

Abstract
Coinfection in RNA virus populations results in two important phenomena, complementation and recombination. Of the two, complementation has a strong effect on selection against deleterious mutations, as has been confirmed in earlier studies. As complementation delays the purging of less-fit mutations, coinfection may be detrimental to the evolution of a virus population. Here we employ both deterministic modeling and stochastic simulation to explore the mechanisms underlying the interactions between complementation and other evolutionary factors, namely, mutation, selection, and epistasis. We find that strong complementation reduces slightly the overall fitness of a virus population but substantially enhances its diversity and robustness, especially when interacting with selection and epistasis.
RNA viruses infect many organisms. They have high mutation rates, compact segmented genomes, very short generation times, huge population sizes, and substantial diversity within populations (Chao 1988; Wilke and Novella 2003; Froissart et al. 2004; Elena et al. 2006). These properties enable RNA viruses to change continuously and adapt easily to adverse environments, posing continual challenges for antiviral therapies and vaccinations. Current research on RNA viruses has turned to theoretical population genetics and evolutionary theory for novel and practical approaches for management of viral diseases (e.g., Turner and Chao 1999, 2003; Burch et al. 2003; Burch and Chao 2004; Dennehy and Turner 2004; Moya et al. 2004; Bretscher et al. 2004; Sanjuán et al. 2005; Boni et al. 2006).
Coinfection, where multiple viruses infect a cell simultaneously, is important in the life cycle of RNA viruses. During coinfection, haploid viral genomes act in a manner similar to those of diploid organisms, exchanging genetic material randomly or preferentially. Reassortment of genome segments may create severely deleterious mutational combinations, thus speeding up the elimination of mutational load. Coinfection may, however, result in complementation, where viruses carrying different deleterious mutations may benefit from the normal products that each can produce, so that both types of viruses can be represented in the offspring. In contrast to recombination, complementation weakens the selection against deleterious mutations. In this way, it contributes to the stability of the whole virus population as it maintains a high level of diversity without sacrificing the overall fitness of the population.
Froissart et al. (2004) investigated whether complementation or random reassortment would have the stronger effect on selection against deleterious mutations in viral life cycles. They proposed a simple model combining the processes of viral replication and free recombination and, via both experiments and simulation, confirmed the major role of complementation in reducing the selective pressure against deleterious mutations. However, their model assumes only two independent loci and does not accommodate such key evolutionary factors as mutation or higher-order epistasis. They simplified their model by ignoring mutation. In reality, as RNA viruses are highly mutable, a viral population does not completely eliminate deleterious mutations after a period of time, contrary to what appears to be the case in Froissart et al. (2004), but reaches a dynamic balance between selection and mutation. When studying the long-term evolution of a viral population, mutation becomes a key factor that should be taken into account appropriately. Epistasis is another important evolutionary factor that affects many evolutionary processes (Desai et al. 2007). Epistasis may be classified as synergistic or antagonistic, which may act on the mutation–selection balance in different ways and also interact with different levels of complementation. Epistasis can also be classified as magnitude and sign epistasis, where magnitude epistasis at a locus indicates that the magnitude of fitness change (loss or gain) of a mutation depends on the genotypic background; sign epistasis exists if the sign, as well as the magnitude, of the fitness cost of a mutation is affected by the genotypic background (Weinreich et al. 2005). To date, the roles of these types of epistasis interacting with coinfection have not been studied systematically. In modeling viral evolution, it may be important to incorporate epistasis and, in particular, to study how the effects exerted by complementation interact with different types of epistasis.
Here we extend Froissart's model to allow for multiple loci as well as mutation and complex interactions among mutations. We investigate how complementation interacts with epistasis and how this interaction affects the evolutionary dynamics of viruses. We utilize both deterministic modeling and stochastic simulation to investigate these interactions between complementation and mutation, selection, and epistasis. To introduce mutation, we divide the process of viral replication into three stages, prereplication preparation, replication, and assembly. For each segment of the viral genome, complementation and epistasis determine the strength of selection, and these occur before replication. Mutation occurs during replication, and random reassortment take place after replication during assembly. Mutation and random reassortment can be considered as a time-reversible process as the process of replication of one segment multiple times and then randomly choosing one copy for each assembly is equivalent to randomly choosing one of the two parental viral segments and replicating that segment, and repeating these two steps multiple times. Therefore, it is reasonable to consider mutation as a separate step. For epistasis, we employ a model of interactions similar to that used by Desai et al. (2007), including higher-order epistasis. Our results indicate that although it has only moderate effects on the overall mean fitness, complementation interacting with epistasis has a strong effect on the composition and diversity of the virus population.
MODEL
We first consider an RNA virus population evolving under selection, complementation, random reassortment, and epistasis. Suppose the virus is haploid with L segments. Without complementation, we assume each segment carrying one or more deleterious mutation incurs a fitness cost s and  . In this setting, following the multiplicative selection model proposed by Desai et al. (2007), a virus with k mutated segments has fitness e−ks.
. In this setting, following the multiplicative selection model proposed by Desai et al. (2007), a virus with k mutated segments has fitness e−ks.
