve Treg activity in AA. We introduced XCL1 in suppression assays and killing assays of Treg against responder T cells from AA subjects. The optimal dose of XCL1 was determined to be 200 ng/ml (Fig. 4A), which was similar to those concentrations of XCL1 used by previous studies (17, 21). For suppression assays, XCL1 was added in cell cultures at the beginning of the 7-day assay. For killing assays, XCL1 was added at the beginning of the 4-day activation period of Treg with IL-2 and plate-bound anti-CD3/CD46 Abs. Suppression assay results showed a significant enhancement of AA Treg suppression of autologous responder T cell proliferation at 1:1 cell ratio in the presence of XCL1 (Fig. 4B). Further cultures of AA Treg/responder T cell alone in the presence of XCL1 indicated no significant effects of XCL1 on either Treg or responder T cell proliferation, suggesting that XCL1 modulated suppressive activity of Treg rather than proliferative potential of responder T cells or Treg (Fig. 4C). Killing assays of AA Treg against autologous responder T cells also showed a significantly elevated level of responder T cell death in cultures with XCL1 (Fig. 4B). This phenomenon was not due to XCL1 effects on cell viability, because analysis of both Treg and responder T cells before and after 4-day cultures by trypan blue exclusion assay or propidium iodide/annexin staining showed no significant signs of XCL1-induced cell death (data not shown).
FIGURE 4.
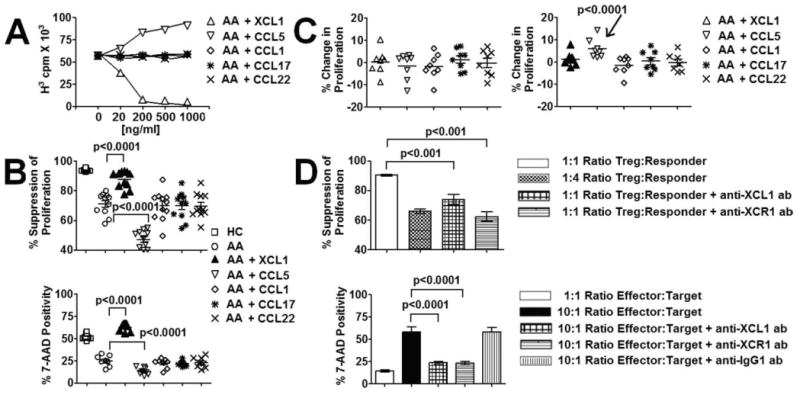
Modulation of Treg-mediated suppression and cytotoxicity by XCL1. A, Effects of exogenous XCL1 and several other chemokines on Treg-mediated suppression of responder T cell proliferation at different concentrations. Representative data of cocultures of Treg and responder T cells at 1:1 ratio comes from an AA subject. Each data point represented the average values of a triplicate sample from an AA subject. B, Effects of XCL1 and several other chemokines on Treg-mediated suppression and cytotoxicity against autologous responder T cells in AA subjects. Optimal concentration of XCL1 and CCL5 (200 ng/ml) were used in these experiments. Chemokines that showed no effects on Treg function were also used at 200 ng/ml. Horizontal bars in dot plot graphs represented median values. p values represented significant differences from paired t tests between assays with AA cells alone and assays with chemokine-primed AA cells (n = 11 for each group). C, Effects of XCL1 and several other chemokines on Treg anergy and responder T cell proliferation in AA subjects. Fold changes in proliferation were compared with 1 (indicating no changes) in statistical tests. Increased proliferation of CCL5-primed responder T cells was statistically significant (p < 0.0001). D, Suppression and killing assays with blocking Abs against XCL1 or XCR1 in HC subjects. Optimal concentrations of blocking Abs were determined to be 25 μg/ml (R&D Systems). Negative control Abs (IgG1 Abs with low endotoxin; BD Biosciences) were used at the same concentration. Bar heights with SD error bars represented the average value of 5 subjects. p values represented significant differences from ANOVA tests (n = 5 for each group).
