

Abstract
Background
Group B Streptococcus (GBS) is the leading cause of bacterial meningitis in newborn infants. As GBS is able to invade, survive and cross the blood-brain barrier (BBB), we sought to identify surface-expressed virulence factors that contribute to BBB penetration and the pathogenesis of meningitis.
Methods
Targeted deletion and insertional mutants were generated in different GBS clinical isolates. Wild type and mutant bacteria were analyzed for their capacity to adhere and invade human brain microvascular endothelial cells (hBMEC) and penetrate the BBB using our model of hematogeneous meningitis.
Results
Analysis of a GBS (serotype V) clinical isolate revealed the presence of a surface anchored serine-rich protein previously designated serine-rich repeat-1 (Srr-1). GBS Srr-1 is a high molecular weight glycosylated protein. Deletion of srr1 in NCTC 10/84 resulted in a significant decrease in adherence and invasion of hBMEC. Additional mutants in other GBS serotypes commonly associated with meningitis showed a similar decrease in hBMEC invasion compared to parental strains. Finally, wild type GBS penetrated the BBB and established meningitis more frequently than mice challenged with the Δsrr1 mutant strain.
Conclusion
Our data suggest that GBS Srr glycoproteins play an important role in crossing the BBB and the development of streptococcal meningitis.
Keywords: Group B Streptococcus, blood, brain barrier, meningitis, invasion, glycoprotein
Introduction
Group B Streptococcus (GBS), also called Streptococcus agalactiae, is the leading cause of bacterial meningitis in newborns [1]. Mortality remains high despite antibiotic therapy, and 25–50% of surviving infants are left with permanent neurological sequelae including cognitive deficits, cerebral palsy, blindness, deafness or seizures [2]. In order to gain access into the central nervous system (CNS), GBS must cross the blood-brain barrier (BBB), which is primarily comprised of a single layer of specialized brain microvascular endothelial cells (BMEC).
Penetration of the BBB by a bacterial pathogen is multi-factorial and reflects a complex interplay between host endothelium and microbial products. Studies to better understand this interaction have become feasible through the availability of tissue culture models of human BMEC (hBMEC) [3]. The hBMEC model system has been used to identify virulence genes in different bacterial species that promote cellular invasion [5–10]. While GBS adheres to and invades hBMEC [11], the factors that contribute to the penetration of the BBB and the development of meningitis are still being elucidated. For GBS, only two specific virulence factors have been shown to contribute to the pathogenesis of meningitis in vivo, the pore-forming β-hemolysin/cytolysin (β-h/c) toxin [12], and the glycosyltransferase, iagA, which is required for proper anchoring of lipoteichoic acid to the cell wall [5]. However, neither of these proteins function directly to mediate GBS attachment to BBB endothelium, the critical first step in CNS invasion and disease progression.
Members of the streptococcal and staphylococcal serine-rich repeat (Srr) protein familyhave been described to function as adhesins in different model systems [13–15], including endothelial cells [16]. Recently it was shown that GBS strains carry one of two srr gene alleles, designated srr1 [14] and srr2 [17], which are similar in architecture but show only limited homology (<20% identity) [17]. Interestingly, expression of the Srr-2 protein seems to be restricted to serotype III-3 and multilocus sequence typing (ST)-17 strains [17], which represents >90% of the serotype III invasive neonatal disease isolates. In addition to serotype III, serotypes Ia, Ib and V are also frequently isolated from neonates, children and adults patients with meningitis [18], suggesting that both Srr-1 and Srr-2 could be relevant for the pathogenesis of meningitis.
Given the described role of Srr homologues in host interactions, we hypothesized that GBS Srr proteins could play a role in the interaction with brain endothelium. In this study, we identify and characterize Srr-1 in the serotype V clinical isolate NCTC 10/84. Using targeted mutagenesis we show that the NCTC 10/84 srr1 as well as other srr genes in GBS serotypes III, Ia and Ib contribute directly to hBMEC invasion in vitro. Finally, we provide evidence that Srr-1 promotes BBB penetration and the development of GBS meningitis in a mouse meningitis model.
Methods
Bacterial strains and growth conditions
The following wild type (WT) GBS clinical isolates were used in this study: NCTC 10/84 (1169-NT1; ATCC 49447) (serotype V) [19], COH1 (serotype III) [20], 515 (serotype Ia) [21], and H36B (serotype Ib) [22]. GBS was grown in Todd-Hewitt broth (THB; Difco, BD Diagnostics) at 37°C. For antibiotic selection, 5 μg/ml of erythromycin (Erm) or 2 μg/ml of chloramphenicol (Cm) was used. Streptococcus gordonii (S. gordonii) strain M99 and the GspB mutant (PS846; M99 ΔgspB::pEVP3) have been described previously [13, 23]. S. gordonii WT and GspB mutant strains were grown in THB, with the addition of 5 μg/ml Cm when appropriate, at 37°C.
Cell lines
The human brain microvascular endothelial cell line hBMEC was obtained from Kwang Sik Kim (Johns Hopkins University, Baltimore, Maryland, USA) and maintained as described previously [3, 4, 24]. A549 cells (ATCC), a human type II alveolar epithelial carcinoma cell line, and Hela cells, a human cervical carcinoma cell line, were maintained and passaged in RPMI 1640 tissue culture medium that contained 10% FBS. Cultures were incubated at 37°C in a humid atmosphere of 5% CO2.