We start with Froissart's model combining complementation, selection, and reassortment and also assume that the frequency of coinfection with more than two virions per cell is negligible (Olkkonen and Bamford 1989; Turner et al. 1999; Froissart et al. 2004). The effect of interactions among mutations is similar to that in Desai et al. (2007), but with additional higher-order interactions. We assume that double mutants have fitness  and triple mutants have fitness
and triple mutants have fitness  , where ɛ is the first-order epistasis parameter and η is the second-order epistasis coefficient. For each viral life cycle, we define F0 to be the frequency of the wild-type viruses in the population at the beginning of this cycle and F(i,j, … ,k) to be the initial frequency of viruses with mutations on segments i, j, … , k, where 1 ≤ i, j, …, k ≤ L. As in our model mutations are deleterious, we assume four or more deleterious mutations are lethal; i.e., the frequencies of haploids carrying more than three mutations are negligible; Fi,j,k,m ≈ 0. We denote by
, where ɛ is the first-order epistasis parameter and η is the second-order epistasis coefficient. For each viral life cycle, we define F0 to be the frequency of the wild-type viruses in the population at the beginning of this cycle and F(i,j, … ,k) to be the initial frequency of viruses with mutations on segments i, j, … , k, where 1 ≤ i, j, …, k ≤ L. As in our model mutations are deleterious, we assume four or more deleterious mutations are lethal; i.e., the frequencies of haploids carrying more than three mutations are negligible; Fi,j,k,m ≈ 0. We denote by 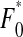 and
and 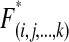 the frequencies of wild type and mutants, respectively, after complementation, epistasis, selection, and reassortment. Denote pc as the fraction of coinfections from the total number of infections occurring per generation. Then the wild-type genotype frequency
the frequencies of wild type and mutants, respectively, after complementation, epistasis, selection, and reassortment. Denote pc as the fraction of coinfections from the total number of infections occurring per generation. Then the wild-type genotype frequency 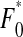 after the action of these four forces is
after the action of these four forces is
 |
(1) |
where I{Φ} is an indicator function and is equal to one if Φ is true; otherwise, it is zero. The complicated formulas for frequencies  ,
, 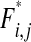 , and
, and 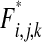 are given in the appendix. Summing these frequency formulas, we obtain the mean fitness
are given in the appendix. Summing these frequency formulas, we obtain the mean fitness 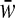 .
.
For viruses, mutations mostly occur during the replication processes. Selection, as a result of complementation and epistasis, occurs mainly during protein synthesis before replication. Random reassortment occurs during assembly of replicates. In our simulation, we can consider mutation and random reassortment as reversible. We can reverse the process of replication of one segment and then randomly choose one copy by randomly choosing one of the two parental viral segments and then replicating the chosen segment, since the two processes are, indeed, equivalent. Thus it is reasonable to separate mutation from the other events. (We also show in the appendix that this separation step does not alter the resulting frequencies, but speeds up the calculation). Denote by 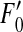 ,
, 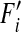 ,
, 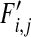 , and
, and 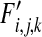 the frequencies of haploids carrying zero, one, two, and three mutations, respectively, after mutation. We now assume that in each segment the wild-type allele can mutate to the deleterious allele at rate μ per individual per generation. We further assume no back mutation occurs and ignore the terms with higher orders of μ as
the frequencies of haploids carrying zero, one, two, and three mutations, respectively, after mutation. We now assume that in each segment the wild-type allele can mutate to the deleterious allele at rate μ per individual per generation. We further assume no back mutation occurs and ignore the terms with higher orders of μ as 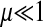 . This produces the following recursions:
. This produces the following recursions:
 |
(2) |
SIMULATION
As these recursions cannot be solved analytically, we conducted both deterministic and stochastic simulations to study the stationary distribution of allele frequencies. For deterministic simulations, we iterated the recursions (1), (2), and (A1)–(A3) 5000 times (corresponding to 5000 generations) with different values of the parameters. For stochastic simulations, we simulated 100 lineages of haploid viruses, with three segments in the genome, independently over 5000 generations. For generation t in lineage l (t = 1, 2, … , 5000; l = 1, 2, … , 100), we used the following sampling procedure:
- According to Froissart et al. (2004), the proportions of bacteria infected by zero, one, and two viruses follow a Poisson distribution with mean equal to the multiplicity-of-infection (MOI) (ratio of phages to bacterial cells). We denote by Pr(0), Pr(1), and Pr(2) the Poisson probabilities that a cell is infected by zero, one, or two viruses, respectively. Assuming no more than two viruses affect one cell simultaneously, the expected proportion of coinfection with two viruses from the total number of infections is
 . Thus each cell is coinfected with probability pc or has a single infection with probability
. Thus each cell is coinfected with probability pc or has a single infection with probability  . The frequency of viruses that coinfect is
. The frequency of viruses that coinfect is  . Suppose we have N0 viruses at the beginning of generation i and whether a virus infects or coinfects occurs as a series of Bernoulli trials with the probability of success equal to fco; i.e., we sample the number of coinfecting viruses kco from a binomial distribution Binom(fco, N0) and need to ensure it is an even and positive number. (If an odd number is sampled, we do an extra sampling step from the uniform distribution on [0, 1]. If the sampled value is <0.5, we deduct one from the odd number; otherwise, we add one to this odd number to make it an even number. We can also discard this odd number and continue to sample until we find an even number.) Within the group of the kco chosen coinfecting viruses, we randomly group them into pairs by iterating the following steps:
. Suppose we have N0 viruses at the beginning of generation i and whether a virus infects or coinfects occurs as a series of Bernoulli trials with the probability of success equal to fco; i.e., we sample the number of coinfecting viruses kco from a binomial distribution Binom(fco, N0) and need to ensure it is an even and positive number. (If an odd number is sampled, we do an extra sampling step from the uniform distribution on [0, 1]. If the sampled value is <0.5, we deduct one from the odd number; otherwise, we add one to this odd number to make it an even number. We can also discard this odd number and continue to sample until we find an even number.) Within the group of the kco chosen coinfecting viruses, we randomly group them into pairs by iterating the following steps:- Sample an individual virus with probability
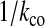 .