Because chemokines possess shared signaling pathways, we also tested the specificity of the XCL1 effect by introducing other chemokines such as CCL1, CCL5, CCL17, and CCL22 into suppression and killing assays. These chemokines were chosen due to their suggested involvement in Treg function and AA. Interestingly, CCL1, CCL17, and CCL22 did not enhance regulatory activities of AA Treg (Fig. 4B). CCL5, a chemokine associated with T cell activation and proliferation (25, 26), as well as asthma exacerbation (27, 28) in similar experimental settings, resulted in further reduction in AA Treg-mediated suppression and cytotoxicity (Fig. 4B). Cultures of CCL5 with Treg or responder T cell alone also suggested that CCL5 possibly reduced Treg function by acting via expansion/activation of responder T cells, because CCL5 incubation induced a significant increase in responder T cell proliferation but not Treg proliferation (Fig. 4C).
To confirm the role of XCL1 in modulating Treg function, we performed suppression assays and killing assays with XCL1 and XCR1 neutralizing Abs. We performed these assays on HC subjects that exhibited normal Treg function. These blocking reagents were added at day 0 in suppression assays and at the beginning of the 4-day activation period of Treg in killing assays. In suppression assays, both neutralizing Abs reduced Treg suppressive function against responder T cell proliferation (Fig. 4D). Killing assays with XCL1 or XCR1 neutralizing Abs also resulted in a reduction in cytotoxicity of Treg against target cells (Fig. 4D). Similar experiments were performed with “negative control” IgG1 Abs (BD Biosciences) that did not show any influences on regulatory activities of Treg (Fig. 4D).
XCL1 up-regulated expression of XCR1/XCL1 and molecules associated with Treg-mediated suppression and cytotoxicity in allergic asthmatics
In association with these functional findings, we examined the effects of recombinant XCL1 on the expression of Foxp3, IL-10, TGF-β, granzyme A, and granzyme B, as well as XCL1/XCR1 by Treg. For Foxp3, IL-10, TGF-β, and XCL1/XCR1 detection, Treg were incubated with XCL1 in C medium with IL-2 supplement for 4 days before analysis. For granzyme A and granzyme B detection, Treg were incubated with XCL1 and anti-CD3/CD46 Abs in C medium with IL-2 supplement for 4 days. Exogenous XCL1 enhanced the expression of Foxp3, IL-10, TGF-β, granzyme A, and granzyme B by Treg (Fig. 5, A–D). On the contrary, similar experiments performed with other chemokines such as CCL1, CCL5, CCL17, and CCL22 did not show any significant modulatory effects of these chemokines on the expression of these proteins. Interestingly, incubation of Treg with XCL1 up-regulated their XCL1 and XCR1 expression (Fig. 5, F and G), suggesting that XCL1 may act on Treg via a positive feedback, autocrine mechanism.
FIGURE 5.
