

Abstract
Soluble oligomers and fibrillar deposits of amyloid beta (A ) are key agents of Alzheimer’s disease pathogenesis. However, the mechanism of amyloid aggregation and its interaction with live cells still remain unclear requiring the preparation of large amounts of pure and different Aβ peptides. Here we describe an E. coli expression system using a fusion protein to obtain either A 1–40 or Aβ1–42 by essentially the same procedure. The fusion protein uses a His-tagged intestinal fatty acid binding protein (IFABP) followed by a six-glycine linker and a Factor Xa cleavage site before the Aβ. The advantages of this system are that the fusion protein can be expressed in large amounts, that the fusion partner, IFABP, has been well characterized in terms of folding, that A or mutated Aβ peptides can be obtained without any extra residues attached to the N-terminus and that the system can be used to incorporate fluorine labeled amino acids. The incorporation of fluorine labeled amino acids using auxotrophic strains is a useful NMR probe of side chain behavior. We obtain final yields of 4 mg/L and 3 mg/L of culture for A 1–40 and A 1–42 respectively.
Keywords: keywords: amyloid-β, fusion protein, fatty acid binding protein, expression
Introduction
Amyloid plaques, which consist of fibrillar aggregates of amyloid- (A) peptides, are a pathological hallmark of Alzheimer’s disease. It is known that A 1–42 is the major and A 1–40 the minor component of the brain plaques [1–3] and it has been proposed that soluble oligomers as well as fibrillar aggregates of A are toxic to neuronal cells [4–10]. In addition, there are indications that interactions of A with several other proteins are important in the progression of the disease [11]. Considerable research has been performed to understand the detailed mechanism of amyloid aggregation, the nature of the toxic species and the cellular pathway to toxicity caused by A. Yet the process of oligomerization or aggregation still remain unclear. Investigation of these mechanisms requires the production of large amounts of pure A peptides as well as the ability to make diverse mutants of the different Aβ peptides. The pure peptides, however, are difficult to isolate from neuronal tissue. Thus, it is advantageous to be able to prepare large amounts of the peptides using biological systems.
The expression of A in E. coli has been difficult due to its toxicity and poor solubility [12]. To overcome these problems a conventional approach is to express A as a suitable fusion protein. The fusion protein helps to solubilize the protein of interest and also prevents toxicity [13–15]. This approach has been successful for several different proteins [13–15]. Expression of A 1–40 or A 1–42 fused with a surface binding protein from the malaria parasite Plasmodium falciparum [16], or with maltose binding protein [17], ubiquitin [18], hen egg lysozyme [19] or glutathione-S-transferase [20] has been reported. In these studies the yield has been good for A 1–40 but less so for A 1–42.
In this paper we present a method using a fusion protein to produce either A 1–40 or A 1–42. The fusion protein consists of a His-tagged intestinal fatty acid binding protein (IFABP) connected by a glycine linker followed by a Factor Xa cleavage site and then the Aβ. IFABP is a 131-residue long 15-kDa protein for which the X-ray [21] and NMR structures have been determined [22]. The protein has been extensively studied with regard to stability, folding and mutations [23–25]. It is highly soluble, folds readily and auxotrophic bacterial strains for the incorporation of fluorine labeled amino acids are available [26]. The high solubility appears to prevent Aβ self-aggregation allowing for easy purification and high yields of the Aβ peptides. Furthermore the use of Factor Xa yields an A peptide with no extra amino acid residues at the N-terminus.