. - Sample the other virus of this pair with probability
 .
. - Let kco = kco − 2.
- Repeat steps 1 and 2 until kco = 2.
Each cell, either infected by a wild-type virus or coinfected by two viruses that can complement each other's defects, produces approximately the same number of offspring, which can be treated as a normal random variable m ∼ N(δ, σ2), where δ is the expected number of offspring produced by a wild-type virus and σ2 is its variance. If a virus carrying one, two, or three mutant segments infects a cell, the number of offspring produced follows m ∼ N(δe−s, σ2 × e−2s), N(δ
 , σ2 ×
, σ2 × 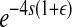 ), and N(δ
), and N(δ , σ2 ×
, σ2 × 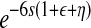 ), respectively, where s is the selection coefficient, ɛ is the pairwise epistasis coefficient, and η is the second-order epistasis coefficient. Similarly, if two mutant viruses coinfect a cell with one, two, or three mutant segments in common, they will produce a number of progeny similar to the case of a single infection above.
), respectively, where s is the selection coefficient, ɛ is the pairwise epistasis coefficient, and η is the second-order epistasis coefficient. Similarly, if two mutant viruses coinfect a cell with one, two, or three mutant segments in common, they will produce a number of progeny similar to the case of a single infection above.When viruses replicate genetic material in coinfected cells, assuming random reassortment, each virion randomly inherits a segment from one of its parents; i.e., each segment is selected from one of its parents with chance 0.5.
During the replication, we assume that deleterious mutations take place at μ = 0.01 per segment per virus generation. According to Drake and Holland (1999) and Drake (2006), the median spontaneous mutation rate of RNA viruses is ∼0.76 per genome per replication cycle. Thus it is reasonable to assume a deleterious mutation rate of 0.01 per segment per generation after accounting for advantageous mutations. We can sample mutations according to this probability and keep track of them. Each mutation occurs on a wild-type segment and may be inherited by offspring. All mutations suffer the same fitness loss s. If a mutation takes place on a mutant segment, we assume this will not have any additional effect on the overall fitness of a virus.
As the virus population size increases exponentially, a sampling step is necessary to keep a constant sample size that is computationally tractable, namely, 10,000. That is, if the population size is >10,000, we apply a Bernoulli sampling process without replacement to generate a sample of 10,000.
RESULTS
To evaluate the effects of interactions between complementation and other factors, we ran both deterministic and stochastic simulation at five levels of coinfection (i.e., pc ∈ {0.1, 0.3, 0.5, 0.7, 0.9}) under nine genetic scenarios (see Table 1). For stochastic modeling, we averaged results across 100 lineages and assessed the variation between lineages. The results from deterministic iterations are mostly similar to those from the stochastic simulation and both are shown in the supporting information. Here we focus mainly on the stationary distribution using the stochastic simulation (as the viral system might get stuck in an unstable equilibrium in the deterministic model; see the discussion) and also show the three-way interactions among complementation, selection, and epistasis based on deterministic modeling.
TABLE 1.
Parameter combinations for nine simulation scenarios
| Scenario | L | μ | s | ɛ | η | Initial frequencies |
|---|---|---|---|---|---|---|
| I | 3 | 0.0 | 0.1 | 0.0 | 0.0 | 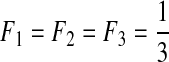 |
| II | 3 | 0.01 | 0.1 | 0.0 | 0.0 |  |
| III | 3 | 0.01 | 0.2 | 0.0 | 0.0 |  |
| IV | 3 | 0.01 | 0.1 | 0.3 | 0.0 |  |
| V | 3 | 0.01 | 0.1 | −0.3 | 0.0 |  |
| VI | 3 | 0.01 | 0.1 | 0.8 | 0.0 | 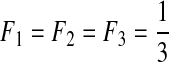 |
| VII | 3 | 0.01 | 0.1 | −0.8 | 0.0 | 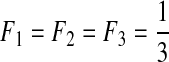 |
| VIII | 3 | 0.01 | 0.1 | 0.1 | 0.8 | 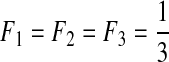 |
| IX | 3 | 0.01 | 0.1 | 0.1 | −0.8 |  |
L, the number of segments in a viral genome; μ, mutation rates per generation per segment; s, selection coefficients; ɛ, epistasis coefficients; η, second-order epistasis coefficients. F1, F2, and F3: the initial frequencies of single mutants.
Interaction of complementation with mutation:
We modified the complementation model proposed by Froissart et al. (2004) to take mutation dynamics into account. In reality, RNA viruses mutate at such high rates that mutations cannot be neglected even over a short time period and, in the long run, a virus population reaches a mutation–selection balance instead of completely eliminating deleterious mutations from the population. The life cycle of an RNA virus is typically composed of five steps, absorption and penetration, synthesis of viral proteins, RNA replication, assembly, and release. Complementation and selection take place during the period of viral protein synthesis, and reassortment happens just before the replication of each segment, whereas nucleotides mutate during the replication process. Therefore, it is reasonable to separate mutation from the other processes in our model.