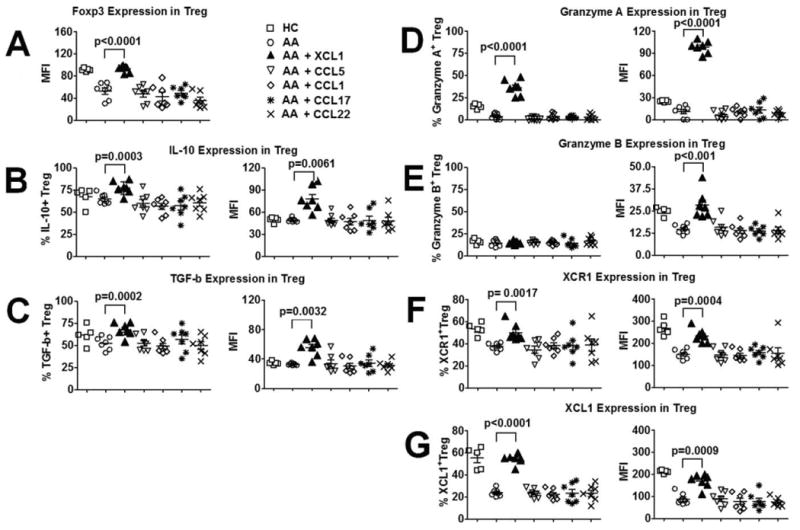
Modulation of expression of effector molecules of Treg function by XCL1. A, Effects of XCL1 and several other chemokines on Foxp3 expression of Treg in AA subjects. B, Effects of XCL1 and several other chemokines on IL-10 expression of Treg in AA subjects. C, Effects of XCL1 and several other chemokines on TGF-β expression of Treg in AA subjects. D, Effects of XCL1 and several other chemokines on granzyme A expression of Treg in AA subjects. E, Effects of XCL1 and several other chemokines on granzyme B expression of Treg in AA subjects. F, Effects of XCL1 and several other chemokines on XCR1 expression of Treg in AA subjects. G, Effects of XCL1 and several other chemokines on XCL1 expression of Treg in AA subjects. Data were presented by means of percentages as well as MFI of cells that expressed candidate molecules. Horizontal bars in dot plot graphs represented median values. p values represented significant differences from paired t tests between assays with AA cells alone and assays with chemokine-primed AA cells (n = 7 for each group).
Regulatory activities and XCL1 expression in allergic asthmatic Treg were enhanced by steroid usage and inhibited by exogenous IL-4
We next examined possible correlations between the reduction in Treg function and disease parameters of asthma such as IgE levels, disease severity, FEV1, disease duration, and allergic status. We found no significant correlation between Treg function and IgE level, disease duration, or allergic status (data not shown). Treg from AA subjects with moderate to severe asthma had lower suppressive and cytolytic function compared with those from mild AA subjects. However, the differences were not statistically significant (data not shown). Degrees of suppression and killing were positively correlated with FEV1 (Table II). We did not analyze the correlation between inhaled corticosteroid usage and Treg function because blood samples from these subjects were obtained after they had been on a 48-h steroid withdrawal. Thus, their medication status might not represent the effects of inhaled corticosteroid on immune cells in peripheral blood.
To test whether steroids could enhance Treg function, and to assess the possible biological and clinical relevance of the ability of XCL1 to enhance Treg function in AA subjects, we examined AA Treg function in AA subjects with asthma exacerbation who were treated with oral corticosteroid (OCS). AA subjects (AA1, AA2, AA3, AA4, and AA5) were effectively treated with OCS, and their blood samples were analyzed at day 2 of 5-day treatment with OCS at 40 mg per day. Studies of Treg function from these samples showed a significant increase in Treg activity (Fig. 6A). We also tested ex vivo incubation of high dose prednisone (1000 ng/ml) with Treg from AA subjects that were on 48-h steroid withdrawal and found that ex vivo treatment of AA Treg with steroids also enhanced their suppressive and cytotoxic activities (Fig. 6A). On the contrary, addition of high-dose IL-4 (1000 U/ml) to suppression and killing assays of Treg obtained from OCS-treated AA subjects reversed the steroid-mediated functional enhancement of Treg (Fig. 6A).
FIGURE 6.