Targeted mutagenesis and complementation vector construction
Using BLAST, PCR and sequence analysis of GBS clinical isolate strain NCTC 10/84 we identified a putative cell-wall anchored protein as a member of the streptococcal and staphylococcal Srr protein family known as Srr-1 [17]. PCR was used to generate in-frame substitution of the srr1 gene with the chloramphenicol acetyltransferase (cat) gene using a method described previously [5]. Briefly, 752 bp and 835 bp immediately upstream and downstream respectively of srr1 was amplified with the primers srr1upF, 5′;-CCGCTCGAGGGCATCTTCCTGAGTAAGTT-3′;, and srr1upR+cat, 5′;-GGTGGTATATCCAGTGATTTTTTTCTCCATGTTTCCTCCATATATAAA-TAT-3′;, and the primers srr1downF+cat, 5′;-TACTGCGATGAGTGGCAGGGCGGGGCGTAATAAACCTACTTTGAATCCTTA-3′, and srr1downR, 5′-GTACTGCAGGTAGGTAGAATAACAACATCCG-3′. The srr1upR+cat and srr1downF+cat primers were constructed with 25-bp 5′; extensions corresponding to the 5′; and 3′; ends of the cat gene, respectively. PCR was used to generate a fragment containing an in-frame substitution of srr1 with cat, which was subcloned to vector pHY304. Allelic exchange mutagenesis in NCTC 10/84 was performed as described previously [25] to generate the mutant NCTC 10/84Δsrr1. Allelic replacement of srr1 with cat in the NCTC 10/84Δsrr1 chromosomal was confirmed by PCR and sequence analysis. For complementation analysis, srr1 plus flanking DNA was PCR amplified from the NCTC 10/84 chromosome using srr1FLF, 5′-CGTGGAATTCGTATCTACGTGCTTAACGG-3′, and srr1FLR, 5′-GCGGGATCCCAAAGTAGGTTTAGTCTTTATC-3′ and cloned into expression vector pDCerm [26], yielding plasmid pSrr1.
Additional srr mutants were generated by plasmid insertional mutagenesis as described previously [25]. PCR was used to amplify a 590-bp fragment of the srr2 gene using primers 5′-CGTGAAGCTTGCAGTTTGGAAACTTTGGTG -3′ and 5′-TCAGCTGCAGGTTGAACTCTAGCGGTCGTTGC - 3′ containing HindIII and PstI restriction sites, respectively. The PCR product was cloned into pTOPO and subcloned to pVE6007 [27]. The construct was transformed into electrocompetent COH1 and plasmid integration confirmed by PCR analysis using primers homologous to plasmid sequences and to sequences upstream of the point of insertion. A similar strategy was used to generate srr-1 mutants in GBS strains 515 and H36B.
Western blot and lectin blot analysis of GBS Srr-1
To determine cross reactivity of GBS Srr-1 with anti-serum raised against Srr-1 homologue GspB in S. gordonii total bacterial lysates were prepared for Western blot analysis. Overnight bacterial cultures were washed and incubated for 3 h at 37°C in Tris buffer (pH 7.0), containing mutanolysin (1 U/μl), DNase I (0.1 U/μl) and complete protease inhibitor (Roche, Indianapolis, USA), before the addition of 3x sample buffer. Samples were separated by SDS-PAGE using 3–8% Tris-acetate gels (Invitrogen, Carlsbad, California, USA) under reducing conditions and transferred to nitrocellulose membranes. Membranes were blocked using Tris buffer (pH 7.5) containing 4 % bovine serum albumin (BSA; Roche). Next, membranes were incubated overnight with goat polyclonal anti-GspB serum (1/500) [28] in TBST 1% BSA at 4°C, followed by detecting with peroxidase-conjugated donkey anti-goat IgG (1/25,000; Jackson Immunoresearch). Lectin blot analysis with the N-acetylglucosamine-specific lectin wheat-germ agglutinin (WGA; Vector labs) was performed as previously described [29]. Western blots were developed with Supersignal West Pico chemiluminescent substrate (Pierce Biotechnology).
Adherence and invasion assays
GBS adherence and invasion assays were performed as described previously with minor modifications [11]. Briefly, confluent hBMEC monolayers were incubated with log-phase grown bacteria at a multiplicity of infection (MOI) of 1, unless indicated otherwise. Plates were centrifuged at 800 × g to synchronize the infection, and incubated at 37°C with 5% CO2. After two hours, the monolayers were washed and 1 ml of RPMI 10% FBS containing 100 μg of gentamicin and 5 μg of penicillin G was added for two additional hours. After washing, monolayers were disrupted and the number of invasive bacteria was quantified by serial dilution plating. To assess the level of surface-adherent (total cell-associated) bacteria, bacteria were quantified from hBMEC monolayers prior to addition of extracellular antibiotics after 30 min of incubation. Parallel invasion experiments were performed in A549 lung epithelial cells and Hela epithelial cells. All cellular adherence and invasion assays were performed in triplicate and repeated at least three times.
Mouse infection studies
All animal experiments were approved by the Committee on the Use and Care of Animals and performed using accepted veterinary standards. A murine model of hematogenous GBS meningitis has been described previously [12]. Briefly, out bred 6- to 8-week-old male CD-1 mice (Charles River Laboratories; n = 10 mice per group) were injected via tail vein with 7.5×107CFU GBS WT or the Δsrr1 mutant. Blood was collected by retro-orbital puncture at indicated times and plated to determine the level of bacteremia. At the experimental endpoint (day 4 p.i.) blood, brain and spleen were collected aseptically from mice upon euthanasia. Bacterial counts in blood and tissue homogenates were determined by plating serial dilutions. Brain and spleen bacterial counts were corrected for differences in organ weight.
Statistical analysis
SPSS (Version 12.0.02 for Windows) was used for statistical analysis. Student’s two-tailed t-test was used to study differences adherence and invasion in hBMEC cells between WT and mutant strains and for differences in bacterial load in mice. Statistical significance was accepted at p < 0.05.