In the absence of mutation, the virus population clears all the initial polymorphisms efficiently at all levels of coinfection, as illustrated in Figure 1, A1–A5, and in the supporting information, Figure S1 and Figure S6. With a mutation rate of 0.01 per generation per segment, mutations always exist in the population and the mean fitness is ∼0.03 lower when the population reaches the mutation–selection equilibrium than in the no mutation case. This is reasonable; according to classical population genetic theory the expected mean fitness of a viral population at the mutation–selection balance is 1 − Lμ = 0.97. We observed drastic changes in the patterns of frequencies of wild type, single mutants, double mutants, and triple mutants. With no mutation, the wild type frequency is fixed at one whereas mutants' frequencies quickly drop to zero. Generally the frequencies of mutants decrease faster with low coinfection rates. When mutation occurs, the wild-type frequency increases until it reaches a limit and this stationary frequency decreases as pc increases. The stationary wild-type frequency drops more at high levels of coinfection than at low coinfection levels. The frequencies of single mutants decrease from one to a lower level under low coinfection rates than with high coinfection, and the difference between two consecutive coinfection levels remains approximately the same irrespective of the coinfection rate. Both double and triple mutants show a similar frequency pattern with their equilibrium frequencies substantially higher with frequent coinfections than with rare coinfections.
Figure 1.—

Interaction patterns at equilibrium under different levels of coinfection ratios, 0.1, 0.3, 0.5, 0.7, and 0.9, respectively. (A1–A5) The interaction between complementation and mutation (μ) under scenarios I and II, with s = 0.1, ɛ = 0.0, η = 0.0, red for μ = 0.0, and light blue for μ = 0.01. (B1–B5) The interaction between complementation and selection (s) under scenarios II and III, with μ = 0.01, ɛ = 0.0, η = 0.0, red for s = 0.1, and light blue for s = 0.2. (C1–C5) The interaction of complementation with magnitude epistasis (ɛ) under scenarios II, IV, and V, with μ = 0.01, s = 0.1, η = 0.0, red for ɛ = 0.3, yellow for ɛ = 0.0, and light blue for ɛ = −0.3. (D1–D5) The interaction between complementation and sign epistasis (ɛ) under scenarios II, VI, and VII, with μ = 0.01, s = 0.1, η = 0.0, red for ɛ = 0.8, yellow for ɛ = 0.0, and light blue for ɛ = −0.8. (E1–E5) The interaction of complementation with second-order epistasis (η) under scenarios VIII and IX, with μ = 0.01, s = 0.1, ɛ = 0.1, red for η = 0.8, and light blue for η = −0.8. The first column of bar plots represents the pattern of the overall mean fitness under various scenarios, the second column shows that for wild-type frequencies, the third column shows that for single-mutant frequencies, the fourth column shows that for double-mutant frequencies, and the fifth column shows that for triple-mutant frequencies. Note that the vertical scales of the last three columns of bar plots are different and vary from 0.02 to 1.0.
Interaction of complementation with selection:
We then investigated how complementation affects the evolutionary dynamics of virus populations when the selection pressure varies. From the mean fitness plot (see Figure 1, B1–B5, Figure S2, and Figure S7), different selection strengths drive the virus population to a similar equilibrium mean fitness, but the composition of this population varies markedly when the selection coefficient increases from 0.1 to 0.2. When selection becomes stronger, more mutants are eliminated from the population; thus the balanced frequencies of wild-type viruses generally increase for all coinfection levels and this increase is larger and more obvious at a high coinfection level. The opposite pattern is observed for single-mutant frequencies, which drop more with rare coinfection than with frequent coinfection. Both double and triple mutants show a drastic frequency drop at high coinfection levels and their frequencies remain close to zero with low coinfection rates when the selection parameter increases to 0.2. We observed that the virus system reaches equilibrium faster under stronger selection, which implies that increasing selection pressure will weaken the effect of complementation on maintaining population diversity and drive the population to purge mutations faster.
Interaction of complementation with pairwise epistasis:
Synergistic epistasis entails that mutations show a stronger effect in combination than the sum of their individual effects. Antagonistic epistasis (ɛ < 0) makes this effect weaker. When the epistatic effect is moderate, e.g., ɛ = ±0.3 as in Figure 1, C1–C5, Figure S3, and Figure S8 [known as magnitude epistasis (Weinreich et al. 2005)], the overall mean fitness is similar at different levels of coinfection, with slight differences among the cases of positive, negative, and no epistasis. Compared with the case of no epistasis, synergistic epistasis increases the frequency of wild type while antagonistic epistasis reduces its frequency, and their effects are generally enhanced as pc increases. For single mutants, both types of epistasis have subtle effects in the presence of strong or weak complementation (e.g., 0.9 or 0.1, respectively), but their effects become stronger at intermediate levels of coinfection (e.g., ∼0.5). Strong complementation has a much larger impact on the stationary frequencies of double and triple mutants when interacting with both types of epistasis as illustrated in Figure 1, C4 and C5. At high pc, the frequencies of double and triple mutants increase more with antagonistic epistasis than they are reduced with synergistic epistasis. When the epistatic effect increases dramatically (| ɛ | > 0.5), say ±0.8, we have sign epistasis (Weinreich et al. 2005); i.e., double and triple mutants can have much higher or lower fitness than single mutants, resulting in remarkably variable frequency trajectories before stabilizing (see Figures 1, D1–D5, and 2 and Figure S4). When mutations interact synergistically, the mean fitness equilibria shift to just above those without epistasis, and the higher the coinfection rate, the greater the mean fitness. Synergistic interaction affects the patterns of frequencies of wild type and single mutants in a manner similar to strong selection. But it has a much stronger effect in eliminating double mutants and triple mutants, as illustrated in Figure 2 where their frequencies are seen to decrease dramatically to close to zero. Antagonistic epistasis, in contrast, drives wild type and single mutants to elimination while double and triple mutants thrive in the population. Strong complementation slows down elimination of the wild type and single mutants. It is reasonable that double and triple mutants constitute the ultimate population as our simulation starts with pure single mutants, and double and triple mutants are superior to single mutants; random reassortment shuffles these segments and generates a lot more combinations of mutations that accumulate over time, while wild types and single mutants are removed. The frequencies of double mutants and triple mutants fluctuate dramatically and it takes a much longer time to reach the balanced state. Frequent coinfection tends to elevate the frequencies of triple mutants over double mutants as triple mutants are easily generated through recombination. As double and triple mutants are frequent and they have a slightly lower fitness than the wild type, the mean fitness with antagonistic epistasis is ∼0.02 lower than that with synergistic interaction.