Materials and methods
Cloning and construction of the recombinant plasmid
The plasmid pQE-80L-6(His)-IFABP was constructed as described elsewhere [21]. The plasmid pQE-80L-6(His)-IFABP contains a EcoR1 restriction site at the 5′ end and a Kpn1 restriction site at the 3′ end of the sequence of 6(His)-IFABP fusion protein. To make the plasmid for the fusion protein, a single stranded DNA containing the sequence for A 1–40 was constructed from two synthetic oligonucleotides (Operon Biotechnologies) using overlapping PCR. From the 5′ end of the PCR product, the first 18 base pairs match the 18 base pairs of the 3′ end of IFABP, the next 18 base pairs encode 6 consecutive glycine resides, the next 9 base pairs encode Factor Xa recognition amino acid sequence IEGR, the 120 base pairs encode 40 residues of A 1–40 and the last 6 base pairs after the A stop codon encode Kpn1 restriction site. The construct for the full length fusion protein was synthesized by PCR using a reverse complement of the synthesized construct Aβ as a reverse primer, the EcoR1 as forward primer and the pQE-80L vector as a template. The vector pQE-80L was purified after cleaving the insert for the 6(His)-IFABP from the plasmid pQE-80L-6(His)-IFABP. The construct of the fusion protein is then inserted into the vector using ligation at the EcoR1 site and the Kpn1 site. The details of the plasmid pQE-80L-6(His)-IFABP-Aβ are described in Figure 1. The recombinant plasmid was transformed into XL-1 Blue cells (New England Biolabs, USA). The IFABP-Aβ insert gene sequence was confirmed using DNA sequencing. For Aβ1–42, a QuikChange site-directed mutagenesis kit (Stratagene, USA) was used to insert two amino acids (isoleucine and alanine) at the 3′ end to make pQE-80L-IFABP-Aβ1–42 from pQE-80L-IFABP-Aβ1–40. The QuikChange site-directed mutagenesis method was performed using PfuTurbo DNA polymerase and a temperature cycler.
Figure 1.

Construct of the pQE-80L-IFABP-Aβ expression vector.
Expression of IFABP-A fusion protein
The standard protocol for expression of IFABP was used for the expression of the fusion protein [21]. Briefly, E. coli cells (MG1655) were transformed with the fusion protein plasmid (either for Aβ1–40 or Aβ1–42) and a single colony chosen to grow a 250 ml starter culture in Luria broth overnight at 37 °C. The next day the culture was refreshed with 100 ml Terrific Broth media and 10 ml taken to initiate a 1 L culture in Terrific Broth. Generally it required about 2.5 hr to reach an OD600 of 3.0. Isopropyl-beta-D-thiogalactopyranoside (IPTG) was then added to 1 mM for induction. The culture was grown for an additional 2 hours and the cells then harvested by centrifugation at 4000 g for 20 minutes. Approximately 10 gram (wet weight) cells were obtained from 1 L culture.
Cell lysis and affinity purification of the fusion protein
After centrifugation, the cell pellet was re-suspended in 10 ml (for 1 L culture) ice-cold buffer (10 mM Tris-Cl, 150 mM NaCl, pH 7.4) and lysed by sonication at ice-cold temperature. The cell extract was then centrifuged at 20,000 g for 30 minutes and the supernatant discarded. The pellet was washed with 25 ml native buffer (10 mM Tris-Cl, 150 mM NaCl, pH 7.4) and then re-suspended in 50 ml Buffer B (8 M urea, 10 mM Tris-Cl, 100 mM NaH2PO4, pH 8.0) and kept at room temperature for 30 minutes. This solution was then centrifuged at 25,000 g for 30 minutes and the supernatant used for affinity purification using nickel-nitriloacetic acid (Ni-NTA) column purification. The Ni-NTA column (5 ml, GE Healthcare, USA) was equilibrated with buffer B prior to loading the sample on the column. The column was washed with buffer B for 5 – 10 column volumes and then washed with native binding buffer (20 mM NaH2PO4, 500 mM NaCl and 30 mM imidazole at pH 8.0) with another 5–10 column volumes. The fusion protein thus folds on the column. The protein is then eluted with the elution buffer (20 mM NaH2PO4, 500 mM NaCl and 500 mM imidazole at pH 7.4).
Estimation of fusion protein amount using absorbance
The absorbance of IFABP, A280, is 1.1/mg/ml. Using this value, the absorbance at 280 nm of the fusion protein is calculated to be 1.4/mg/ml. This value was used to estimate the amount of the fusion protein obtained in each step of purification when the purity of the fusion protein was greater than 90%.