Results
Genetic characterization of highly-conserved serine-rich repeat protein Srr-1 in GBS strain NCTC 10/84
To characterize new GBS virulence factors that are critical for penetration of the BBB, we focused on GBS genes predicted to encode surface-anchored proteins. Through BLAST analysis of published GBS genome sequences, we identified a locus encoding a putative RofA-like transcriptional regulator protein (RALP), followed by a protein containing a LP(X)TG cell-well anchoring motif, a SecY2/A2 secretion system, accessory secretion proteins and multiple glycosyltransferases (Fig. 1A). This locus represents one of a group “genomic islands” that are highly conserved and identical among different GBS strains [30]. Also, similar loci have been identified in other Gram-positive streptococcal and staphylococcal species [31], including Streptococcus gordonii [28], Streptococcus pneumoniae [32] and Staphylococcus aureus [15]. PCR and sequence analyses confirmed the presence of this locus in serotype V GBS clinical isolate NCTC 10/84, a highly virulent strain in a mouse model of hematogenous meningitis [12].
Figure 1.

(A) Schematic diagram of the GBS srr1 locus. RofA-like is a putative transcriptional regulator, srr1 is a putative cell-surface anchored serine-rich repeat protein, nss is similar to nucleotide sugar synthetases, asp are accessory secretory proteins, Gly and gtf are putative glycosyltransferases and secA2/Y2 is a putative sec translocation system. (B) Protein identity between Srr-1 in serotype V GBS strains NCTC 10/84 (deduced from determined nucleotide sequence) and 2603V/R, and between GBS Srr-1 and GspB homologue in S. gordonii. Alignment was performed using COFFEE-T alignment program. N.d. not determined. SP, signal peptide; SRR1, serine-rich repeat domain 1; NRD, non-repeat domain; SRR2, serine-rich repeat domain 2; CWAD, cell wall anchoring domain.
The putative cell-wall anchored protein was identified as a member of the streptococcal and staphylococcal serine-rich repeat (Srr) protein family known as Srr-1 [14, 17]. Gene amplification showed that the NCTC 10/84 srr1 gene is larger, approximately 4300 bp, than other srr1 genes in sequenced GBS strains which range from 2730 bp to 3981 bp. DNA sequence analysis of the NCTC 10/84 srr1 gene revealed >99% sequence identity between the N- and C-terminal serine rich regions (SRR1 and SRR2) compared to another serotype V GBS strain 2603V/R (Fig. 1B). We found the same repeat pattern SAS(T/M) in the long SRR2 domain, which is present in the majority of sequenced GBS strains and is responsible for the observed size variation among GBS Srr-1 proteins [14]. In addition, protein alignment demonstrated a high sequence similarity between GBS Srr-1 and Srr homologues in other species, such as GspB in oral S. gordonii (Fig. 1B).
GBS Srr-1 is a high-molecular weight glycoprotein
To characterize Srr-1 in GBS strain NCTC 10/84, we analyzed bacterial cell lysates by Western blot analysis and lectin staining. Using an anti-serum reactive to the native form of S. gordonii GspB [28], we observed multiple cross-reactive proteins in the WT GBS NCTC 10/84 sample including a protein of similar molecular weight (> 460 kD) as GspB (Fig. 2A). In contrast, cross-reactive species were not observed with anti-serum raised against a recombinant (nonglycosylated) Srr1-family protein, SraP from S. aureus [15] (data not shown). Western blot analysis of the isogenic Δsrr1 mutant revealed loss of the > 460 kD glycoprotein band reactive to S. gordonii GspB antisera (Fig. 2A); complementation of the Δsrr1 mutant with the WT GBS srr1 gene on a plasmid expression vector (pSrr1) restored expression of the glycoprotein (Fig. 2A).
Figure 2.

(A) NCTC 10/84 Srr-1 is cross-reactive with GspB-specific anti-serum. NCTC 10/84 Srr-1 migrates as a > 460 kD protein. Specificity was determined using Δsrr1 mutant and Δsrr1+pSrr1 strains as control for protein band in NCTC 10/84 WT protein lysate, whereas S. gordonii WT and ΔgspB strains were used as positive and negative control for staining with GspB-specific anti-serum, respectively. (B) Detection of carbohydrate modifications on NCTC 10/84 Srr-1 protein using the N-acetylglucosamine-specific lectin WGA. Proteins extracts were separated by gel electrophoresis using 3–8% Tris-acetate gradient gels.
Proteins of the Srr family are usually of much higher molecular weight than predicted by amino acid content due to extensive glycosylation [15, 28, 33]. Similarly, NCTC 10/84 Srr-1 was impressively larger than the expected size of approximately 140 kD. We performed lectin blot analysis using the N-acetylglucosamine-specific lectin WGA, which has been shown to bind S. gordonii GspB [34]. As shown in Fig. 2B, WGA reacted with the same high-molecular weight protein in the WT GBS strain which was not present in the Δsrr1 mutant but was restored in the complemented strain. Taken together these results demonstrate that GBS Srr-1 is post-translationally modified by glycosylation.