Figure 2.—

(a) Trajectories of mean fitness changes under scenarios II, VI, and VII using stochastic simulation. (b, c, d, and e) Under scenarios II, VI, and VII the frequency trajectories of wild type, single mutants, double mutants, and triple mutants, respectively. Dashed lines represent the results assuming epistasis coefficient ɛ = 0.8. Dotted lines represent the results assuming no epistasis. Solid lines represent the results assuming epistasis coefficient ɛ = −0.8. Each color represents a coinfection fraction as labeled in the inset (a).
Interaction of complementation with higher-order epistasis:
We also compared the results from scenarios VIII and IX (described in Table 1) to assess how higher-order epistasis interacts with complementation and how this interaction affects the virus population. Second-order epistasis tends to have a much milder effect on mean fitness and frequencies of wild type and single mutants (see Figure 1, E1–E5, Figure S5, and Figure S9). The difference in wild-type proportions and double-mutant proportions between positive and negative second-order epistasis becomes slightly apparent as the coinfection fraction increases to 0.9. Second-order epistasis has a fairly strong effect on the fraction of triple mutants when the fraction of coinfections is high. This is reasonable as the initial virus population is composed of single mutants, and double mutants are less fit than single mutants or wild types. Few double mutants exist, leading to a low number of triple mutants even though they are strongly favored by selection.
Three-way interaction among complementation, selection, and epistasis:
To observe evolutionary trajectories under the simultaneous influence of complementation, selection, and epistasis, we iterated deterministically for 5000 generations, assuming mutation rate 0.01 and initial frequencies as in Table 1, using a series of parameters, pc ∈ {0.1, 0.3, 0.5, 0.7, 0.9}, s ∈ {0.0, 0.01, … , 0.49, 0.50}, and ɛ ∈ {−0.50, −0.48, … , 0.48, 0.50} (only magnitude epistasis is considered here; i.e., fitness decreases monotonically with increasing k, the number of mutant segments in a virus). From Figure S10, we observe that the overall mean fitness at equilibrium decreases as selection intensity increases and epistasis decreases, and strong complementation tends to weaken the effect of selection and synergistic epistasis on mean fitness. The equilibrium frequency of wild type increases gradually with the strength of selection, while frequent coinfection and antagonistic epistasis substantially reduce the speed at which the wild-type frequency increases, as shown in Figure 3. Figure 4 shows an intriguing frequency pattern of single mutants that starts at zero in the absence of selection against deleterious mutations, then sharply increases and attains its maximum when the selection coefficient increases to 0.05– ∼0.1, but gradually decreases as selection becomes stronger. Strong complementation and negative epistasis generally slow down the reduction of the single-mutant frequencies. Double mutants have an equilibrium frequency pattern similar to that of single mutants, “start low–increase–decrease,” although they disappear more quickly as the selection coefficient increases, as in Figure S11. Triple mutants take over the virus population when the selection coefficient is zero, as mutations without fitness cost continually accumulate. Then the frequencies of triple mutants decrease dramatically as stronger selection is applied (see Figure S12). Even strong complementation and antagonistic epistasis have moderate effects on selection against triple mutants. Through these three-way interaction patterns, we find that both selection and synergistic epistasis tend to eliminate mutations and combinations of mutations, whereas strong complementation and antagonistic epistasis drive the virus population in the opposite direction.
Figure 3.—

Wild-type frequency pattern under the three-way interaction among complementation, selection, and epistasis. (a, b, c, d, and e) The interaction patterns between selection and epistasis with the coinfection ratios 0.1, 0.3, 0.5, 0.7, and 0.9, respectively. Colors represent the values of wild-type frequency: red = 0.00, yellow 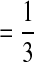 , green
, green 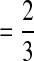 , and blue = 1.00.
, and blue = 1.00.
Figure 4.—

Single-mutant frequency pattern under the three-way interaction among complementation, selection, and epistasis. (a, b, c, d, and e) The interaction patterns between selection and epistasis with the coinfection ratios, 0.1, 0.3, 0.5, 0.7, and 0.9, respectively. Colors are as in Figure 3.
DISCUSSION
Coinfection generates two important phenomena in the viral life cycle, reassortment and complementation. It is commonly acknowledged that reassortment benefits the virus population by generating combinations of deleterious mutations, thus accelerating the removal of these mutations, whereas complementation is detrimental to the overall fitness of the group as it allows the population to harbor some deleterious mutations for long periods of time. Froissart et al. (2004) showed that complementation, rather than reassortment, is a major force in the evolution of viruses. What is the real role of complementation in viral evolution that allows viruses to sacrifice higher mean fitness, especially when complementation interacts with mutation, selection, or epistasis?
Here we extend the complementation model of Froissart et al. (2004) to multiple loci and incorporate the key evolutionary factors, mutation and epistasis, into the model. This more realistic model enables the assessment of the evolutionary impact of the interactions between complementation and mutation, selection, and epistasis as well as higher-order epistasis. Our simulation has confirmed Froissart's result that in the absence of mutation, a high coinfection ratio delays the clearance of deleterious mutants in a virus population. We also find that with mutation, the virus population achieves almost the same overall mean fitness at equilibrium irrespective of the levels of coinfection. Scrutinizing the population composition, we observe that strong complementation lowers the wild-type frequencies and increases the frequencies of mutants. Similar results are found in the case of selection and epistasis; complementation has a moderate effect on the overall ability of the virus population to reproduce, but plays a major role in the population composition. In this way, complementation increases the diversity and stability of a virus population substantially while maintaining similar or slightly lower fitness, which in turn improves the chance of survival of a virus population when coping with adverse environments.