Size exclusion chromatography prior to Factor Xa cleavage
Size exclusion chromatography was used to transfer the Ni-NTA purified fusion protein into the Factor Xa cleavage buffer. A Sephacryl S-200, 320 ml column (GE Healthcare, USA), equilibrated with 20 mM Tris-Cl, 100 mM NaCl at pH 7.4 buffer, was used. The Ni-NTA column purified fusion protein, which is collected at high concentration, is found to contain some small oligomers (data not shown). To dissociate these forms, the protein is dissolved in 6 M GdnCl. This solution is then passed through the Sephacryl S-200 column and the major fraction, representing the monomeric protein, was used in the following step. Since there is good separation between the fusion protein and the salts, a large sample volume could be loaded in this step.
Factor Xa cleavage of the fusion protein
For the cleavage reaction the must be kept between 0.3 to 0.4, A280 corresponding to a concentration of ~0.42 to 0.56 mg/ml. At lower fusion protein concentrations Factor Xa does not cleave efficiently and at higher concentrations the fusion protein forms oligomers which also are not cleaved well. Thus, it is critical to use a concentration of the fusion protein under conditions where it is monomeric. CaCl2 is added to a concentration of 2 mM and 10 μg of Factor Xa (Haematologic Technologies Inc., USA) is used per mg of fusion protein. Generally 5 hours at room temperature is sufficient for the complete cleavage. After 5 hours the reaction is terminated with 20 μM 4-amidinophenylmethanesulfonyl fluoride hydrochloride (APMSF) (Sigma, USA).
Separation of A from the cleaved fusion protein using aggregation
After completion of the cleavage reaction, the Aβ peptide is allowed to aggregate overnight at room temperature with gentle shaking. In this step, 100 μM ZnCl2 is added to enhance aggregation of Aβ1–40 but is not required for Aβ1–42. By the next day the sample appears turbid. The solution is centrifuged and the supernatant discarded. Although IFABP alone does not aggregate at this concentration it does co-aggregate with A. Thus, the precipitate contains Aβ and some IFABP. A measurement of A280 of the supernatant shows about 40% percent of the IFABP remains in the supernatant while the rest remains in the precipitate.
HPLC purification
To solubilize the Aβ the precipitate is dissolved in 6 M GdnCl, 10 mM EDTA and 0.1%-mercaptoethanol. Generally 5 ml buffer is sufficient to dissolve the precipitate obtained from the cleavage of 30 mg fusion protein. Immediately prior to injecting the sample into the HPLC, 10% formic acid is added. The sample is purified with HPLC on a C18 column (C18 column, 5 μm, 250mm×25mm, Vydac, USA). After loading the sample, the column is washed for 5 minutes with solvent A (0.05% TFA water), then a gradient of solvent B (100% acetonitrile) from 15% to 50% is applied. The solvent flow rate is kept at 8 –10 ml/min. A is eluted as a single peak at 32–35% acetonitrile while IFABP elutes at 40–50% acetonitrile (data not shown). The A containing fractions are collected in a glass vial and lyophilized. The lyophilized powder is stored at −80 °C.
Gel electrophoresis
SDS-PAGE gel elctrophoresis is performed with 10–20% Tris-HCl gels (precast gel, BioRad, USA) using the Tris-Glycine SDS running buffer (BioRad, USA). The gels are stained with Simple Blue for 1 hour (Invitrogen, USA) and then washed with de-ionized water. To check for fusion protein induction, a 1-ml cell sample was centrifuged and the cell pellet was resuspended in 1 ml Bugbuster reagent (Novagen, USA) in presence of 2.5 unit of Benzonase (Novagen, USA) and kept on ice for additional 30 minutes. This solution was centrifuged at 10,000g for 15 minutes. Fifteen μl of the supernatant was taken for SDS-PAGE gel electrophoresis. The precipitate was dissolved in 8 M urea, centrifuged at 13,000 g for 20 minutes and 15 μl of the supernatant taken for gel electrophoresis.
To check the purity of the final product a small amount (~50 μg) of the lyophilized peptide (for both Aβ1–40 and Aβ1–42) is dissolved in 100 μl 100 mM NaOH. This solution is immediately taken for SDS-PAGE gel electrophoresis.