Srr-1 promotes hBMEC adherence and invasion
GBS Srr-1 and Srr-1 homologues function as adhesins in different cell model systems [14, 15, 28]. Therefore, we addressed the role of Srr-1 in the GBS-BBB interaction using our well-characterized in vitro hBMEC model [11, 12]. GBS WT bacteria efficiently adhered to and invaded hBMEC while the GBSΔsrr1 mutant exhibited up to a 50% reduction in adherence and 70% reduction in invasion (Fig. 3A, B). Growth kinetics and sensitivity to antibiotics used in our invasion assays did not differ between the WT and Δsrr1 mutant strains (data not shown). Additionally, WT levels of invasion were restored by complementation of the GBS Δsrr1 mutant with Srr-1 cloned on expression vector pSrr1 (Fig. 3C). We performed additional invasion assays using the human lung epithelial A549 cell line and the human cervical Hela cell line. Invasion in these cells was significantly attenuated in the Δsrr1 mutant (Fig. 3D), demonstrating that GBS Srr-1 contributes to penetration of different host cell barriers.
Figure 3.

Contribution of Srr-1 to (A) adherence and (B) invasion of hBMEC. (C) Attenuation of invasion is rescued by expressing Srr-1 on expression vector pSrr1 (MOI 1). (D) NCTC 10/84 Srr-1 contributes to invasion in A549 and Hela epithelial cells (MOI 1). Contribution of Srr proteins to (E) adherence and (F) invasion of hBMEC in different GBS serotypes. Serotype III expresses Srr-2, whereas serotypes Ia and Ib express the Srr-1 protein. For adherence, bacteria were enumerated after 30 min incubation, whereas invasion was quantified following 2 h of incubation with hBMEC and 2 h of incubation with penicillin/gentamicin to kill extracellular bacteria. All experiments were repeated at least three times, data from a representative experiment are shown. The error bars indicate 95% confidence intervals of the means of three wells. * p < 0.05, ** p < 0.01, *** p < 0.005.
Srr-1 and Srr-2 in other GBS serotypes also contribute to hBMEC adherence and invasion
GBS strains contain either one of two srr genes, srr-1 or srr-2. To address the potential role of Srr proteins in other GBS serotypes associated with meningitis, we generated plasmid insertional mutations in srr-2 in COH1 (serotype III-3), and srr-1 in 515 (serotype Ia) and H36B (serotype Ib). All of these isogenic mutants exhibited decreased hBMEC invasion compared to the parental strain (Fig. 3E and F), demonstrating that Srr proteins generally contribute to interaction with brain endothelium.
GBS Srr-1 does not contribute to survival in whole blood
Several studies report an association between the levels of bacteremia and the development of meningitis [35–37]. To reach this critical bacterial threshold, bacteria must subvert host defense mechanisms in order to proliferate and survive in the bloodstream [38]. We therefore tested whether Srr-1 affected bacterial survival in human blood. GBS WT or Δsrr1 bacteria were incubated with human blood obtained from a healthy donor and sampled for surviving bacteria at multiple time points. Both the GBS WT and Δsrr1 strains were killed at similar rate indicating that Srr-1 does not contribute to GBS survival in whole human blood (data not shown).
Role of Srr-1 in BBB penetration in a murine model of GBS meningitis
To test whether our in vitro phenotype translated into a diminished ability to breach the BBB in vivo, we used a murine model of hematogenous GBS meningitis [12]. Groups of CD-1 mice (n=10) were infected intravenously with GBS WT or the Δsrr1 mutant strain. Similar to observation in human whole blood bactericidal assays, mice developed similar levels of bacteremia at 24 h and 48 h post infection (Fig. 4A, B). However, mice infected with the GBS WT strain had significantly higher bacterial counts in the brain than mice infected with the Δsrr1 mutant at the experimental endpoint (Fig. 4C). Few bacteria were recovered from the blood or spleen at the experimental endpoint (Fig. 4C), indicating that the bacteria recovered from the brain were not due to blood present in the microvasculature of the brain.
Figure 4.

Deletion of srr1 does not affect survival of GBS in mouse blood at (A) 24 h or (B) 48 h after infection compared to survival of WT bacteria. Line indicates median number of bacteria in the group of ten mice. One square represents one mouse. Filled squares represent mice infected with WT bacteria, open squares represent mice infected with Δsrr1 bacteria. (C) Bacterial counts (CFU) in brains, spleens and blood of mice infected with NCTC 10/84 WT or Δsrr1 mutant bacteria at day 4 after infection. Line indicates median number of bacteria in the group of ten mice. One square represents one mouse. Filled squares represent mice infected with WT bacteria, open squares represent mice infected with Δsrr1 bacteria. * p < 0.05
To confirm the development of meningitis in these GBS-infected mice, the brains were examined by microscopy. Representative histopathologies of brains from infected mice are shown in Fig. 5A–E. In mice with a high bacterial load in the brain, histopathology revealed clear evidence of GBS meningitis with associated neutrophil infiltration in the meninges and CNS (Fig 5B, C) accompanied by tissue destruction (Fig 5D, E). Mice without bacterial penetration did not show signs of inflammation (Fig 5A), although occasionally hemorrhage was observed. Histopathology was not different between mice that developed meningitis as a consequence of GBS WT or Δsrr1 bacteria. These data demonstrate that Srr-1 significantly contributes to the penetration of the BBB and the development of GBS meningitis in vivo.
Figure 5.