In our modeling of recombination, we assume implicitly that reassortment is always random during replication and segments of two viruses are randomly incorporated into offspring genomes with probability 0.5 when coinfecting a cell. These assumptions are not necessarily met in reality. Preferential associations among RNA segments have been observed in several types of RNA viruses (Graham et al. 1987; Urquidi and Bishop 1992). And the abundance of offspring segments may also vary due to the lengths of virus segments, the concentration of RNA polymerase, etc. Although the effect of recombination is critical when interacting with epistasis, as shown in Bretscher et al. (2004), we focus here on the interaction between complementation and other factors, thus simplifying our model but without loss of generality. We also assume a fixed selection coefficient for all mutations in each simulation scenario, which, although not necessarily true in reality, helps to reveal the mechanism underlying the interaction between complementation and different types of epistasis.
To model sign epistasis, which has been detected in RNA viruses [e.g., HIV-1 (Mammano et al. 2000)] and is mostly induced by antiviral drugs, we modify the epistasis model of Desai et al. (2007) by taking the product of the number of mutations and the epistatic effect (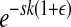 ) instead of raising the number of mutations to the power of the epistatic effect (
) instead of raising the number of mutations to the power of the epistatic effect (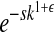 ). This enables us to contrast magnitude epistasis with sign epistasis.
). This enables us to contrast magnitude epistasis with sign epistasis.
In the deterministic simulation with sign epistasis, we observe that the frequencies of the wild type and the single mutants reach an equilibrium above zero when coinfection rates are >0.7, as illustrated in Figure S4, while in the stochastic simulation, these frequencies decrease to zero. We conducted further simulations to investigate this phenomenon under different initial conditions and observe that initial frequencies affect the equilibrium state of the deterministic system (see Figure S13), which clearly exhibits more than one equilibrium state when pc > 0.7. In the stochastic simulation, the stationary distribution with wild type absent appears to be strongly attracting, which produces the results in Figure 2. Experiments with other initial conditions also appear to result in distributions very similar to Figure 2.
We also observe that it takes a much longer time for a virus system to reach an equilibrium with antagonistic sign epistasis in the stochastic simulation than in the deterministic model. This may result from the strong fluctuations produced in the stochastic case that add variation to the frequencies of double and triple mutants. The variance of frequencies among the 100 simulated lineages is generally quite low (data not shown), probably owing to the congruent initial frequencies for all simulation replications.
Complementation with synergistic epistasis functions differently from that with antagonistic epistasis. Synergistic epistasis tends to strengthen the selection against the combinations of less-fit mutations, while frequent coinfection generally weakens the selection against all mutants, with the result that wild types and single mutants constitute a major proportion of the virus population, and the stronger the complementation is, the lower the frequency of wild types. In the presence of antagonistic epistasis, the pattern of frequency variation becomes complex. With magnitude antagonistic epistasis, [i.e., the magnitude of their effect may depend on the genetic background although the sign of the fitness effect of mutations is unconditional (Weinreich et al. 2005)], wild types and single mutants are slightly advantageous over double mutants. At equilibrium, wild type, single mutants, and double mutants all constitute a major fraction of the population when coinfection is frequent. With sign epistasis, i.e., both wild type and combinations of mutations are more advantageous than single mutants in terms of selection (Weinreich et al. 2005), at the beginning we observe a sharp increase in the frequencies of both wild type and double mutants and then a quick drop in single mutants through random reassortment and selection. As mutations accumulate over time in the population, the wild-type frequency decreases gradually until it is close to zero. The frequency of single mutants continues to drop as these are the least fit genotypes. The number of double mutants increases to a peak and decreases sharply as coinfection begins to play a major role, which generates a sharp rise in the frequency of triple mutants, although double mutants are slightly superior to triple mutants. The fluctuations in the frequencies of double and triple mutants before equilibrium are mainly due to stochasticity, which does not appear in the deterministic simulation. Higher coinfection rates lead to higher frequencies of triple mutants at stationarity, because with high pc, triple mutants have a greater chance of sharing normal gene products and transmitting their mutant genes to offspring; thus the viral system tends to produce more triple-mutant offspring. Therefore, the overall mean fitness of the population reaches almost the same equilibrium value at all levels of coinfection with antagonistic epistasis.
When complementation, selection, and epistasis interact simultaneously, the overall mean fitness decreases as selection, i.e., fitness cost, increases. Increase in the coinfection rate and decrease of the epistasis coefficient also reduce the mean fitness since both coinfection and antagonistic epistasis (ɛ < 0) result in accumulation of mutations, and double and triple mutants have lower fitness than wild type or single mutants although the effects are greater than additive. The “low–high–low” trajectories of single-mutant frequencies along the selection axis occur because when selection against deleterious mutations is very weak, e.g., 0.01, triple mutants become a major part of the population. As the selection strength increases to ∼0.1, the fitness of single mutants is slightly less than that of wild type but the fitness cost increases exponentially with the number of mutations; thus double and triple mutants are inferior and the frequency of single mutants reaches a peak. High levels of coinfection tend to expand this stage until the selection coefficient increases to 0.2, as complementation and random reassortment can mitigate the increased fitness cost for single mutants. Strong negative epistasis retards the increase in frequency of single mutants as it reduces the fitness cost of double and triple mutants. As selection strength continues to increase, the wild type becomes much more superior to single mutants, resulting in a gradual reduction in single-mutant frequencies. The frequency pattern of double mutants is similar to that of single mutants although double mutants are more sensitive to the increase of either selection strength or synergistic epistasis. Triple mutants start at the highest frequency with weak selection and then their frequency drops sharply as selection increases. Coinfection and strong negative epistasis tend to exert mild effects on the decrease of triple-mutant frequencies.