Mass spectrometry
The A 1–40 or A 1–42 sample collected after HPLC purification was directly injected into the electrospray ionization (ESI) mass spectrometer. The m/z spectrum recorded between 200–2000 Dalton was analyzed with 1.0 Dalton resolution using the maximum entropy method.
Results
An outline of the purification scheme for Aβ is illustrated in Figure 2. The requirement for each step is discussed in this section. Table 1 gives a summary of A 1–40 and A 1–42 purification from 1 L E. coli culture.
Figure 2.

Diagrammatic scheme of the purification protocol.
Table 1.
Summary of A 1–40 and A 1–42 purification from 1 L E. coli culture†
| For A 1–40 preparation |
For A 1–42 preparation |
|||||||
|---|---|---|---|---|---|---|---|---|
| Purification step | Volume (ml) |
IFABP-A 1–40 (mg) |
A 1–40 (mg) |
Recovery (%) |
Volume (ml) |
IFABP-A 1–42 (mg) |
A 1–42 (mg) |
Recovery (%) |
| Inclusion bodies dissolved in urea (25,000g supernatant) | 50 | NDa | NDa | NDa | 50 | NDa | NDa | NDa |
| Ni-NTA chromatography | 40 | 40 | 8.2b | 100 | 40 | 40 | 8.6b | 100 |
| Sephacryl S-200 size exclusion chromatography | 80 | 32 | 6.6b | 80 | 80 | 30 | 6.4b | 75 |
| Factor Xa cleavage | 80 | NDa | NDa | NDa | 80 | NDa | NDa | NDa |
| Overnight aggregation, ppt dissolved in GdnCl | 5 | 22 | 6.6b,c | NDa | 5 | 21 | 6.4b,c | NDa |
| C18 reverse phase chromatography | 9 | NDa | 4 | 49 | 9 | NDa | 3 | 35 |
10 g cell (wet weight) cell is obtained from 1 L culture.
Not detemined.
The amount of A is calculated from the mass ratio of A and the fusion protein.
The precipitate is expected to contain the full amount of the A initially present.
After the cells were harvested with centrifugation, the supernatant solution and the inclusion bodies were checked for the extent of induction and whether the fusion protein appeared in the supernatant solution or in inclusion bodies. Figure 3a (for Aβ1–40 fusion protein) and Figure 3b (for Aβ1–42 fusion protein) show SDS-PAGE gels of the proteins from the supernatant and the pellet of the cell extract. A large protein band of approximate mass of 21 kDa, that of the fusion protein, is seen in the pellet of the cell extract (Lane 2 in both Figure 3a and 3b). The supernatant of the cell extract shows a prominent band of approximately 19 kDa which is probably a fragment of the full length fusion protein. Hence the supernatant is discarded and the inclusion bodies used for further purification.
Figure 3.

SDS PAGE gels of cell extracts. a) IFABP-A 1–40. Lane 1, cell extract supernatant. Lane 2, pellet of the cell extract of dissolved in 8 M urea. Lane 3, molecular weight markers. b) IFABP-A 1–42. Lane 1, cell extract supernatant. Lane 2, pellet of cell extract dissolved in 8 M urea. Lane 3, molecular weight markers. The fusion protein band is circled in Figure 3a and 3b.
The pellet of the cell extract is washed with Tris-Cl buffer, dissolved in 8 M urea and then purified by Ni-NTA affinity chromatography. An SDS-PAGE gel of the eluted protein is shown in Lane 1 of Figure 4a (for the Aβ1–40 fusion protein) and Figure 4b (for the Aβ1–42 fusion protein). At this point, the fusion protein is mostly free of other contaminants although a small amount of the 19 kDa fragment remains. Following affinity purification more than 40 mg of fusion protein is obtained from 1 L culture (Table 1).
Figure 4.