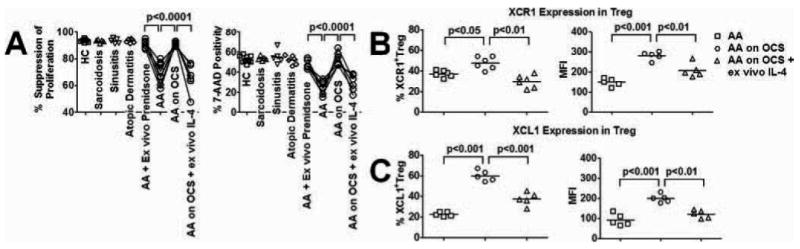
Modulation of Treg function and XCL1 expression by steroids and exogenous IL-4. A, Suppression assays and killing assays of Treg against responder T cells in HC subjects (n = 11), sarcoidosis subjects (n = 5), sinusitis on high-dose OCS subjects (n = 5), atopic dermatitis subjects (n = 6), AA on ex vivo prednisone (n = 6, ex vivo experiments), AA on 48 h steroid withdrawal before blood drawn (n = 11), AA on high-dose OCS (n = 6), AA on high-dose OCS and ex vivo high-dose IL-4 (n = 6). B, XCR1 expression by Treg in AA on 48-h steroid withdrawal before blood was drawn, AA on high-dose OCS, AA on high-dose OCS and ex vivo high-dose IL-4 (n = 5 for each group). C, XCL1 expression by Treg in AA on 48 h steroid withdrawal before blood drawn, AA on high-dose OCS, and AA on high-dose OCS and ex vivo high-dose IL-4 (n = 5 for each group). Data were presented by means of percentages as well as MFI of cells that expressed candidate molecules. Horizontal bars in dot plot graphs represented median values. p values represented significant differences from paired t tests among assays with AA cells alone and assays with AA cells subjected to different treatments and experimental conditions.
To test whether steroids and IL-4 might enhance Treg function via modulation of their XCL1/XCR1 expression, we examined XCL1/XCR1 levels in Treg from AA subjects before and after OCS treatment, as well as Treg from OCS treated subjects that were subsequently incubated with high dose IL-4. XCL1 and XCR1 expression in Treg from AA subjects were significantly elevated after OCS treatment (Fig. 6, B and B). In contrast, exogenous IL-4 suppressed XCL1/XCR1 expression by Treg (Fig. 6, B and C). Altogether, these findings suggested opposite roles of steroids and IL-4 on the maintenance of Treg function, possibly via modulation of XCL1/XCR1 expression of Treg.
Discussion
Even though chemokines have been implicated in immune tolerance by Treg, their effects have mostly been associated with enhanced motility of total Treg or a Treg subset in response to chemokines produced during inflammation in a particular disease. Data presented in this study provided a direct linkage between XCL1 and Treg function in humans. We found that the reduction in peripheral Treg function was associated with decreased XCL1 expression by Treg in AA, and airway inflammation in AA was associated with decreased XCL1 levels from BAL and BAL Treg. Furthermore, we showed the potent modulatory effect of recombinant human XCL1 in rescuing the impaired Treg function in AA. Mechanistically, our data suggested that XCL1 neither affected the proliferative capacity of responder T cells and Treg nor induced their apoptosis. In fact, AA Treg incubated with XCL1 up-regulated their expression of chief effector molecules of their cytotoxicity and suppressive function. Foxp3, granzyme A, and granzyme B expression were up-regulated in AA Treg that were incubated with XCL1. IL-10 and TGF-β levels in AA Treg were not decreased compared with control subjects but were also up-regulated by exogenous XCL1 treatment. These results suggested that the XCL1 might act on (a) common regulatory molecule(s) which is (are) upstream of these effector molecules of Treg function. Investigation of candidate regulatory molecules, such as STAT5 (previously implicated in regulating cytotoxic enzyme expression; Ref. 29), Foxp3 expression (30), and IL-10 expression (31) in several lymphocyte subsets (including Treg) is underway in our laboratory. Furthermore, exogenous XCL1 also up-regulated the expression of XCL1 and XCR1 by Treg, suggesting that it might act via an autocrine, positive feedback mechanism. This hypothesis was strengthened by data from blocking XCL1 and XCR1 experiments as neutralizing Abs impeded Treg-mediated suppression and cytotoxicity. Because neutralizing XCL1 signaling only resulted in a partial reduction but not a complete abrogation of Treg suppressive function and cytotoxicity, it is likely that XCL1 signaling is not the one and only determinant, but is one of several signaling networks that control Treg activity. Nevertheless, these results suggested a potential role for XCL1 in optimal functional maintenance of Treg.