We anticipate that our results will have broad implications in viral disease management. Strong complementation results in weaker selection against deleterious mutations generally and may even favor mutants over wild type when complementation interacts with antagonistic epistasis. A large number of mutations persist in the population, and through random reassortment, multiple combinations of mutant segments can be generated. Both magnitude and sign antagonistic epistasis can exist (Mammano et al. 2000; Weinreich et al. 2005), which substantially reduces the efficacy of antiviral drugs. Therefore, reducing the coinfection ratio is critical in controlling the spread and variation of viruses. Isolation of virus-infected patients or organisms and alternating drug usage over a period of time are commonly used to reduce the coinfection level. We also suggest using multiple antiviral drugs simultaneously instead of one at a time. As antiviral drugs are designed mainly according to the structure of viral proteins, there is a greater chance that not all viruses are eliminated in a population with high pc when these kinds of therapies are applied, as some mutated viral molecules will not be affected by these specifically designed drugs. In addition, as viruses mutate with high frequency and have short generation times, these drug-resistant viruses will, with high probability, either mutate into fitter individuals or recombine with those carrying strongly favored genes, thus spreading their drug-resistant genes rapidly. This implies that using a single type of antiviral therapy will not work efficiently. It is more rational to design antiviral drugs for both wild-type proteins and potential mutant proteins and apply them simultaneously. In this way, the number of viruses will be reduced dramatically even if it is a stable system with strong complementation. When the density of viruses is very low, the chance of coinfection becomes minimal and the effect of complementation in reducing the selection against deleterious mutations is ameliorated.
Acknowledgments
We thank Marcel Salathé, Daniel Weissman, and Hua Tang for constructive comments on the manuscript. We are grateful to Raul Andino and Adam Lauring for discussions that stimulated us to work on this problem. We also thank Yidong Lei for optimizing the C code for stochastic simulation. He is supported by the National Natural Science Foundation of China (grant 40771084). This research project is supported in part by National Institutes of Health (NIH) grant GM28016 to M.W.F. and NIH grant RO1 GM073059 to Hua Tang.
APPENDIX: DETAILS OF DETERMINISTIC MODEL
Frequencies of mutants:
The single-mutant frequency  after the action of complementation, epistasis, selection, and reassortment is
after the action of complementation, epistasis, selection, and reassortment is
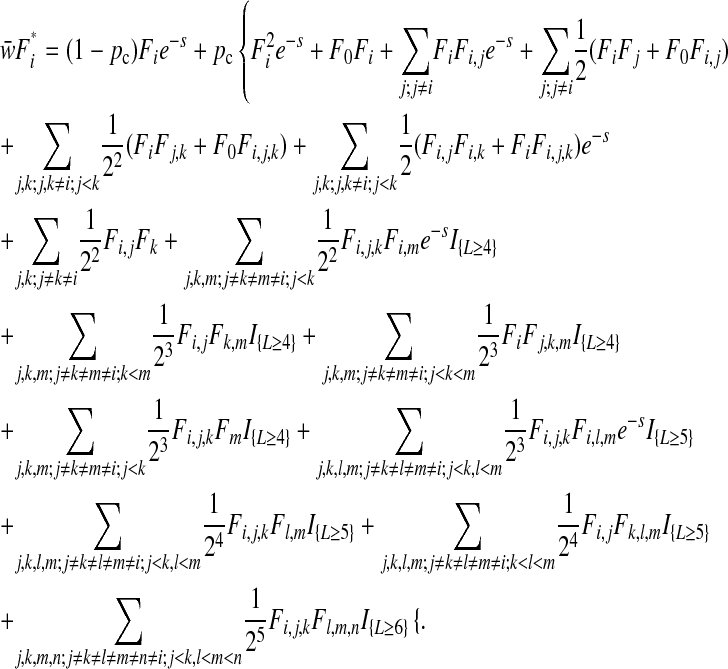 |
(A1) |
The double-mutant frequency 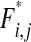 after the action of complementation, epistasis, selection, and reassortment is
after the action of complementation, epistasis, selection, and reassortment is
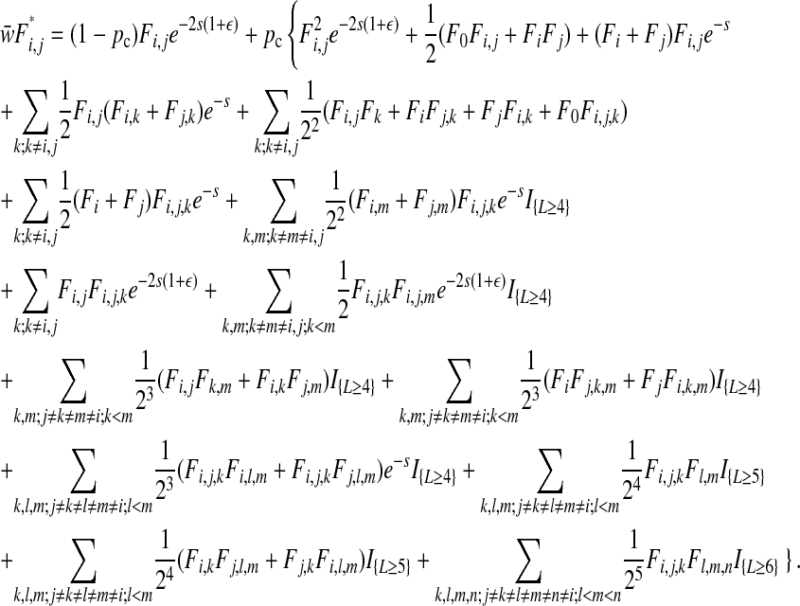 |
(A2) |
The triple-mutant frequency  after the action of complementation, epistasis, selection, and reassortment is
after the action of complementation, epistasis, selection, and reassortment is
 |
(A3) |
Integrated deterministic model:
We propose a model to integrate the effects of complementation, epistasis, selection, mutation, and random recombination and show it is equivalent to the previous deterministic model with the separation step. We denote the mean fitness of viral populations using this integrated model  and frequencies of mutants with F**. The frequency of wild-type genotype at the end of the viral replicating cycle is
and frequencies of mutants with F**. The frequency of wild-type genotype at the end of the viral replicating cycle is
 |
(A4) |
Similarly, the frequencies of mutants are
 |
(A5) |
After simple arithmetic operations, it is clear that F**'s are equal to F's.