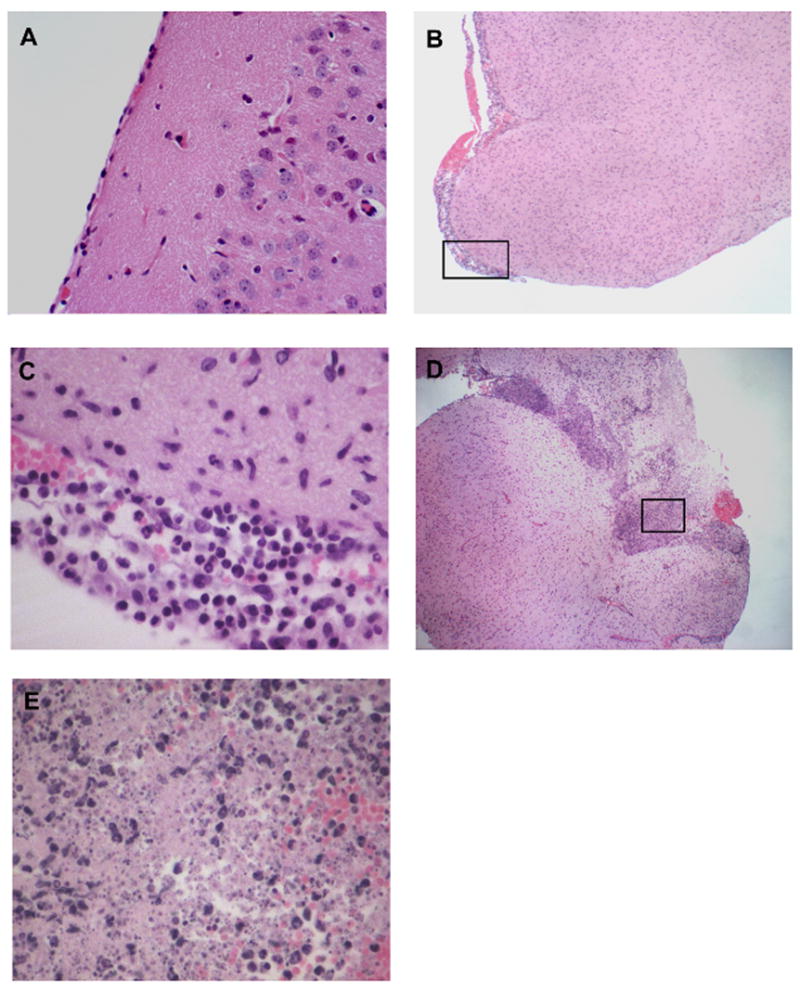
Histopathology of H&E-stained brain tissues of GBS-infected mice. (A) Represents a sample from a mouse infected with the Δsrr1 mutant showing normal brain histopathology (10x). (B–E) Samples from mice infected with WT GBS show meningeal thickening (B), neutrophil infiltration (C), and tissue destruction (D, E). C and E are inserts at higher magnifications (400x) of B (4x) and D (4x).
Discussion
Our studies indicate that the surface-anchored GBS Srr glycoproteins contribute to hBMEC invasion. Furthermore, Srr-1 represents the first “invasin” known to directly promote GBS BBB penetration in vivo. Additional in vitro studies have described a role for GBS surface-expressed proteins in promoting hBMEC interactions, including PilB, the major backbone protein for pilus formation [6], fibrinogen-binding protein FbsA [39] and laminin-binding protein Lmb [9]; however, none of these proteins have been examined for their potential contribution to GBS meningitis in vivo.
Srr family proteins were first characterized in oral streptococci as serine-rich (>35%) high-molecular mass glycosylated proteins that are transported across the membrane by a dedicated SecA2/Y2 secretion system [28, 40, 41]. In GBS, two types of Srr proteins have been identified, termed Srr-1 and Srr-2 [17]. Expression of Srr-1 seems to be widespread among GBS isolates [14], whereas Srr-2 expression seems to be restricted to serotype III-3 and ST-17 strain [17]. Srr-1 is surface-exposed [14] and highly conserved (>85% nucleotide identity and amino acid identity) among published genomes of GBS strains belonging to different serotypes. In contrast there is < 20% sequence identity with Srr-2. GBS Srr-1 proteins contain approximately 30% serines, showing 48% identity and 60% similarity to S. gordonii GspB. Comparison of protein profiles of GBS WT and Δsrr1 mutant bacteria, demonstrated that Srr-1 runs as a protein of > 460 kD and reacts with the glycan-specific lectin WGA, confirming the post-translational modification of the protein. Additionally, GBS Srr-1 cross-reacts with GspB-specific anti-serum, which is known to recognize the carbohydrate moieties on GspB (data not shown). This suggests that the post-translational modification of Srr-1 may share similarities with the glycosylation of GspB. Although glycosylation was a suggested modification of Srr-2 [17], this is the first experimental proof of Srr-1 glycosylation in GBS. The pathway responsible for this extensive glycosylation of GBS Srr-1 is currently not known, but likely involves several genes encoding putative glycosyltransferases in the srr1 locus.
GBS Srr homologues have been described to function as adhesins in different model systems. GspB in S. gordonii mediates binding to platelets interacting with sialylated moieties of the platelet glycoprotein Ibα[42], and Fap1 in S. parasanguis is required for expression of long fimbriae mediating bacterial attachment to saliva-coated hydroxyapatite [33, 43]. In the present work, we demonstrate a role for Srr glycoproteins in different GBS serotypes in invasion of hMBEC and a role of Srr-1 in the pathogenesis of meningitis. This mechanism could also be relevant for other meningeal pathogens such as S. pneumoniae, the leading cause of meningitis after the neonatal period and the elderly, as S. pneumoniae encodes a GBS Srr protein homologue, PsrP [44]. Interestingly, PsrP resides in one of two S. pneumoniae pathogenicity islands whose presence appears to correlate with an invasive phenotype [32]. Although a recent study did not show a role for PsrP in adherence hBMEC, the strains were not tested for differences in invasive capacity [45]. Additional studies of the role of PsrP in interaction of S. pneumoniae with the BBB are required but may reveal a common pathway for the pathogenesis of streptococcal meningitis in humans.