It is well-established that chemokine responses in lymphocytes, specifically in T cells, occur via shared pathways. Therefore, it is likely that other chemokines could also enhance Treg function in a similar manner. We have tested several chemokines, such as CCL1, CCL5, CCL17, and CCL22, and found that unlike XCL1, these chemokines were not able to enhance the defective Treg function in AA. In addition, the specificity of XCL1 activity was confined within the Treg population, but not responder T cells. Even though responder T cells express lower levels of XCR1, which could be a potential explanation for its lower responsiveness to XCL1, it is still possible that there is differential intracellular connectivity between Treg and responder T cells downstream of XCL1. Altogether, these observations suggested that distinct signaling pathways and/or regulatory mechanisms might be available to XCL1 and Treg. Investigation of key signaling cascades associated with Treg function and GPCR/XCL1 signaling, such as the AKT/PTEN/mTOR and STAT5 pathways, is underway in our laboratory to further explore the mechanism of action of XCL1 on Treg.
In contrast with previous studies that demonstrated that decrease in Treg function is only limited to a subset of AA subjects (5, 6, 32), our study used a more stringent gating method for flow sorting procedure to avoid contamination of CD25high activated effector T cells (particularly present in high frequency in peripheral blood of severe AA subjects). This difference in methodology could perhaps be the reason why those studies did not find a consistent decrease in Treg function in AA. Furthermore, we found no abnormality in suppressive and killing function of Treg in subjects with other allergic and pulmonary diseases, which were consistent with previous reports (Fig. 6A). Vukmanovic-Stejic and colleagues reported that circulating Treg from atopic dermatitis subjects were able to suppress responder T cell proliferation (33). Miyara and colleagues found that circulating Treg from sarcoidosis failed to suppress TNF-α production by responder T cells but indeed possessed potently suppressive activities against responder T cell proliferation (34). Cytotoxic potential of Treg was not examined in these studies. Altogether, these data suggested that suboptimal cytotoxicity and suppressive function of Treg are specific to AA but not other allergic and pulmonary diseases. However, our data did not rule out the possibility that Treg function in other allergic and pulmonary diseases exhibits distinct suboptimal regulatory functions. An explanation for distinct abnormalities in Treg function observed in several allergic and pulmonary diseases could be that each disease possesses a signature inflammatory environment. Thus, the effects of systemic and/or local cytokine/chemokine milieu of each disease on Treg function might very well be different.
Our studies did not rule out the possibility that XCL1/Treg dysfunction is the primary defect in AA; however, data presented in this study suggested that the defective Treg function in AA might be secondary to abnormalities in other immune responses in the disease. First, OCS, an immunosuppressive reagent that effectively reduced the predominant Th2 cytokine environment in AA, could correct the impaired Treg function in AA. In addition, this functional enhancement was associated with the up-regulation of XCL1. Second, Treg from OCS-treated subjects (with normalized suppressive and cytotoxic activities) incubated with high-dose IL-4 in vitro exhibited significantly lower regulatory activities, which associated with their lower expression of XCL1. These results were consistent with a previous report that also found that XCL1 expression in lymphocytes could be suppressed by high-dose IL-4, a signature cytokine in AA pathogenesis (18). Thus, it is plausible to suggest that decreased XCL1 level in Treg and their associated suboptimal suppression and cytotoxicity could result from the presence of IL-4 at high concentration both systemically and locally (i.e., in the airway) in AA. Furthermore, it is also possible that OCS might indirectly modulate Treg function in AA by suppressing the inflammatory manifestation driven by IL-4. Collectively, these results suggested that altered inflammatory environment in AA is a possible cause of the defective Treg function and XCL1 expression in the disease.
In conclusion, our study described a reduction in Treg function in AA that is associated with dysregulated XCL1 expression. More importantly, data presented in this study pointed to a novel role of the chemokine XCL1 in the functional maintenance of Treg. Thus, XCL1 is a promising molecular target for therapeutic development to rescue Treg-mediated immune regulation in AA and possibly other diseases.
Acknowledgments
We thank the asthmatic and healthy subjects who provided samples for the study.
Footnotes
This work was supported by grants from the Mary Hewitt Loveless Foundation and the Parker B. Francis Foundation.