Supporting information is available online at http://www.genetics.org/cgi/content/full/genetics.108.099796/DC1.
References
- Boni, M., J. Gog, V. Andreasen and M. M. Feldman, 2006. Epidemic dynamics and antigenic evolution in a single season of influenza A. Proc. Biol. Sci. 273 1307–1316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bretscher, M., C. Althaus, V. Müller and S. Bonhoeffer, 2004. Recombination in HIV and the evolution of drug resistance: For better or for worse? BioEssays 26 180–188. [DOI] [PubMed] [Google Scholar]
- Burch, C., and L. Chao, 2004. Epistasis and its relationship to canalization in the RNA virus phi6. Genetics 167 559–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burch, C., P. Turner and K. Hanley, 2003. Patterns of epistasis in RNA viruses: a review of the evidence from vaccine design. J. Evol. Biol. 16 1223–1235. [DOI] [PubMed] [Google Scholar]
- Chao, L., 1988. Evolution of sex in RNA viruses. J. Theor. Biol. 133 99–112. [DOI] [PubMed] [Google Scholar]
- Dennehy, J., and P. P. Turner, 2004. Reduced fecundity is the cost of cheating in RNA virus phi6. Proc. Biol. Sci. 271 2275–2282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desai, M., D. Weissman and M. Feldman, 2007. Evolution can favor antagonistic epistasis. Genetics 177 1001–1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drake, J., 2006. Chaos and order in spontaneous mutation. Genetics 173 1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drake, J., and J. Holland, 1999. Mutation rates among RNA viruses. Proc. Nat. Acad. Sci. USA 96 13910–13913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elena, S., P. Carrasco, J. Daròs and R. Sanjuán, 2006. Mechanisms of genetic robustness in RNA viruses. EMBO Rep. 7 168–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Froissart, R., C. Wilke, R. Montville, S. Remold, L. Chao et al., 2004. Co-infection weakens selection against epistatic mutations in RNA viruses. Genetics 168 9–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham, A., G. Kudesia, A. M. Allen and U. Desselberger, 1987. Reassortment of human rotavirus possessing genome rearrangements with bovine rotavirus: evidence for host cell selection. J. Gen. Virol. 68 115–122. [DOI] [PubMed] [Google Scholar]
- Mammano, F., V. Trouplin, V. Zennou and F. Clavel, 2000. Retracing the evolutionary pathways of human immunodeficiency virus type 1 resistance to protease inhibitors: virus fitness in the absence and in the presence of drugs. J. Virol. 74 8524–8531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moya, A., E. Holmes and F. Gonzalez-Candelas, 2004. The population genetics and evolutionary epidemiology of RNA viruses. Nat. Rev. Microbiol. 2 279–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olkkonen, V., and D. Bamford, 1989. Quantitation of the absorption and penetration stages of bacteriophage phi6 infection. Virology 171 229–238. [DOI] [PubMed] [Google Scholar]
- Sanjuán, R., J. Cuevas, A. Moya and S. Elena, 2005. Epistasis and the adaptability of an RNA virus. Genetics 170 1001–1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner, P., and L. Chao, 1999. Prisoner's dilemma in an RNA virus. Nature 398 441–443. [DOI] [PubMed] [Google Scholar]
- Turner, P., and L. Chao, 2003. Escape from prisoner's dilemma in RNA phage phi6. Am. Nat. 161 497–505. [DOI] [PubMed] [Google Scholar]
- Turner, P., C. Burch, K. Hanley and L. Chao, 1999. Hybrid frequencies confirm limit to coinfection in the RNA bacteriophage phi6. J. Virol. 73 2420–2424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urquidi, V., and D. H. L. Bishop, 1992. Non-random reassortment between the tripartite RNA genomes of La Crosse and snowshoe hare viruses. J. Gen. Virol. 73 2255–2265. [DOI] [PubMed] [Google Scholar]
- Weinreich, D., R. Watson and L. Chao, 2005. Sign epistasis and genetic constraint on evolutionary trajectories. Evolution 59 1165–1174. [PubMed] [Google Scholar]
- Wilke, C., and I. Novella, 2003. Phenotypic mixing and hiding may contribute to memory in viral quasispecies. BMC Microbiol. 3 11. [DOI] [PMC free article] [PubMed] [Google Scholar]