‘Invasion genes’ often mediate entry and passage through a number of host cellular barriers, and evidence indicates that adherence to and invasion of other cell types such as chorion epithelial cells [46] and lung epithelial cells [47] are likely to be important for earlier stages of GBS neonatal disease pathogenesis. Deletion of GBS iagA, for example, not only affects invasion of hBMEC but also results in attenuated invasion of lung and chorion epithelial cells [5]. Similarly, we found that deletion of Srr-1 attenuates GBS invasion in multiple cell types including both endothelial and epithelial cell lines. Samen et al. have also recently reported that Srr-1 in another GBS clinical isolate strain contributes to adherence to Hep-2 cells [14]. It will be of interest to determine the role of carbohydrate modification to the ability of Srr-1 to mediate host cell adherence and invasion.
The human BBB endothelium serves as a critical barrier to protect the CNS against microbial invasion, and penetration of the BBB is likely to be a primary and essential step in the pathogenesis of neonatal meningitis [38]. While it is conceivable that the neonatal BBB displays unique features that render it more susceptible to penetration by neonatal pathogens like GBS, previous studies comparing this hBMEC cell line to hBMEC derived from humans and rats at different ages (ranging from fetal to geriatric) demonstrated no differences in susceptibilty to bacterial infection and invasion [48]. These results corroborate the usefulness of this in vitro BBB model in the study of host-pathogen interactions, specifically the identification and characterization of GBS genes and gene products that are responsible for the interaction of GBS with brain endothelium. This report represents the first identification of a GBS surface expressed protein that directly interacts with the BBB to promote CNS invasion and the development of GBS meningitis. Our data also suggest that Srr proteins may represent a target for pharmacologic or vaccine strategies to prevent the development of meningitis in the vulnerable neonate.
Acknowledgments
We thank Kwang Sik Kim and Monique Stins (Johns Hopkins University, Baltimore, MD) for providing the hBMEC cell line. This work was supported by the Department of Veterans Affairs and grants RO1AI41513 and RO1AI057433 from NIH to P.M.S., an American Heart Association Established Investigator Award to V.N., and by a Burroughs Wellcome Fund Career Award and grant RO1NS051247 from the NINDS/NIH to K.S.D.
Footnotes
Kelly S. Doran, Department of Biology and Center for Microbial Sciences, San Diego State University 5500 Campanile Drive San Diego, California 92182, USA. Phone: 619-594-1867, Fax: 619-594-5676E-mail: kdoran@sciences.sdsu.edu
The authors report no conflict of interest.
This work was supported by the Department of Veterans Affairs and grants RO1AI41513 and RO1AI057433 from NIH to P.M.S., an American Heart Association Established Investigator Award to V.N., and by a Burroughs Wellcome Fund Career Award and grant RO1NS051247 from the NINDS/NIH to K.S.D.
This work will be presented as a poster (abstract no. B-379) at the American Society of Microbiology Meeting, June 2008, Boston, MA, USA.
References
- 1.Doran KS, Nizet V. Molecular pathogenesis of neonatal group B streptococcal infection: no longer in its infancy. Mol Microbiol. 2004;54:23–31. doi: 10.1111/j.1365-2958.2004.04266.x. [DOI] [PubMed] [Google Scholar]
- 2.Edwards MS, Rench MA, Haffar AA, Murphy MA, Desmond MM, Baker CJ. Long-term sequelae of group B streptococcal meningitis in infants. J Pediatr. 1985;106:717–22. doi: 10.1016/s0022-3476(85)80342-5. [DOI] [PubMed] [Google Scholar]
- 3.Stins MF, Prasadarao NV, Ibric L, Wass CA, Luckett P, Kim KS. Binding characteristics of S fimbriated Escherichia coli to isolated brain microvascular endothelial cells. Am J Pathol. 1994;145:1228–36. [PMC free article] [PubMed] [Google Scholar]
- 4.Stins MF, Prasadarao NV, Zhou J, Arditi M, Kim KS. Bovine brain microvascular endothelial cells transfected with SV40-large T antigen: development of an immortalized cell line to study pathophysiology of CNS disease. In Vitro Cell Dev Biol Anim. 1997;33:243–7. doi: 10.1007/s11626-997-0042-1. [DOI] [PubMed] [Google Scholar]
- 5.Doran KS, Engelson EJ, Khosravi A, et al. Blood-brain barrier invasion by group B Streptococcus depends upon proper cell-surface anchoring of lipoteichoic acid. J Clin Invest. 2005;115:2499–507. doi: 10.1172/JCI23829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Maisey HC, Hensler M, Nizet V, Doran KS. Group B streptococcal pilus proteins contribute to adherence to and invasion of brain microvascular endothelial cells. J Bacteriol. 2007;189:1464–7. doi: 10.1128/JB.01153-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Prasadarao NV, Wass CA, Weiser JN, Stins MF, Huang SH, Kim KS. Outer membrane protein A of Escherichia coli contributes to invasion of brain microvascular endothelial cells. Infect Immun. 1996;64:146–53. doi: 10.1128/iai.64.1.146-153.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ring A, Weiser JN, Tuomanen EI. Pneumococcal trafficking across the blood-brain barrier. Molecular analysis of a novel bidirectional pathway. J Clin Invest. 1998;102:347–60. doi: 10.1172/JCI2406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tenenbaum T, Spellerberg B, Adam R, Vogel M, Kim KS, Schroten H. Streptococcus agalactiae invasion of human brain microvascular endothelial cells is promoted by the laminin-binding protein Lmb. Microbes Infect. 2007;9:714–20. doi: 10.1016/j.micinf.2007.02.015. [DOI] [PubMed] [Google Scholar]