Abbreviations used in the article: Treg, regulatory T cell; AA, allergic asthma; HC, healthy control; FEV1, forced expiratory volume in 1 s; NA, nonallergic asthmatic; BAL, bronchoalveolar lavage fluid; MFI, mean fluorescent intensity; OCS, oral corticosteroid; 7-AAD, 7-aminoactinomycin D; QT-PCR, quantitative PCR.
Disclosures The authors have no financial conflict of interest.
References
- 1.Lee TH. Interactions between alveolar macrophages, monocytes, and granulocytes: implications for airway inflammation. Am Rev Respir Dis. 1987;135:S14–S17. doi: 10.1164/arrd.1987.135.6P2.S14. [DOI] [PubMed] [Google Scholar]
- 2.Robinson DS, Hamid Q, Ying S, Tsicopoulos A, Barkans J, Bentley AM, Corrigan C, Durham SR, Kay AB. Predominant TH2-like bronchoalveolar T-lymphocyte population in atopic asthma. N Engl J Med. 1992;326:298–304. doi: 10.1056/NEJM199201303260504. [DOI] [PubMed] [Google Scholar]
- 3.Akbari O, Stock P, DeKruyff RH, Umetsu DT. Role of regulatory T cells in allergy and asthma. Curr Opin Immunol. 2003;15:627–633. doi: 10.1016/j.coi.2003.09.012. [DOI] [PubMed] [Google Scholar]
- 4.Baecher-Allan C, Viglietta V, Hafler DA. Human CD4+CD25+ regulatory T cells. Semin Immunol. 2004;16:89–98. doi: 10.1016/j.smim.2003.12.005. [DOI] [PubMed] [Google Scholar]
- 5.Grindebacke H, Wing K, Andersson AC, Suri-Payer E, Rak S, Rudin A. Defective suppression of Th2 cytokines by CD4CD25 regulatory T cells in birch allergics during birch pollen season. Clin Exp Allergy. 2004;34:1364–1372. doi: 10.1111/j.1365-2222.2004.02067.x. [DOI] [PubMed] [Google Scholar]
- 6.Ling EM, Smith T, Nguyen XD, Pridgeon C, Dallman M, Arbery J, Carr VA, Robinson DS. Relation of CD4+CD25+ regulatory T-cell suppression of allergen-driven T-cell activation to atopic status and expression of allergic disease. Lancet. 2004;363:608–615. doi: 10.1016/S0140-6736(04)15592-X. [DOI] [PubMed] [Google Scholar]
- 7.Strickland DH, Stumbles PA, Zosky GR, Subrata LS, Thomas JA, Turner DJ, Sly PD, Holt PG. Reversal of airway hyperresponsiveness by induction of airway mucosal CD4+CD25+ regulatory T cells. J Exp Med. 2006;203:2649–2660. doi: 10.1084/jem.20060155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lewkowich IP, Herman NS, Schleifer KW, Dance MP, Chen BL, Dienger KM, Sproles AA, Shah JS, Köhl J, Belkaid Y, Wills-Karp M. CD4+CD25+ T cells protect against experimentally induced asthma and alter pulmonary dendritic cell phenotype and function. J Exp Med. 2005;202:1549–1561. doi: 10.1084/jem.20051506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shevach EM. CD4+CD25+ suppressor T cells: more questions than answers. Nat Rev Immunol. 2002;2:389–400. doi: 10.1038/nri821. [DOI] [PubMed] [Google Scholar]
- 10.Fontenot JD, Gavin MA, Rudensky AY. Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nat Immunol. 2003;4:330–336. doi: 10.1038/ni904. [DOI] [PubMed] [Google Scholar]