SDS Page gels showing Factor Xa cleavage of the fusion proteins. a) IFABP-A 1–40. Lane 1, Ni-NTA column purified fusion protein. Lane 2, after purification with size exclusion chromatography and prior to cleavage with Factor Xa, Lane 3, after cleavage with Factor Xa. Lane 4, molecular weight markers. b) IFABP-A 1–42. Lane 1, Ni-NTA column purified fusion protein. Lane 2, after purification with size exclusion chromatography and prior to cleavage with Factor Xa, Lane 3, after cleavage with Factor Xa. Lane 4, molecular weight markers.
The fusion protein forms small oligomers at high concentrations in native buffer (data not shown) which cannot be cleaved efficiently with Factor Xa. To dissociate these oligomers, the fusion protein is treated with 6 M GdnCl and re-purified by size exclusion chromatography. In this step the fusion protein is transferred to the Factor Xa cleavage buffer and obtained in monomeric form. The loss of the fusion protein in this step is less than 20% for Aβ1–40 and less than 25% for Aβ1–42 (Table 1). The monomeric fusion protein is then cleaved with Factor Xa. The SDS-PAGE gel of the fusion protein before and after the Factor Xa cleavage reaction is shown in Figure 4a (for Aβ1–40 fusion protein) and in Figure 4b (for Aβ1–42 fusion protein). Lane 2, which corresponds to the fusion protein prior to cleavage in both Figures 4a and 4b, shows the expected 21 kDa band. Lane 3, which corresponds to the protein solution after cleavage in both Figures 4a and 4b, shows the fragments of the respective fusion proteins. It can be seen that the 21 kDa band is almost absent and a new band of approximate mass of 16 kDa appears in Lane 3. The Aβ bands in the figures are not visualized well due to the low sensitivity of detection of small peptide by the SDS-PAGE gel.
After the fusion protein is cleaved, IFABP and shorter fragments of A remain. To remove these impurities the A is allowed to aggregate. The aggregate which contains A and about 60% of the total IFABP is then dissolved in 6 M GdnCl. This solution is further purified with HPLC. The SDS-PAGE gel of the purified A is shown in Figure 5, Lane 1 (for A 1–40) and Lane 2 (for A 1–42). Both lanes show a single band corresponding to the mass less than 10 kDa. The absence of any other contaminating band in the gel indicates good purity of the sample.
Figure 5.
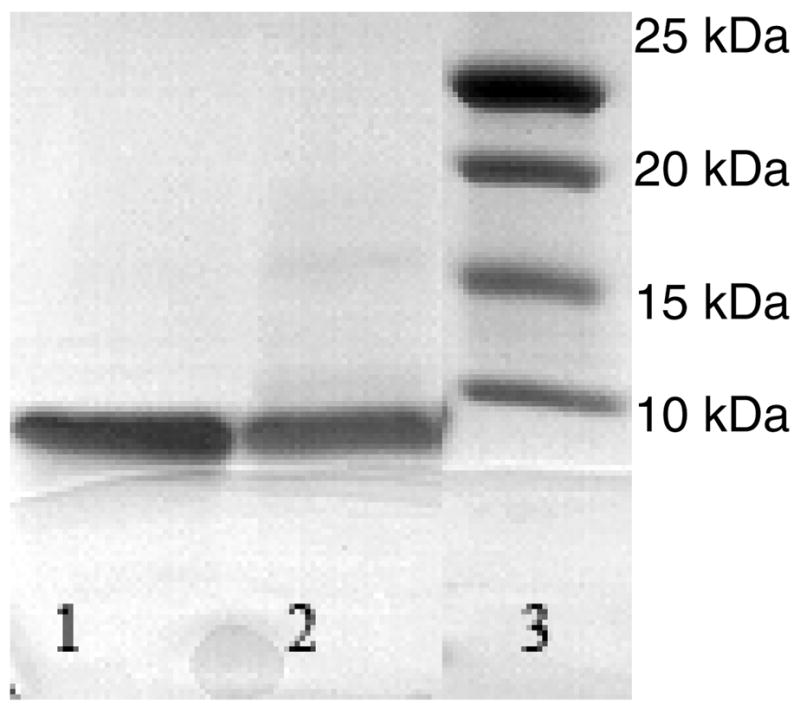
SDS-PAGE gel of purified A 1–40 and A 1–42. Purified A 1–40 (Lane 1), and A 1–42 (Lane 2). Markers shown in Lane 3.