- 10.Turner DP, Marietou AG, Johnston L, et al. Characterization of MspA, an immunogenic autotransporter protein that mediates adhesion to epithelial and endothelial cells in Neisseria meningitidis. Infect Immun. 2006;74:2957–64. doi: 10.1128/IAI.74.5.2957-2964.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nizet V, Kim KS, Stins M, et al. Invasion of brain microvascular endothelial cells by group B streptococci. Infect Immun. 1997;65:5074–81. doi: 10.1128/iai.65.12.5074-5081.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Doran KS, Liu GY, Nizet V. Group B streptococcal beta-hemolysin/cytolysin activates neutrophil signaling pathways in brain endothelium and contributes to development of meningitis. J Clin Invest. 2003;112:736–44. doi: 10.1172/JCI17335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bensing BA, Takamatsu D, Sullam PM. Determinants of the streptococcal surface glycoprotein GspB that facilitate export by the accessory Sec system. Mol Microbiol. 2005;58:1468–81. doi: 10.1111/j.1365-2958.2005.04919.x. [DOI] [PubMed] [Google Scholar]
- 14.Samen U, Eikmanns BJ, Reinscheid DJ, Borges F. The surface protein Srr-1 of Streptococcus agalactiae binds human keratin 4 and promotes adherence to epithelial HEp-2 cells. Infect Immun. 2007;75:5405–14. doi: 10.1128/IAI.00717-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Siboo IR, Chambers HF, Sullam PM. Role of SraP, a serine-rich surface protein of Staphylococcus aureus, in binding to human platelets. Infect Immun. 2005;73:2273–80. doi: 10.1128/IAI.73.4.2273-2280.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Stinson MW, Alder S, Kumar S. Invasion and killing of human endothelial cells by viridans group streptococci. Infect Immun. 2003;71:2365–72. doi: 10.1128/IAI.71.5.2365-2372.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Seifert KN, Adderson EE, Whiting AA, Bohnsack JF, Crowley PJ, Brady LJ. A unique serine-rich repeat protein (Srr-2) and novel surface antigen (epsilon) associated with a virulent lineage of serotype III Streptococcus agalactiae. Microbiology. 2006;152:1029–40. doi: 10.1099/mic.0.28516-0. [DOI] [PubMed] [Google Scholar]
- 18.Phares CR, Lynfield R, Farley MM, et al. Epidemiology of invasive group B streptococcal disease in the United States, 1999–2005. JAMA. 2008;299:2056–65. doi: 10.1001/jama.299.17.2056. [DOI] [PubMed] [Google Scholar]
- 19.Wilkinson HW. Nontypable group B streptococci isolated from human sources. J Clin Microbiol. 1977;6:183–4. doi: 10.1128/jcm.6.2.183-184.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wilson CB, Weaver WM. Comparative susceptibility of group B streptococci and Staphylococcus aureus to killing by oxygen metabolites. J Infect Dis. 1985;152:323–9. doi: 10.1093/infdis/152.2.323. [DOI] [PubMed] [Google Scholar]
- 21.Wessels MR, Paoletti LC, Rodewald AK, et al. Stimulation of protective antibodies against type Ia and Ib group B streptococci by a type Ia polysaccharide-tetanus toxoid conjugate vaccine. Infect Immun. 1993;61:4760–6. doi: 10.1128/iai.61.11.4760-4766.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lancefield RC, McCarty M, Everly WN. Multiple mouse-protective antibodies directed against group B streptococci. Special reference to antibodies effective against protein antigens. J Exp Med. 1975;142:165–79. doi: 10.1084/jem.142.1.165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sullam PM, Valone FH, Mills J. Mechanisms of platelet aggregation by viridans group streptococci. Infect Immun. 1987;55:1743–50. doi: 10.1128/iai.55.8.1743-1750.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kim KS. Escherichia coli translocation at the blood-brain barrier. Infect Immun. 2001;69:5217–22. doi: 10.1128/IAI.69.9.5217-5222.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pritzlaff CA, Chang JC, Kuo SP, Tamura GS, Rubens CE, Nizet V. Genetic basis for the beta-haemolytic/cytolytic activity of group B Streptococcus. Mol Microbiol. 2001;39:236–47. doi: 10.1046/j.1365-2958.2001.02211.x. [DOI] [PubMed] [Google Scholar]
- 26.Jeng A, Sakota V, Li Z, Datta V, Beall B, Nizet V. Molecular genetic analysis of a group A Streptococcus operon encoding serum opacity factor and a novel fibronectin-binding protein, SfbX. J Bacteriol. 2003;185:1208–17. doi: 10.1128/JB.185.4.1208-1217.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Maguin E, Duwat P, Hege T, Ehrlich D, Gruss A. New thermosensitive plasmid for gram-positive bacteria. J Bacteriol. 1992;174:5633–8. doi: 10.1128/jb.174.17.5633-5638.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bensing BA, Sullam PM. An accessory sec locus of Streptococcus gordonii is required for export of the surface protein GspB and for normal levels of binding to human platelets. Mol Microbiol. 2002;44:1081–94. doi: 10.1046/j.1365-2958.2002.02949.x. [DOI] [PubMed] [Google Scholar]