- 11.Wei S, Kryczek I, Zou W. Regulatory T-cell compartmentalization and trafficking. Blood. 2006;108:426–431. doi: 10.1182/blood-2006-01-0177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hasegawa H, Inoue A, Muraoka M, Yamanouchi J, Miyazaki T, Yasukawa M. Therapy for pneumonitis and sialadenitis by accumulation of CCR2-expressing CD4+CD25+ regulatory T cells in MRL/lpr mice. Arthritis Res Ther. 2007;9:R15. doi: 10.1186/ar2122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mizukami Y, Kono K, Kawaguchi Y, Akaike H, Kamimura K, Sugai H, Fujii H. CCL17 and CCL22 chemokines within tumor microenvironment are related to accumulation of Foxp3+ regulatory T cells in gastric cancer. Int J Cancer. 2008;122:2286–2293. doi: 10.1002/ijc.23392. [DOI] [PubMed] [Google Scholar]
- 14.Ishida T, Ueda R. CCR4 as a novel molecular target for immunotherapy of cancer. Cancer Sci. 2006;97:1139–1116. doi: 10.1111/j.1349-7006.2006.00307.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hasegawa H, Inoue A, Kohno M, Lei J, Miyazaki T, Yoshie O, Nose M, Yasukawa M. Therapeutic effect of CXCR3-expressing regulatory T cells on liver, lung and intestinal damages in a murine acute GVHD model. Gene Ther. 2008;15:171–182. doi: 10.1038/sj.gt.3303051. [DOI] [PubMed] [Google Scholar]
- 16.Kelner GS, Kennedy J, Bacon KB, Kleyensteuber S, Largaespada DA, Jenkins NA, Copeland NG, Bazan JF, Moore KW, Schall TJ. Lymphotactin: a cytokine that represents a new class of chemokine. Science. 1994;266:1395–1399. doi: 10.1126/science.7973732. [DOI] [PubMed] [Google Scholar]
- 17.Emtage PC, Xing Z, Wan Y, Zlotnik A, Graham FL, Gauldie J. Adenoviral-mediated gene transfer of lymphotactin to the lungs of mice and rats results in infiltration and direct accumulation of CD4+, CD8+, and NK cells. J Interferon Cytokine Res. 2002;5:573–582. doi: 10.1089/10799900252982052. [DOI] [PubMed] [Google Scholar]
- 18.Huang H, Li F, Gordon JR, Xiang J. Synergistic enhancement of antitumor immunity with adoptively transferred tumor-specific CD4+ and CD8+ T cells and intratumoral lymphotactin transgene expression. Cancer Res. 2002;62:2043–2051. [PubMed] [Google Scholar]
- 19.Huang H, Xiang J. Synergistic effect of lymphotactin and interferon γ-inducible protein-10 transgene expression in T-cell localization and adoptive T-cell therapy of tumors. Int J Cancer. 2004;109:817–825. doi: 10.1002/ijc.20043. [DOI] [PubMed] [Google Scholar]
- 20.Cerdan C, Devilard E, Xerri L, Olive D. The C-class chemokine lymphotactin costimulates the apoptosis of human CD4+ T cells. Blood. 2001;97:2205–2212. doi: 10.1182/blood.v97.8.2205. [DOI] [PubMed] [Google Scholar]
- 21.Rumsaeng V, Vliagoftis H, Oh CK, Metcalfe DD. Lymphotactin gene expression in mast cells following Fc∊ receptor I aggregation: modulation by TGF-β, IL-4, dexamethasone, and cyclosporin A. J Immunol. 1997;158:1353–1360. [PubMed] [Google Scholar]
- 22.Mekori YA, Metcalfe DD. Mast cell-T cell interactions. J Allergy Clin Immunol. 1999;104:517–523. doi: 10.1016/s0091-6749(99)70316-7. [DOI] [PubMed] [Google Scholar]
- 23.Kemper C, Chan AC, Green JM, Brett KA, Murphy KM, Atkinson JP. Activation of human CD4+ cells with CD3 and CD46 induces a T-regulatory cell 1 phenotype. Nature. 2003;421:388–392. doi: 10.1038/nature01315. [DOI] [PubMed] [Google Scholar]