Mass spectrometry is then performed to determine the actual mass of the purified peptides and to detect the presence of any contaminating peptides. The results from the mass spectrometry of the purified Aβ1–40 and Aβ1–42 peptides are shown in Figure 6a and 6b, respectively. The theoretical mass of Aβ1–40 is 4329 while that of Aβ1–42 is 4514. In Figure 6a, peaks with decreasing abundances at 4351, 4373 and 4393 are presumably due to presence of 1, 2 and 3 sodium atoms respectively. Similarly, in Figure 6b, the peak at 4536 indicates a single sodium atom bound to Aβ1–42. It is evident from Figures 5 and 6 that the Aβ finally obtained is essentially pure. We obtain approximately 4.0 mg Aβ1–40 or 3.0 mg Aβ1–42 from 1 L culture (Table 1).
Figure 6.

Mass spectrometry of purified A 1–40 and A 1–42. a) Electrospray mass spectrometry of A 1–40. The theoretical mass is 4329 daltons. The peaks at 4351, 4373 and 4393 daltons correspond to one, two and three sodium atoms bound to A 1–40. b) Mass spectrometry of A 1–42. The theoretical mass is 4514 daltons. The peak at 4536 daltons corresponds to presence of one sodium atom bound to A 1–42
Discussion
The original intent of this work was to have the ability to incorporate fluorine labeled amino acids into Aβ peptides since chemical synthesis, which would involve using only the L-isomer of the amino acid, would be prohibitively expensive. In the process of developing the protocol for that work, it became apparent that the same method could be used to produce large amounts of unlabeled Aβ.
As previous reports have noted, the use of fusion proteins to express and purify Aβ from E. coli provides amyloid peptides of high purity to be obtained. Here we present a protocol for the purification of either Aβ1–40 or Aβ1–42 as a fusion protein which should be applicable to other lengths (i.e., 39–43) of the Aβ peptide. It should be noted that the purification procedure is slightly different depending on the length of the Aβ. For Aβ1–40, Zn2+ is added to enhance the rate of aggregation while this step is not necessary for Aβ1–42 which aggregates rapidly.
The system used has several advantages over previous reports. First, the Aβ that is obtained does not have any extra residues at the N-terminal end in contrast to some other reports [20, 27]. This is consequence of the fact that Factor Xa cleaves between the C-terminal arginine residue of the recognition site (IEGR) and the first residue of the Aβ. Second, IFABP can be expressed with high yields providing for the expression of large amounts of peptide. Although the fusion protein is found in inclusion bodies, it is soluble after dissolving in denaturant and remains so even after removing the denaturant. Third, IFABP is a well characterized protein which refolds easily after mutating various residues, allowing mutation of the Aβ without concerns about the expression of the fusion protein. Thus, our method should be applicable to purifying other mutants as well. Finally, we have used auxotrophic strains of the IFABP expression system for the fusion protein to incorporate fluorine labeled residues for NMR studies of Aβ (data not shown).
Acknowledgments
This work was supported by National Institute of Health Grant DK13332. The authors thank Mr. Robert Horton and Ms. Berevan Baban for excellent technical assistance. Mass spectrometry of was provided by the A 1–42 Washington University Mass Spectrometry Resource, an NIH Research Source (Grant No. P41RR0954).