- 29.Takamatsu D, Bensing BA, Sullam PM. Four proteins encoded in the gspB-secY2A2 operon of Streptococcus gordonii mediate the intracellular glycosylation of the platelet-binding protein GspB. J Bacteriol. 2004;186:7100–11. doi: 10.1128/JB.186.21.7100-7111.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tettelin H, Masignani V, Cieslewicz MJ, et al. Complete genome sequence and comparative genomic analysis of an emerging human pathogen, serotype V Streptococcus agalactiae. Proc Natl Acad Sci U S A. 2002;99:12391–6. doi: 10.1073/pnas.182380799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Takamatsu D, Bensing BA, Sullam PM. Genes in the accessory sec locus of Streptococcus gordonii have three functionally distinct effects on the expression of the platelet-binding protein GspB. Mol Microbiol. 2004;52:189–203. doi: 10.1111/j.1365-2958.2004.03978.x. [DOI] [PubMed] [Google Scholar]
- 32.Obert C, Sublett J, Kaushal D, et al. Identification of a candidate Streptococcus pneumoniae core genome and regions of diversity correlated with invasive pneumococcal disease. Infect Immun. 2006;74:4766–77. doi: 10.1128/IAI.00316-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wu H, Mintz KP, Ladha M, Fives-Taylor PM. Isolation and characterization of Fap1, a fimbriae-associated adhesin of Streptococcus parasanguis FW213. Mol Microbiol. 1998;28:487–500. doi: 10.1046/j.1365-2958.1998.00805.x. [DOI] [PubMed] [Google Scholar]
- 34.Bensing BA, Gibson BW, Sullam PM. The Streptococcus gordonii platelet binding protein GspB undergoes glycosylation independently of export. J Bacteriol. 2004;186:638–45. doi: 10.1128/JB.186.3.638-645.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Petersdorf RG, Swarner DR, Garcia M. Studies on the pathogenesis of meningitis. II Development of meningitis during pneumococcal bacteremia. J Clin Invest. 1962;41:320–7. doi: 10.1172/JCI104485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bell LM, Alpert G, Campos JM, Plotkin SA. Routine quantitative blood cultures in children with Haemophilus influenzae or Streptococcus pneumoniae bacteremia. Pediatrics. 1985;76:901–4. [PubMed] [Google Scholar]
- 37.Dietzman DE, Fischer GW, Schoenknecht FD. Neonatal Escherichia coli septicemia--bacterial counts in blood. J Pediatr. 1974;85:128–30. doi: 10.1016/s0022-3476(74)80308-2. [DOI] [PubMed] [Google Scholar]
- 38.Kim KS. Pathogenesis of bacterial meningitis: from bacteraemia to neuronal injury. Nat Rev Neurosci. 2003;4:376–85. doi: 10.1038/nrn1103. [DOI] [PubMed] [Google Scholar]
- 39.Tenenbaum T, Bloier C, Adam R, Reinscheid DJ, Schroten H. Adherence to and invasion of human brain microvascular endothelial cells are promoted by fibrinogen-binding protein FbsA of Streptococcus agalactiae. Infect Immun. 2005;73:4404–9. doi: 10.1128/IAI.73.7.4404-4409.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Takahashi Y, Konishi K, Cisar JO, Yoshikawa M. Identification and characterization of hsa, the gene encoding the sialic acid-binding adhesin of Streptococcus gordonii DL1. Infect Immun. 2002;70:1209–18. doi: 10.1128/IAI.70.3.1209-1218.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chen Q, Wu H, Fives-Taylor PM. Investigating the role of secA2 in secretion and glycosylation of a fimbrial adhesin in Streptococcus parasanguis FW213. Mol Microbiol. 2004;53:843–56. doi: 10.1111/j.1365-2958.2004.04116.x. [DOI] [PubMed] [Google Scholar]
- 42.Bensing BA, Lopez JA, Sullam PM. The Streptococcus gordonii surface proteins GspB and Hsa mediate binding to sialylated carbohydrate epitopes on the platelet membrane glycoprotein Ibalpha. Infect Immun. 2004;72:6528–37. doi: 10.1128/IAI.72.11.6528-6537.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Stephenson AE, Wu H, Novak J, Tomana M, Mintz K, Fives-Taylor P. The Fap1 fimbrial adhesin is a glycoprotein: antibodies specific for the glycan moiety block the adhesion of Streptococcus parasanguis in an in vitro tooth model. Mol Microbiol. 2002;43:147–57. doi: 10.1046/j.1365-2958.2002.02725.x. [DOI] [PubMed] [Google Scholar]
- 44.Takamatsu D, Bensing BA, Cheng H, et al. Binding of the Streptococcus gordonii surface glycoproteins GspB and Hsa to specific carbohydrate structures on platelet membrane glycoprotein Ibalpha. Mol Microbiol. 2005;58:380–92. doi: 10.1111/j.1365-2958.2005.04830.x. [DOI] [PubMed] [Google Scholar]
- 45.Rose L, Shivshankar P, Hinojosa E, Rodriguez A, Sanchez CJ, Orihuela CJ. Antibodies against PsrP, a novel Streptococcus pneumoniae adhesin, block adhesion and protect mice against pneumococcal challenge. J Infect Dis. 2008;198:375–83. doi: 10.1086/589775. [DOI] [PubMed] [Google Scholar]
- 46.Winram SB, Jonas M, Chi E, Rubens CE. Characterization of group B streptococcal invasion of human chorion and amnion epithelial cells in vitro. Infect Immun. 1998;66:4932–41. doi: 10.1128/iai.66.10.4932-4941.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Doran KS, Chang JC, Benoit VM, Eckmann L, Nizet V. Group B streptococcal beta-hemolysin/cytolysin promotes invasion of human lung epithelial cells and the release of interleukin-8. J Infect Dis. 2002;185:196–203. doi: 10.1086/338475. [DOI] [PubMed] [Google Scholar]
- 48.Stins MF, Nemani PV, Wass C, Kim KS. Escherichia coli binding to and invasion of brain microvascular endothelial cells derived from humans and rats of different ages. Infect Immun. 1999;67:5522–5. doi: 10.1128/iai.67.10.5522-5525.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]