- 24.Grossman WJ, Verbsky JW, Barchet W, Colonna M, Atkinson JP, Ley TJ. Human T regulatory cells can use the perforin pathway to cause autologous target cell death. Immunity. 2004;21:589–601. doi: 10.1016/j.immuni.2004.09.002. [DOI] [PubMed] [Google Scholar]
- 25.Bacon KB, Premack BA, Gardner P, Schall TJ. Activation of dual T cell signaling pathways by the chemokine RANTES. Science. 1995;269:1727–1730. doi: 10.1126/science.7569902. [DOI] [PubMed] [Google Scholar]
- 26.Makino Y, Cook DN, Smithies O, Hwang OY, Neilson EG, Turka LA, Sato H, Wells AD, Danoff TM. Impaired T cell function in RANTES-deficient mice. Clin Immunol. 2002;102:302–309. doi: 10.1006/clim.2001.5178. [DOI] [PubMed] [Google Scholar]
- 27.Schuh JM, Blease K, Hogaboam CM. The role of CC chemokine receptor 5 (CCR5) and RANTES/CCL5 during chronic fungal asthma in mice. FASEB J. 2002;16:228–230. doi: 10.1096/fj.01-0528fje. [DOI] [PubMed] [Google Scholar]
- 28.Castro M, Bloch SR, Jenkerson MV, DeMartino S, Hamilos DL, Cochran RB, Zhang XE, Wang H, Bradley JP, Schechtman KB, Holtzman MJ. Asthma exacerbation after glucocorticoid withdrawal reflects T cell recruitment to the airway. Am J Respir Crit Care Med. 2004;169:842–849. doi: 10.1164/rccm.200208-960OC. [DOI] [PubMed] [Google Scholar]
- 29.Morishima N, Owaki T, Asakawa M, Kamiya S, Mizuguchi J, Yoshimoto T. Augmentation of effector CD8+ T cell generation with enhanced granzyme B expression by IL-27. J Immunol. 2005;175:1686–1693. doi: 10.4049/jimmunol.175.3.1686. [DOI] [PubMed] [Google Scholar]
- 30.Zorn E, Nelson EA, Mohseni M, Porcheray F, Kim H, Litsa D, Bellucci R, Raderschall E, Canning C, Soiffer RJ, et al. IL-2 regulates FOXP3 expression in human CD4+CD25+ regulatory T cells through a STAT-dependent mechanism and induces the expansion of these cells in vivo. Blood. 2006;108:1571–1579. doi: 10.1182/blood-2006-02-004747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tsuji-Takayama K, Suzuki M, Yamamoto M, Harashima A, Okochi A, Otani T, Inoue T, Sugimoto A, Motoda R, Yamasaki F, et al. IL-2 activation of STAT5 enhances production of IL-10 from human cytotoxic regulatory T cells, HOZOT. Exp Hematol. 2007;36:181–192. doi: 10.1016/j.exphem.2007.09.010. [DOI] [PubMed] [Google Scholar]
- 32.Bellinghausen I, Klostermann B, Knop J, Saloga J. Human CD4+CD25+ T cells derived from the majority of atopic donors are able to suppress TH1 andTH2 cytokine production. J Allergy Clin Immunol. 2003;111:862–868. doi: 10.1067/mai.2003.1412. [DOI] [PubMed] [Google Scholar]
- 33.Vukmanovic-Stejic M, McQuaid A, Birch KE, Reed JR, Macgregor C, Rustin MH, Akbar AN. Relative impact of CD4+CD25+ regulatory T cells and tacrolimus on inhibition of T-cell proliferation in patients with atopic dermatitis. Br J Dermatol. 2005;153:750–757. doi: 10.1111/j.1365-2133.2005.06675.x. [DOI] [PubMed] [Google Scholar]
- 34.Miyara M, Amoura Z, Parizot C, Badoual C, Dorgham K, Trad S, Kambouchner M, Valeyre D, Chapelon-Abric C, Debré P, et al. The immune paradox of sarcoidosis and regulatory T cells. J Exp Med. 2006;203:359–370. doi: 10.1084/jem.20050648. [DOI] [PMC free article] [PubMed] [Google Scholar]