Abbreviations used
- IFABP
intestinal fatty acid binding protein
- TFA
trifluoroacetic acid
- GdnCl
guanidine-HCl
- ppt
precipitate
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Selkoe DJ. The molecular pathology of Alzheimer’s disease. Neuron. 1991;6:487–498. doi: 10.1016/0896-6273(91)90052-2. [DOI] [PubMed] [Google Scholar]
- 2.Selkoe DJ. Alzheimer’s disease: genes, proteins, and therapy. Physiol Rev. 2001;81:741–766. doi: 10.1152/physrev.2001.81.2.741. [DOI] [PubMed] [Google Scholar]
- 3.Teplow DB. Structural and kinetic features of amyloid beta-protein fibrillogenesis. Amyloid. 1998;5:121–142. doi: 10.3109/13506129808995290. [DOI] [PubMed] [Google Scholar]
- 4.Lorenzo A, Yankner BA. Beta-amyloid neurotoxicity requires fibril formation and is inhibited by Congo Red. Proc Natl Acad Sci U S A. 1994;91:12243–12247. doi: 10.1073/pnas.91.25.12243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Walsh DM, Hartley DM, Kusumoto Y, Fezoui Y, Condron MM, Lomakin A, Benedek GB, Selkoe DJ, Teplow DB. Amyloid beta-protein fibrillogenesis. Structure and biological activity of protofibrillar intermediates. J Biol Chem. 1999;274:25945–25952. doi: 10.1074/jbc.274.36.25945. [DOI] [PubMed] [Google Scholar]
- 6.Klein WL, Krafft GA, Finch CE. Targeting small Abeta oligomers: the solution to an Alzheimer’s disease conundrum? Trends Neurosci. 2001;24:219–224. doi: 10.1016/s0166-2236(00)01749-5. [DOI] [PubMed] [Google Scholar]
- 7.Lashuel HA, Hartley D, Petre BM, Walz T, Lansbury PT., Jr Neurodegenerative disease: amyloid pores from pathogenic mutations. Nature. 2002;418:291. doi: 10.1038/418291a. [DOI] [PubMed] [Google Scholar]
- 8.Hoshi M, Sato M, Matsumoto S, Noguchi A, Yasutake K, Yoshida N, Sato K. Spherical aggregates of beta-amyloid (amylospheroid) show high neurotoxicity and activate tau protein kinase I/glycogen synthase kinase-3beta. Proc Natl Acad Sci U S A. 2003;100:6370–6375. doi: 10.1073/pnas.1237107100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hartley DM, Walsh DM, Ye CP, Diehl T, Vasquez S, Vassilev PM, Teplow DB, Selkoe DJ. Protofibrillar intermediates of amyloid beta-protein induce acute electrophysiological changes and progressive neurotoxicity in cortical neurons. J Neurosci. 1999;19:8876–8884. doi: 10.1523/JNEUROSCI.19-20-08876.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cleary JP, Walsh DM, Hofmeister JJ, Shankar GM, Kuskowski MA, Selkoe DJ, Ashe KH. Natural oligomers of the amyloid-beta protein specifically disrupt cognitive function. Nat Neurosci. 2005;8:79–84. doi: 10.1038/nn1372. [DOI] [PubMed] [Google Scholar]
- 11.Holtzman DM. In vivo effects of ApoE and clusterin on amyloid-beta metabolism and neuropathology. J Mol Neurosci. 2004;23:247–254. doi: 10.1385/JMN:23:3:247. [DOI] [PubMed] [Google Scholar]
- 12.Sharpe S, Yau WM, Tycko R. Expression and purification of a recombinant peptide from the Alzheimer’s beta-amyloid protein for solid-state NMR. Protein Expr Purif. 2005;42:200–210. doi: 10.1016/j.pep.2005.03.005. [DOI] [PubMed] [Google Scholar]
- 13.LaVallie ER, DiBlasio EA, Kovacic S, Grant KL, Schendel PF, McCoy JM. A thioredoxin gene fusion expression system that circumvents inclusion body formation in the E. coli cytoplasm. Biotechnology (N Y) 1993;11:187–193. doi: 10.1038/nbt0293-187. [DOI] [PubMed] [Google Scholar]
- 14.Morrow JA, Arnold KS, Weisgraber KH. Functional characterization of apolipoprotein E isoforms overexpressed in Escherichia coli. Protein Expr Purif. 1999;16:224–230. doi: 10.1006/prep.1999.1069. [DOI] [PubMed] [Google Scholar]
- 15.Austin BP, Nallamsetty S, Waugh DS. Hexahistidine-tagged maltose-binding protein as a fusion partner for the production of soluble recombinant proteins in Escherichia coli. Methods Mol Biol. 2009;498:157–172. doi: 10.1007/978-1-59745-196-3_11. [DOI] [PubMed] [Google Scholar]
- 16.Dobeli H, Draeger N, Huber G, Jakob P, Schmidt D, Seilheimer B, Stuber D, Wipf B, Zulauf M. A biotechnological method provides access to aggregation competent monomeric Alzheimer’s 1–42 residue amyloid peptide. Biotechnology (N Y) 1995;13:988–993. doi: 10.1038/nbt0995-988. [DOI] [PubMed] [Google Scholar]
- 17.Hortschansky P, Schroeckh V, Christopeit T, Zandomeneghi G, Fandrich M. The aggregation kinetics of Alzheimer’s beta-amyloid peptide is controlled by stochastic nucleation. Prot Sci. 2005;14:1753–1759. doi: 10.1110/ps.041266605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lee EK, Hwang JH, Shin DY, Kim DI, Yoo YJ. Production of recombinant amyloid-beta peptide 42 as an ubiquitin extension. Protein Expr Purif. 2005;40:183–189. doi: 10.1016/j.pep.2004.12.014. [DOI] [PubMed] [Google Scholar]
- 19.Nagata-Uchiyama M, Yaguchi M, Hirano Y, Ueda T. Expression and purification of uniformly (15)N-labeled amyloid beta peptide 1–40 in Escherichia coli. Protein Pept Lett. 2007;14:788–792. doi: 10.2174/092986607781483741. [DOI] [PubMed] [Google Scholar]
- 20.Zhang L, Yu H, Song C, Lin X, Chen B, Tan C, Cao G, Wang Z. Expression, purification, and characterization of recombinant human beta-amyloid42 peptide in Escherichia coli. Protein Expr Purif. 2009;64:55–62. doi: 10.1016/j.pep.2008.10.007. [DOI] [PubMed] [Google Scholar]
- 21.Sacchettini JC, Banaszak LJ, Gordon JI. Expression of rat intestinal fatty acid binding protein in E. coli and its subsequent structural analysis: a model system for studying the molecular details of fatty acid-protein interaction. Mol Cell Biochem. 1990;98:81–93. doi: 10.1007/BF00231371. [DOI] [PubMed] [Google Scholar]
- 22.Hodsdon ME, Toner JJ, Cistola DP. 1H, 13C and 15N assignments and chemical shift-derived secondary structure of intestinal fatty acid-binding protein. J Biomol NMR. 1995;6:198–210. doi: 10.1007/BF00211784. [DOI] [PubMed] [Google Scholar]
- 23.Jiang N, Frieden C. Intestinal fatty acid binding protein: characterization of mutant proteins containing inserted cysteine residues. Biochemistry. 1993;32:11015–11021. doi: 10.1021/bi00092a010. [DOI] [PubMed] [Google Scholar]
- 24.Kim K, Frieden C. Turn scanning by site-directed mutagenesis: application to the protein folding problem using the intestinal fatty acid binding protein. Prot Sci. 1998;7:1821–1828. doi: 10.1002/pro.5560070818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chattopadhyay K, Zhong S, Yeh SR, Rousseau DL, Frieden C. The intestinal fatty acid binding protein: the role of turns in fast and slow folding processes. Biochemistry. 2002;41:4040–4047. doi: 10.1021/bi012042l. [DOI] [PubMed] [Google Scholar]
- 26.Frieden C, Hoeltzli SD, Bann JG. The preparation of 19F-labeled proteins for NMR studies. Methods Enzymol. 2004;380:400–415. doi: 10.1016/S0076-6879(04)80018-1. [DOI] [PubMed] [Google Scholar]
- 27.Macao B, Hoyer W, Sandberg A, Brorsson AC, Dobson CM, Hard T. Recombinant amyloid beta-peptide production by coexpression with an affibody ligand BMC. Biotechnol. 2008;8:82. doi: 10.1186/1472-6750-8-82. [DOI] [PMC free article] [PubMed] [Google Scholar]